
The Role of Tripartite Motif Family Proteins in Chronic Liver Diseases: Molecular Mechanisms and Therapeutic Potential

Abstract
:1. Introduction
2. TRIM Family
2.1. Structure of TRIM Proteins
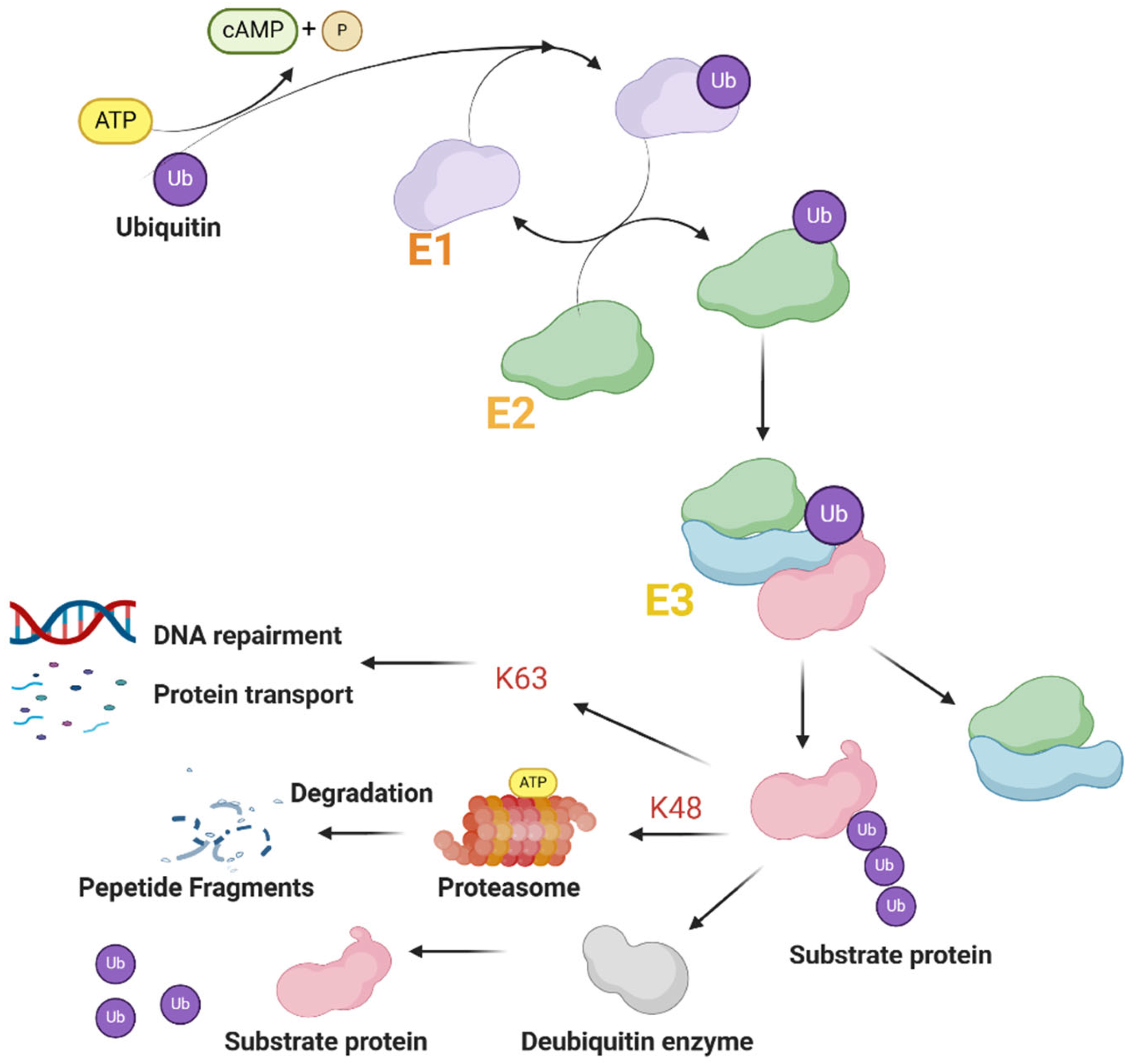
2.2. Function of TRIM Proteins
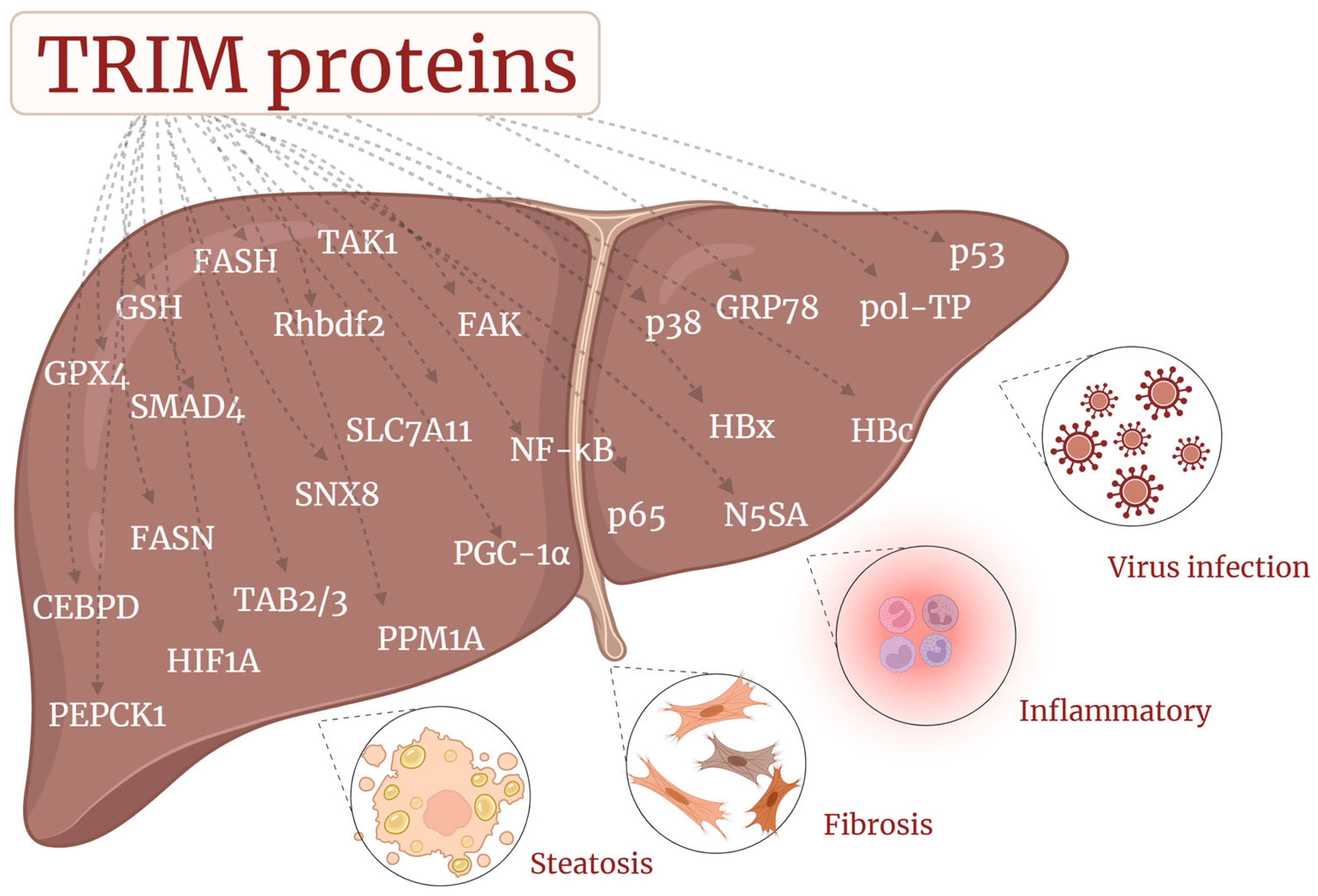
3. TRIM Proteins in CLD
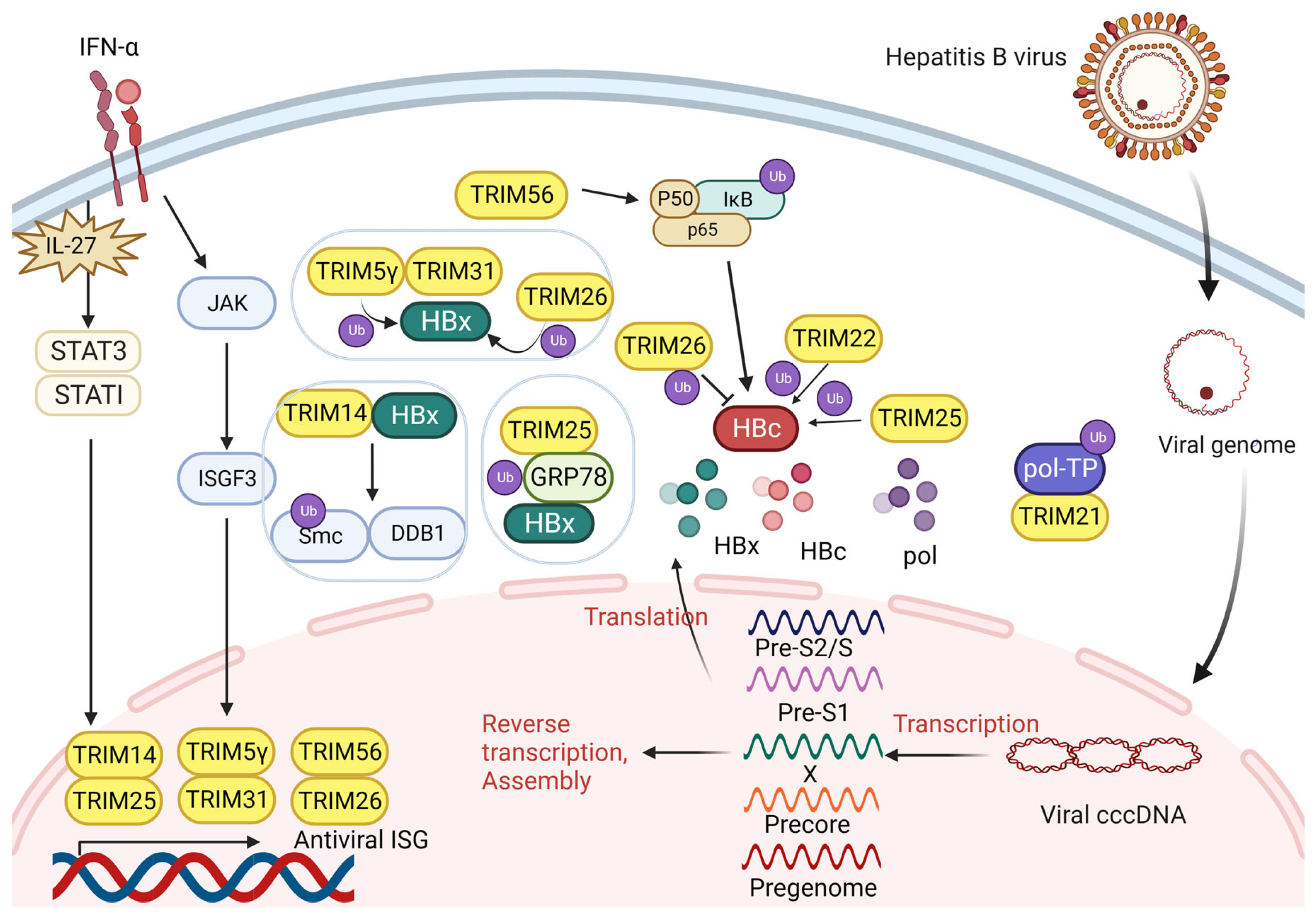
3.1. TRIM and Hepatitis B Virus Infection
3.1.1. TRIM Proteins Interact with HBx
3.1.2. TRIM Proteins Interact with HBV Pol
3.1.3. TRIM Proteins Interact with HBc
3.2. TRIM and Hepatitis C Virus Infection
3.3. TRIM and MAFLD/MASH
3.4. TRIM and Liver Fibrosis
4. Clinical Perspective
4.1. TRIM in HBV
4.2. TRIM in HCV
4.3. TRIM in MAFLD/MASH
4.4. TRIM in Liver Fibrosis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef]
- Sharma, A.; Nagalli, S. Chronic Liver Disease. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2024. [Google Scholar]
- Post, A.; Nagendra, S. Reactivation of hepatitis B: Pathogenesis and clinical implications. Curr. Infect. Dis. Rep. 2009, 11, 113–119. [Google Scholar] [CrossRef]
- Gower, E.; Estes, C.; Blach, S.; Razavi-Shearer, K.; Razavi, H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef]
- Chisari, F.V.; Isogawa, M.; Wieland, S.F. Pathogenesis of hepatitis B virus infection. Pathol. Biol. 2010, 58, 258–266. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Lai, J.C.; Wong, G.L.; Wong, V.W.; Yip, T.C. Can we use old NAFLD data under the new MASLD definition? J. Hepatol. 2024, 80, e54–e56. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Dufour, J.F.; Anstee, Q.M.; Bugianesi, E.; Harrison, S.; Loomba, R.; Paradis, V.; Tilg, H.; Wong, V.W.; Zelber-Sagi, S. Current therapies and new developments in NASH. Gut 2022, 71, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D. Liver fibrosis: Common mechanisms and antifibrotic therapies. Clin. Res. Hepatol. Gastroenterol. 2015, 39 (Suppl. S1), S51–S59. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Wan, T.; Li, X.; Li, Y. The role of TRIM family proteins in autophagy, pyroptosis, and diabetes mellitus. Cell Biol. Int. 2021, 45, 913–926. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, L.; Cui, S.; Wu, S. Tripartite motif protein 6 promotes hepatocellular carcinoma progression via multiple pathways. Turk. J. Med. Sci. 2023, 53, 1032–1044. [Google Scholar]
- Vali, Y.; Lee, J.; Boursier, J.; Petta, S.; Wonders, K.; Tiniakos, D.; Bedossa, P.; Geier, A.; Francque, S.; Allison, M.; et al. Biomarkers for staging fibrosis and non-alcoholic steatohepatitis in non-alcoholic fatty liver disease (the LITMUS project): A comparative diagnostic accuracy study. Lancet Gastroenterol. Hepatol. 2023, 8, 714–725. [Google Scholar] [CrossRef]
- Honma, Y.; Shimizu, S.; Takehara, T.; Harada, M. Sorafenib enhances proteasome inhibitor-induced cell death via inactivation of Akt and stress-activated protein kinases. J. Gastroenterol. 2014, 49, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Massiah, M.A.; Simmons, B.N.; Short, K.M.; Cox, T.C. Solution structure of the RBCC/TRIM B-box1 domain of human MID1: B-box with a RING. J. Mol. Biol. 2006, 358, 532–545. [Google Scholar] [CrossRef]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of 'single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Zeng, X.; Deng, X.; Ni, Y.; Bi, H.; Jiang, M.; Wang, D.; Dong, P.; Xiao, Y.; Jiang, M. LPS inhibits TRIM65 expression in macrophages and C57BL/6J mouse by activating the ERK1/2 signaling pathway. Exp. Ther. Med. 2023, 25, 188. [Google Scholar] [CrossRef]
- Minucci, S.; Maccarana, M.; Cioce, M.; De Luca, P.; Gelmetti, V.; Segalla, S.; Di Croce, L.; Giavara, S.; Matteucci, C.; Gobbi, A.; et al. Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol. Cell 2000, 5, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM gene expression in response to interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef]
- Liu, S.; Bi, H.; Jiang, M.; Chen, Y.; Jiang, M. An update on the role of TRIM/NLRP3 signaling pathway in atherosclerosis. Biomed. Pharmacother. 2023, 160, 114321. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liang, L.; Jin, Y.; Yin, Y. The TRIM14 PRYSPRY domain mediates protein interaction via its basic interface. FEBS Lett. 2019, 593, 1122–1129. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Heikel, G.; Trubitsyna, M.; Kubik, P.; Nowak, J.S.; Webb, S.; Granneman, S.; Spanos, C.; Rappsilber, J.; Castello, A.; et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 2017, 15, 105. [Google Scholar] [CrossRef]
- Short, K.M.; Cox, T.C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 2006, 281, 8970–8980. [Google Scholar] [CrossRef]
- Jain, K.; Fraser, C.S.; Marunde, M.R.; Parker, M.M.; Sagum, C.; Burg, J.M.; Hall, N.; Popova, I.K.; Rodriguez, K.L.; Vaidya, A.; et al. Characterization of the plant homeodomain (PHD) reader family for their histone tail interactions. Epigenet. Chromatin. 2020, 13, 3. [Google Scholar] [CrossRef]
- Zapata, J.M.; Martínez-García, V.; Lefebvre, S. Phylogeny of the TRAF/MATH domain. Adv. Exp. Med. Biol. 2007, 597, 1–24. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Asmamaw, M.D.; Liu, Y.; Zheng, Y.C.; Shi, X.J.; Liu, H.M. Skp2 in the ubiquitin-proteasome system: A comprehensive review. Med. Res. Rev. 2020, 40, 1920–1949. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, F.; Zhang, X.; Lin, H.K.; Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct. Target Ther. 2021, 6, 422. [Google Scholar] [CrossRef]
- Song, L.; Luo, Z.Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Chen, Y.; Wu, Y.; Wang, Z.; Zhou, A.; Zhang, S.; Lin, K.; Aldape, K.; Majumder, S.; Lu, Z.; et al. Tumour suppressor TRIM33 targets nuclear β-catenin degradation. Nat. Commun. 2015, 6, 6156. [Google Scholar] [CrossRef]
- Rajsbaum, R.; García-Sastre, A.; Versteeg, G.A. TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 2014, 426, 1265–1284. [Google Scholar] [CrossRef] [PubMed]
- van Huizen, M.; Kikkert, M. The Role of Atypical Ubiquitin Chains in the Regulation of the Antiviral Innate Immune Response. Front. Cell Dev. Biol. 2019, 7, 392. [Google Scholar] [CrossRef]
- Evans, J.D.; Seeger, C. Differential effects of mutations in NS4B on West Nile virus replication and inhibition of interferon signaling. J. Virol. 2007, 81, 11809–11816. [Google Scholar] [CrossRef] [PubMed]
- Penna, A.; Chisari, F.V.; Bertoletti, A.; Missale, G.; Fowler, P.; Giuberti, T.; Fiaccadori, F.; Ferrari, C. Cytotoxic T lymphocytes recognize an HLA-A2-restricted epitope within the hepatitis B virus nucleocapsid antigen. J. Exp. Med. 1991, 174, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef] [PubMed]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Zhang, N.; Yu, T.; Ma, L.; Li, Q.; Wang, X.; Yuan, D.; Kong, D.; Liu, X.; Hu, W.; et al. Hepatitis B virus X protein promotes MAN1B1 expression by enhancing stability of GRP78 via TRIM25 to facilitate hepatocarcinogenesis. Br. J. Cancer 2023, 128, 992–1004. [Google Scholar] [CrossRef]
- Tan, G.; Xiao, Q.; Song, H.; Ma, F.; Xu, F.; Peng, D.; Li, N.; Wang, X.; Niu, J.; Gao, P.; et al. Type I IFN augments IL-27-dependent TRIM25 expression to inhibit HBV replication. Cell. Mol. Immunol. 2018, 15, 272–281. [Google Scholar] [CrossRef]
- Tan, G.; Xu, F.; Song, H.; Yuan, Y.; Xiao, Q.; Ma, F.; Qin, F.X.; Cheng, G. Identification of TRIM14 as a Type I IFN-Stimulated Gene Controlling Hepatitis B Virus Replication by Targeting HBx. Front. Immunol. 2018, 9, 1872. [Google Scholar] [CrossRef]
- Tan, G.; Yi, Z.; Song, H.; Xu, F.; Li, F.; Aliyari, R.; Zhang, H.; Du, P.; Ding, Y.; Niu, J.; et al. Type-I-IFN-Stimulated Gene TRIM5γ Inhibits HBV Replication by Promoting HBx Degradation. Cell Rep. 2019, 29, 3551–3563.e3. [Google Scholar] [CrossRef]
- Clark, D.N.; Flanagan, J.M.; Hu, J. Mapping of Functional Subdomains in the Terminal Protein Domain of Hepatitis B Virus Polymerase. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef]
- Mu, T.; Zhao, X.; Zhu, Y.; Fan, H.; Tang, H. The E3 Ubiquitin Ligase TRIM21 Promotes HBV DNA Polymerase Degradation. Viruses 2020, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, Y.; Nishizawa, T.; Nishitsuji, H.; Morita, H.; Yamagata, T.; Onomura, D.; Murata, K. TRIM26 positively affects hepatitis B virus replication by inhibiting proteasome-dependent degradation of viral core protein. Sci. Rep. 2023, 13, 13584. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Duan, Z.; Xu, W.; Xiong, S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 2009, 50, 424–433. [Google Scholar] [CrossRef]
- Tian, X.; Dong, H.; Lai, X.; Ou, G.; Cao, J.; Shi, J.; Xiang, C.; Wang, L.; Zhang, X.; Zhang, K.; et al. TRIM56 impairs HBV infection and replication by inhibiting HBV core promoter activity. Antiviral. Res. 2022, 207, 105406. [Google Scholar] [CrossRef]
- Lin, Y.C.; Hsu, E.C.; Ting, L.P. Repression of hepatitis B viral gene expression by transcription factor nuclear factor-kappaB. Cell Microbiol. 2009, 11, 645–660. [Google Scholar] [CrossRef]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef]
- Suzuki, T.; Aizaki, H.; Murakami, K.; Shoji, I.; Wakita, T. Molecular biology of hepatitis C virus. J. Gastroenterol. 2007, 42, 411–423. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Li, C.; Wu, Y.; Guo, L.; Peng, C.; Huang, Y.; Cheng, G.; Qin, F.X. TRIM14 inhibits hepatitis C virus infection by SPRY domain-dependent targeted degradation of the viral NS5A protein. Sci. Rep. 2016, 6, 32336. [Google Scholar] [CrossRef]
- Qashqari, H.; Al-Mars, A.; Chaudhary, A.; Abuzenadah, A.; Damanhouri, G.; Alqahtani, M.; Mahmoud, M.; El Sayed Zaki, M.; Fatima, K.; Qadri, I. Understanding the molecular mechanism(s) of hepatitis C virus (HCV) induced interferon resistance. Infect. Genet. Evol. 2013, 19, 113–119. [Google Scholar] [CrossRef]
- Yang, C.; Zhao, X.; Sun, D.; Yang, L.; Chong, C.; Pan, Y.; Chi, X.; Gao, Y.; Wang, M.; Shi, X.; et al. Interferon alpha (IFNα)-induced TRIM22 interrupts HCV replication by ubiquitinating NS5A. Cell. Mol. Immunol. 2016, 13, 94–102. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, G.; Li, Q.; Han, L.; Hu, X.; Guo, Y.; Tao, W.; Zhao, X.; Guo, M.; Gan, T.; et al. TRIM26 is a critical host factor for HCV replication and contributes to host tropism. Sci. Adv. 2021, 7, eabd9732. [Google Scholar] [CrossRef]
- Khan, R.; Ali, A.; Bibi, S.; Rafique, S.; Idrees, M.; Halim, S.A.; Waqas, M.; Bahadar, H.; Uddin, J.; Khan, A.; et al. Expression Profiling of the Tripartite Motif Family Genes in Chronic Hepatitis C Patients. ACS Omega 2023, 8, 25370–25377. [Google Scholar] [CrossRef]
- Chitturi, S.; Farrell, G.C. Etiopathogenesis of nonalcoholic steatohepatitis. Semin. Liver Dis. 2001, 21, 27–41. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, H.; Yao, J.; Jin, W.; Pan, H.; Pan, Z.; Xie, D.; Xie, D. TRIM59 promotes steatosis and ferroptosis in non-alcoholic fatty liver disease via enhancing GPX4 ubiquitination. Hum. Cell 2023, 36, 209–222. [Google Scholar] [CrossRef]
- Jiang, M.X.; Hong, X.; Liao, B.B.; Shi, S.Z.; Lai, X.F.; Zheng, H.Y.; Xie, L.; Wang, Y.; Wang, X.L.; Xin, H.B.; et al. Expression profiling of TRIM protein family in THP1-derived macrophages following TLR stimulation. Sci. Rep. 2017, 7, 42781. [Google Scholar] [CrossRef]
- An, Y.; Ni, Y.; Xu, Z.; Shi, S.; He, J.; Liu, Y.; Deng, K.Y.; Fu, M.; Jiang, M.; Xin, H.B. TRIM59 expression is regulated by Sp1 and Nrf1 in LPS-activated macrophages through JNK signaling pathway. Cell Signal 2020, 67, 109522. [Google Scholar] [CrossRef]
- Jin, Z.; Zhu, Z.; Liu, S.; Hou, Y.; Tang, M.; Zhu, P.; Tian, Y.; Li, D.; Yan, D.; Zhu, X. TRIM59 Protects Mice From Sepsis by Regulating Inflammation and Phagocytosis in Macrophages. Front. Immunol. 2020, 11, 263. [Google Scholar] [CrossRef]
- Tang, T.; Li, P.; Zhou, X.; Wang, R.; Fan, X.; Yang, M.; Qi, K. The E3 Ubiquitin Ligase TRIM65 Negatively Regulates Inflammasome Activation through Promoting Ubiquitination of NLRP3. Front. Immunol. 2021, 12, 741839. [Google Scholar] [CrossRef]
- Yan, F.J.; Zhang, X.J.; Wang, W.X.; Ji, Y.X.; Wang, P.X.; Yang, Y.; Gong, J.; Shen, L.J.; Zhu, X.Y.; Huang, Z.; et al. The E3 ligase tripartite motif 8 targets TAK1 to promote insulin resistance and steatohepatitis. Hepatology 2017, 65, 1492–1511. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Lin, Z.B.; Yang, P.J.; Xu, H.; Duan, J.L.; Ruan, B.; Song, P.; Liu, J.J.; Yue, Z.S.; et al. Tripartite motif 16 ameliorates nonalcoholic steatohepatitis by promoting the degradation of phospho-TAK1. Cell Metab. 2021, 33, 1372–1388.e7. [Google Scholar] [CrossRef]
- Yao, X.; Dong, R.; Hu, S.; Liu, Z.; Cui, J.; Hu, F.; Cheng, X.; Wang, X.; Ma, T.; Tian, S.; et al. Tripartite motif 38 alleviates the pathological process of NAFLD-NASH by promoting TAB2 degradation. J. Lipid Res. 2023, 64, 100382. [Google Scholar] [CrossRef]
- Zhang, K.; Yang, C.; Zhou, X.; Liang, J.; Guo, J.; Li, M.; Zhang, Y.; Shao, S.; Sun, P.; Li, K.; et al. TRIM21 ameliorates hepatic glucose and lipid metabolic disorders in type 2 diabetes mellitus by ubiquitination of PEPCK1 and FASN. Cell Mol. Life Sci. 2023, 80, 168. [Google Scholar] [CrossRef]
- Jiang, S.; Minter, L.C.; Stratton, S.A.; Yang, P.; Abbas, H.A.; Akdemir, Z.C.; Pant, V.; Post, S.; Gagea, M.; Lee, R.G.; et al. TRIM24 suppresses development of spontaneous hepatic lipid accumulation and hepatocellular carcinoma in mice. J. Hepatol. 2015, 62, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K.; Sharan, S.; Klarmann, K.D.; Zhang, Y.; Coppola, V.; Summers, G.H.; Roger, T.; Morrison, D.K.; Keller, J.R.; Sterneck, E. FBXW7α attenuates inflammatory signalling by downregulating C/EBPδ and its target gene Tlr4. Nat. Commun. 2013, 4, 1662. [Google Scholar] [CrossRef]
- Xu, M.; Tan, J.; Liu, X.; Han, L.; Ge, C.; Zhang, Y.; Luo, F.; Wang, Z.; Xue, X.; Xiong, L.; et al. Tripartite motif containing 26 prevents steatohepatitis progression by suppressing C/EBPδ signalling activation. Nat. Commun. 2023, 14, 6384. [Google Scholar] [CrossRef]
- Hu, Y.; He, W.; Huang, Y.; Xiang, H.; Guo, J.; Che, Y.; Cheng, X.; Hu, F.; Hu, M.; Ma, T.; et al. Fatty Acid Synthase-Suppressor Screening Identifies Sorting Nexin 8 as a Therapeutic Target for NAFLD. Hepatology 2021, 74, 2508–2525. [Google Scholar] [CrossRef]
- Xu, M.; Tan, J.; Dong, W.; Zou, B.; Teng, X.; Zhu, L.; Ge, C.; Dai, X.; Kuang, Q.; Zhong, S.; et al. The E3 ubiquitin-protein ligase Trim31 alleviates non-alcoholic fatty liver disease by targeting Rhbdf2 in mouse hepatocytes. Nat. Commun. 2022, 13, 1052. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, C.; Huang, M.; Lin, J.; Fan, X.; Ni, T. TRIM26 Induces Ferroptosis to Inhibit Hepatic Stellate Cell Activation and Mitigate Liver Fibrosis through Mediating SLC7A11 Ubiquitination. Front. Cell Dev. Biol. 2021, 9, 644901. [Google Scholar] [CrossRef]
- Lee, E.J.; Hwang, I.; Lee, J.Y.; Park, J.N.; Kim, K.C.; Kim, I.; Moon, D.; Park, H.; Lee, S.Y.; Kim, H.S.; et al. Hepatic stellate cell-specific knockout of transcriptional intermediary factor 1γ aggravates liver fibrosis. J. Exp. Med. 2020, 217, e20190402. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lan, Q.; Li, W.; Yang, L.; You, J.; Zhang, Y.M.; Ni, W. Tripartite motif protein 52 (TRIM52) promoted fibrosis in LX-2 cells through PPM1A-mediated Smad2/3 pathway. Cell Biol. Int. 2020, 44, 108–116. [Google Scholar] [CrossRef]
- Xie, H.; Xie, D.; Zhang, J.; Jin, W.; Li, Y.; Yao, J.; Pan, Z.; Xie, D. ROS/NF-κB Signaling Pathway-Mediated Transcriptional Activation of TRIM37 Promotes HBV-Associated Hepatic Fibrosis. Mol. Ther. Nucleic Acids 2020, 22, 114–123. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, R.; Li, F.; Lin, S.; Wang, J. Integrated analysis and validation of TRIM23/p53 signaling pathway in hepatic stellate cells ferroptosis and liver fibrosis. Dig. Liver Dis. 2024, 56, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Wang, J.; Zhang, T.; Ding, Q.; Jia, X.; Zhao, X.; Dong, J.; Yang, X.; Lu, S.; Zhang, C.; et al. Targeting Src family kinase member Fyn by Saracatinib attenuated liver fibrosis in vitro and in vivo. Cell Death Dis. 2020, 11, 118. [Google Scholar] [CrossRef]
- Yang, C.; Jin, X.; Liu, X.; Wu, G.; Yang, W.; Pang, B.; Jiang, J.; Liao, D.; Zhang, Y. TRIM15 forms a regulatory loop with the AKT/FOXO1 axis and LASP1 to modulate the sensitivity of HCC cells to TKIs. Cell Death Dis. 2023, 14, 47. [Google Scholar] [CrossRef]
- Medrano, L.M.; Rallón, N.; Berenguer, J.; Jiménez-Sousa, M.A.; Soriano, V.; Aldámiz-Echevarria, T.; Fernández-Rodríguez, A.; García, M.; Tejerina, F.; Martínez, I.; et al. Relationship of TRIM5 and TRIM22 polymorphisms with liver disease and HCV clearance after antiviral therapy in HIV/HCV coinfected patients. J. Transl. Med. 2016, 14, 257. [Google Scholar] [CrossRef]
- Cornberg, M.; Lok, A.S.; Terrault, N.A.; Zoulim, F. Guidance for design and endpoints of clinical trials in chronic hepatitis B—Report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology 2019, 71, 1070–1092. [Google Scholar] [CrossRef]
- Luo, H.; Tan, G.; Hu, X.; Li, Y.; Lei, D.; Zeng, Y.; Qin, B. Triple motif proteins 19 and 38 correlated with treatment responses and HBsAg clearance in HBeAg-negative chronic hepatitis B patients during peg-IFN-α therapy. Virol. J. 2023, 20, 161. [Google Scholar] [CrossRef]
- Luo, M.; Hou, J.; Mai, H.; Chen, J.; Chen, H.; Zhou, B.; Hou, J.; Jiang, D.K. TRIM26 inhibits hepatitis B virus replication by promoting HBx degradation and TRIM26 genetic polymorphism predicts PegIFNα treatment response of HBeAg-positive chronic hepatitis B Patients. Aliment. Pharmacol. Ther. 2022, 56, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Bourlière, M.; Gordon, S.C.; Flamm, S.L.; Cooper, C.L.; Ramji, A.; Tong, M.; Ravendhran, N.; Vierling, J.M.; Tran, T.T.; Pianko, S.; et al. Sofosbuvir, Velpatasvir, and Voxilaprevir for Previously Treated HCV Infection. N. Engl. J. Med. 2017, 376, 2134–2146. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Gonçales, F.L., Jr.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef]
- Sadeghi, F.; Bokharaei-Salim, F.; Salehi-Vaziri, M.; Monavari, S.H.; Alavian, S.M.; Salimi, S.; Vahabpour, R.; Keyvani, H. Associations between human TRIM22 gene expression and the response to combination therapy with Peg-IFNα-2a and ribavirin in Iranian patients with chronic hepatitis C. J. Med. Virol. 2014, 86, 1499–1506. [Google Scholar] [CrossRef]
- Naveed, M.; Ali, A.; Sheikh, N.; Rafique, S.; Idrees, M. Expression of TRIM22 mRNA in chronic hepatitis C patients treated with direct-acting antiviral drugs. Apmis 2020, 128, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, S.; Irani, N.; Sepahi, A.A.; Sakhaee, F.; Jamnani, F.R.; Vaziri, F.; Siadat, S.D.; Fateh, A. Evaluation of TRIM5 and TRIM22 polymorphisms on treatment responses in Iranian patients with chronic hepatitis C virus infection. Gene 2018, 676, 95–100. [Google Scholar] [CrossRef]
- Yang, P.; Liang, Y.; Luo, Y.; Li, Z.; Wen, Y.; Shen, J.; Li, R.; Zheng, H.; Gu, H.F.; Xia, N. Liraglutide ameliorates nonalcoholic fatty liver disease in diabetic mice via the IRS2/PI3K/Akt signaling pathway. Diabetes Metab. Syndr. Obes. 2019, 12, 1013–1021. [Google Scholar] [CrossRef]
- Syed-Abdul, M.M.; Parks, E.J.; Gaballah, A.H.; Bingham, K.; Hammoud, G.M.; Kemble, G.; Buckley, D.; McCulloch, W.; Manrique-Acevedo, C. Fatty Acid Synthase Inhibitor TVB-2640 Reduces Hepatic de Novo Lipogenesis in Males with Metabolic Abnormalities. Hepatology 2020, 72, 103–118. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, P.; Zhou, J.; Liao, R.; Che, Y.; Gao, M.M.; Sun, J.; Cai, J.; Cheng, X.; Huang, Y.; et al. TNFAIP3 Interacting Protein 3 Overexpression Suppresses Nonalcoholic Steatohepatitis by Blocking TAK1 Activation. Cell Metab. 2020, 31, 726–740.e8. [Google Scholar] [CrossRef]
- Lee, E.J.; Hwang, I.; Lee, J.Y.; Park, J.N.; Kim, K.C.; Kim, G.H.; Kang, C.M.; Kim, I.; Lee, S.Y.; Kim, H.S. Hepatocyte Growth Factor Improves the Therapeutic Efficacy of Human Bone Marrow Mesenchymal Stem Cells via RAD51. Mol. Ther. 2018, 26, 845–859. [Google Scholar] [CrossRef]
- Aldrovandi, M.; Conrad, M. Ferroptosis: The Good, the Bad and the Ugly. Cell Res. 2020, 30, 1061–1062. [Google Scholar] [CrossRef]
- Pan, Q.; Luo, Y.; Xia, Q.; He, K. Ferroptosis and Liver Fibrosis. Int. J. Med. Sci. 2021, 18, 3361–3366. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Ren, Z.; Yan, D.; Li, G. The role of TRIM family in metabolic associated fatty liver disease. Front. Endocrinol. 2023, 14, 1210330. [Google Scholar] [CrossRef]
- Hu, X.; Tang, Z.; Ma, S.; Yu, Y.; Chen, X.; Zang, G. Tripartite motif-containing protein 7 regulates hepatocellular carcinoma cell proliferation via the DUSP6/p38 pathway. Biochem. Biophys. Res. Commun. 2019, 511, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wei, W.; Sun, Y. PROTAC Degraders with Ligands Recruiting MDM2 E3 Ubiquitin Ligase: An Updated Perspective. Acta Mater. Med. 2022, 1, 244–259. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases | TRIM Members | Functions | Molecular Mechanisms |
|---|---|---|---|
| HBV | TRIM26 |
|
|
| TRIM11 |
|
| |
| HCV | TRIM59 |
|
|
| MAFLD/MASH | TRIM8 |
|
|
| TRIM67 |
|
| |
| TRIM15 |
|
| |
| Liver fibrosis | TRIM52 |
|
|
| TRIM37 |
|
| |
| TRIM23 |
|
| |
| TRIM47 |
|
|
| Diseases | TRIM Members | Functions | Molecular Mechanisms |
|---|---|---|---|
| HBV | TRIM25 |
|
|
| TRIM21 |
|
| |
| TRIM14 |
|
| |
| TRIM5γ |
|
| |
| TRIM22 |
|
| |
| TRIM56 |
|
| |
| HCV | TRIM14 |
|
|
| TRIM22 |
|
| |
| TRIM26 |
|
| |
| MAFLD/MASH | TRIM24 |
| Unclear |
| TRIM28 |
|
| |
| TRIM16 |
|
| |
| TRIM26 |
|
| |
| TRIM31 |
|
| |
| TRIM38 |
|
| |
| TRIM65 |
|
| |
| Liver fibrosis | TRIM26 |
|
|
| TRIM33 |
|
| |
| TRIM24 |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Chen, Y.; Chen, Y.; Jiang, M. The Role of Tripartite Motif Family Proteins in Chronic Liver Diseases: Molecular Mechanisms and Therapeutic Potential. Biomolecules 2024, 14, 1038. https://doi.org/10.3390/biom14081038
Cao X, Chen Y, Chen Y, Jiang M. The Role of Tripartite Motif Family Proteins in Chronic Liver Diseases: Molecular Mechanisms and Therapeutic Potential. Biomolecules. 2024; 14(8):1038. https://doi.org/10.3390/biom14081038
Chicago/Turabian StyleCao, Xiwen, Yinni Chen, Yuanli Chen, and Meixiu Jiang. 2024. "The Role of Tripartite Motif Family Proteins in Chronic Liver Diseases: Molecular Mechanisms and Therapeutic Potential" Biomolecules 14, no. 8: 1038. https://doi.org/10.3390/biom14081038