

Gene Therapy for Retinitis Pigmentosa: Current Challenges and New Progress


Abstract
1. Introduction
1.1. Retinitis Pigmentosa
1.2. Gene-Editing Technologies and CRISPR-Cas Systems
2. Mutation Type and Compatible Therapeutic Approaches
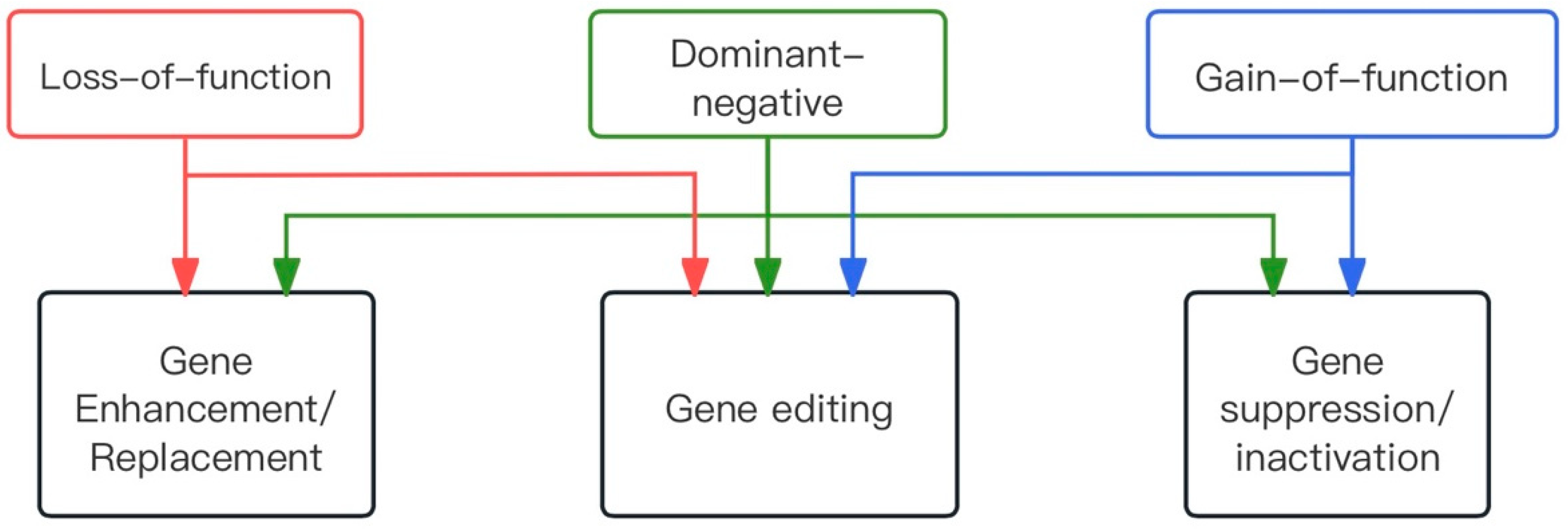
2.1. Types of Dominant Mutations
2.2. Gene Therapy Approaches
2.2.1. Therapeutic Strategy
2.2.2. Viral Vectors
2.2.3. Tools for Gene Editing
2.2.4. Mode of Administration of Vectors
3. Genome Editing for ADRP
3.1. Rhodopsin P23H (RHO-P23H)
3.2. Pre-mRNA Processing Factor 31 (PRPF31)
3.3. Retinitis Pigmentosa 1 (RP1)
3.4. Cone-Rod Homeobox (CRX)
4. Genome Editing for XLRP
4.1. Retinitis Pigmentosa GTPase Regulator (RPGR)
4.2. RP2
5. Genome Editing for ARRP
5.1. Phosphodiesterase 6 (PDE6) Gene Complex
5.1.1. Phosphodiesterase 6A (PDE6A)

{kind=link}
{kind=link}
| Target Gene | Type of Method | Outcome | Sample Size | References |
|---|---|---|---|---|
| PDE6A | Serotype 8 capsid mutant adeno-associated viruses was used to deliver Pde6a | First successful amplification of Pde6a in a large animal model of RP | 10 | [83] |
| Whole mount analysis Immunoblot analysis Electroretinograms | AAV2/8(Y733F)-Rho-Pde6α is an effective gene therapy for mid-stage patients with partial peripheral vision loss | (-) * | [85] | |
| PDE6B | Pde6b-associated RP mouse retinas were edited in vivo using a dual AAV system with PESpRY through a non-NGG PAM (GTG) | Vision loss from RP-related gene mutations can be prevented by unconstrained in vivo prime editing in degenerating retinas. Family history of affected females with RP does not exclude X-linked disease | (-) * | [86] |
| USH2A | Locus-specific RNA-Cas9 ribonucleoproteins were used with subsequent homologous recombination repair induced by an engineered template supply | Successful in vitro mutation repair | (-) * | [87,88] |
| RPE65 | Subretinal injections of the optimized dual-AAV split-PE3 were administered to rd12 mice with the inherited retinal disease Leber congenital amaurosis | Prime editors corrected the pathogenic mutation with up to 16% efficiency, precisely restoring RPE65 expression, rescuing retinal and visual function, and preserving photoceptors, without detectable off-target edits | (-) * | [89,90] |
5.1.2. Phosphodiesterase 6B (PDE6B)
5.2. USH2A
5.3. Retinal Pigment Epithelium 65 (RPE65) Gene
6. Conclusions/Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Neal, T.B.; Luther, E.E. Retinitis Pigmentosa. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yang, J.; Xiong, Y.; Zhu, X.; Zhang, M.; Zhou, D. A novel genomic rearrangement on chr19q13.42 leads to PRPF31-associated retinitis pigmentosa. Clin. Exp. Ophthalmol. 2024, 52, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Barwick, S.R.; Smith, S.B. Comparison of Mouse Models of Autosomal Dominant Retinitis Pigmentosa Due to the P23H Mutation of Rhodopsin. Adv. Exp. Med. Biol. 2023, 1415, 341–345. [Google Scholar] [CrossRef]
- Lath, Y.V.; Thool, A.R.; Jadhav, I. Regeneration of the Retina Using Pluripotent Stem Cells: A Comprehensive Review. Cureus 2024, 16, e53479. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A., Jr.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Akiyama, M.; Nishiguchi, K.M.; Momozawa, Y.; Kamatani, Y.; Takata, S.; Inai, C.; Iwasaki, Y.; Kumano, M.; Murakami, Y.; et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. J. Med. Genet. 2019, 56, 662–670. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Manghwar, H.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas System: Recent Advances and Future Prospects for Genome Editing. Trends Plant Sci. 2019, 24, 1102–1125. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Yao, Y.; Li, L.; Cai, L.; Zhang, H.; Zhang, S.; Xiao, Q.; Wang, X.; Zuo, E.; Xu, C.; et al. Treatment of autosomal dominant retinitis pigmentosa caused by RHO-P23H mutation with high-fidelity Cas13X in mice. Mol. Ther. Nucleic Acids 2023, 33, 750–761. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Banerjee, P.; Kleyn, P.W.; Knowles, J.A.; Lewis, C.A.; Ross, B.M.; Parano, E.; Kovats, S.G.; Lee, J.J.; Penchaszadeh, G.K.; Ott, J.; et al. TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat. Genet. 1998, 18, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Maw, M.A.; Kennedy, B.; Knight, A.; Bridges, R.; Roth, K.E.; Mani, E.J.; Mukkadan, J.K.; Nancarrow, D.; Crabb, J.W.; Denton, M.J. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat. Genet. 1997, 17, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Yadav, A.; Yadav, M.; Tanwar, M. Genetic dissection of non-syndromic retinitis pigmentosa. Indian J. Ophthalmol. 2022, 70, 2355–2385. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Peng, J.; Ying, D.; Peng, Q.H. A Brief Review on the Pathological Role of Decreased Blood Flow Affected in Retinitis Pigmentosa. J. Ophthalmol. 2018, 2018, 3249064. [Google Scholar] [CrossRef]
- Deutschbauer, A.M.; Jaramillo, D.F.; Proctor, M.; Kumm, J.; Hillenmeyer, M.E.; Davis, R.W.; Nislow, C.; Giaever, G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 2005, 169, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Veitia, R.A. Exploring the molecular etiology of dominant-negative mutations. Plant Cell 2007, 19, 3843–3851. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Mohand-Saïd, S.; Dhaenens, C.M.; Germain, A.; Orhan, E.; Antonio, A.; Hamel, C.; Sahel, J.A.; Bhattacharya, S.S.; Zeitz, C. RP1 and autosomal dominant rod-cone dystrophy: Novel mutations, a review of published variants, and genotype-phenotype correlation. Hum. Mutat. 2012, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Lai, T.Y.; Tam, P.O.; Chiang, S.W.; Zhang, X.; Lam, S.; Lai, R.Y.; Lam, D.S.; Pang, C.P. Compound heterozygosity of two novel truncation mutations in RP1 causing autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2236–2242. [Google Scholar] [CrossRef]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef]
- Colella, P.; Cotugno, G.; Auricchio, A. Ocular gene therapy: Current progress and future prospects. Trends Mol. Med. 2009, 15, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Pichi, F.; Morara, M.; Veronese, C.; Nucci, P.; Ciardella, A.P. Multimodal imaging in hereditary retinal diseases. J. Ophthalmol. 2013, 2013, 634351. [Google Scholar] [CrossRef] [PubMed]
- Auricchio, A.; Smith, A.J.; Ali, R.R. The Future Looks Brighter after 25 Years of Retinal Gene Therapy. Hum. Gene Ther. 2017, 28, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.H.; Wensel, T.G. The nature of dominant mutations of rhodopsin and implications for gene therapy. Mol. Neurobiol. 2003, 28, 149–158. [Google Scholar] [CrossRef]
- Mao, H.; James, T., Jr.; Schwein, A.; Shabashvili, A.E.; Hauswirth, W.W.; Gorbatyuk, M.S.; Lewin, A.S. AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum. Gene Ther. 2011, 22, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Price, B.A.; Sandoval, I.M.; Chan, F.; Nichols, R.; Roman-Sanchez, R.; Wensel, T.G.; Wilson, J.H. Rhodopsin gene expression determines rod outer segment size and rod cell resistance to a dominant-negative neurodegeneration mutant. PLoS ONE 2012, 7, e49889. [Google Scholar] [CrossRef] [PubMed]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Sanges, D.; Marrocco, E.; Bonetti, C.; Di Vicino, U.; Marigo, V.; Auricchio, A.; Meroni, G.; Surace, E.M. Zinc-finger-based transcriptional repression of rhodopsin in a model of dominant retinitis pigmentosa. EMBO Mol. Med. 2011, 3, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Botta, S.; Marrocco, E.; de Prisco, N.; Curion, F.; Renda, M.; Sofia, M.; Lupo, M.; Carissimo, A.; Bacci, M.L.; Gesualdo, C.; et al. Rhodopsin targeted transcriptional silencing by DNA-binding. eLife 2016, 5, e12242. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Pang, J.J.; Thomas, J., Jr.; Hauswirth, W.W.; Lewin, A.S. Knockdown of wild-type mouse rhodopsin using an AAV vectored ribozyme as part of an RNA replacement approach. Mol. Vis. 2005, 11, 648–656. [Google Scholar]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef] [PubMed]
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N.; et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2016, 24, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Burnight, E.R.; Gupta, M.; Wiley, L.A.; Anfinson, K.R.; Tran, A.; Triboulet, R.; Hoffmann, J.M.; Klaahsen, D.L.; Andorf, J.L.; Jiao, C.; et al. Using CRISPR-Cas9 to Generate Gene-Corrected Autologous iPSCs for the Treatment of Inherited Retinal Degeneration. Mol. Ther. 2017, 25, 1999–2013. [Google Scholar] [CrossRef] [PubMed]
- Drenser, K.A.; Timmers, A.M.; Hauswirth, W.W.; Lewin, A.S. Ribozyme-targeted destruction of RNA associated with autosomal-dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1998, 39, 681–689. [Google Scholar]
- Murray, S.F.; Jazayeri, A.; Matthes, M.T.; Yasumura, D.; Yang, H.; Peralta, R.; Watt, A.; Freier, S.; Hung, G.; Adamson, P.S.; et al. Allele-Specific Inhibition of Rhodopsin with an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6362–6375. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, D.L.; Cashman, S.M.; Kumar-Singh, R. Engineered zinc finger nuclease-mediated homologous recombination of the human rhodopsin gene. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6374–6380. [Google Scholar] [CrossRef] [PubMed]
- Foltz, L.P.; Howden, S.E.; Thomson, J.A.; Clegg, D.O. Functional Assessment of Patient-Derived Retinal Pigment Epithelial Cells Edited by CRISPR/Cas9. Int. J. Mol. Sci. 2018, 19, 4127. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.I.; Giraud, C. Biology of adeno-associated virus. Curr. Top. Microbiol. Immunol. 1996, 218, 1–23. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- MacLaren, R.E.; Bennett, J.; Schwartz, S.D. Gene Therapy and Stem Cell Transplantation in Retinal Disease: The New Frontier. Ophthalmology 2016, 123, S98–S106. [Google Scholar] [CrossRef] [PubMed]
- Issa, S.S.; Shaimardanova, A.A.; Solovyeva, V.V.; Rizvanov, A.A. Various AAV serotypes and their applications in gene therapy: An overview. Cells 2023, 12, 785. [Google Scholar] [CrossRef] [PubMed]
- Darrow, J.J. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov. Today 2019, 24, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Dalkara, D.; Goureau, O.; Marazova, K.; Sahel, J.A. Let There Be Light: Gene and Cell Therapy for Blindness. Hum. Gene Ther. 2016, 27, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.; Strauss, R.W.; Singh, M.S.; Dalkara, D.; Roska, B.; Picaud, S.; Sahel, J.A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016, 8, 368rv6. [Google Scholar] [CrossRef]
- Liu, Y.; Tai, J.; Yu, C.; Xu, D.; Xiao, D.; Pang, J. Unlocking therapeutic potential: Dual gene therapy for ameliorating the disease phenotypes in a mouse model of RPE65 Leber congenital amaurosis. Front. Med. 2024, 10, 1291795. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J. Taking Stock of Retinal Gene Therapy: Looking Back and Moving Forward. Mol. Ther. 2017, 25, 1076–1094. [Google Scholar] [CrossRef]
- Millington-Ward, S.; Chadderton, N.; O’Reilly, M.; Palfi, A.; Goldmann, T.; Kilty, C.; Humphries, M.; Wolfrum, U.; Bennett, J.; Humphries, P.; et al. Suppression and replacement gene therapy for autosomal dominant disease in a murine model of dominant retinitis pigmentosa. Mol. Ther. 2011, 19, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Wert, K.J.; Davis, R.J.; Sancho-Pelluz, J.; Nishina, P.M.; Tsang, S.H. Gene therapy provides long-term visual function in a pre-clinical model of retinitis pigmentosa. Hum. Mol. Genet. 2013, 22, 558–567. [Google Scholar] [CrossRef]
- Chadderton, N.; Palfi, A.; Millington-Ward, S.; Gobbo, O.; Overlack, N.; Carrigan, M.; O’Reilly, M.; Campbell, M.; Ehrhardt, C.; Wolfrum, U.; et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2013, 21, 62–68. [Google Scholar] [CrossRef]
- Mookherjee, S.; Hiriyanna, S.; Kaneshiro, K.; Li, L.; Li, Y.; Li, W.; Qian, H.; Li, T.; Khanna, H.; Colosi, P.; et al. Long-term rescue of cone photoreceptor degeneration in retinitis pigmentosa 2 (RP2)-knockout mice by gene replacement therapy. Hum. Mol. Genet. 2015, 24, 6446–6458. [Google Scholar] [CrossRef] [PubMed]
- LaVail, M.M.; Yasumura, D.; Matthes, M.T.; Yang, H.; Hauswirth, W.W.; Deng, W.T.; Vollrath, D. Gene Therapy for MERTK-Associated Retinal Degenerations. Adv. Exp. Med. Biol. 2016, 854, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Zhao, J.; Duan, D.; Lai, Y. Design of AAV Vectors for Delivery of Large or Multiple Transgenes. Methods Mol. Biol. 2019, 1950, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Maddalena, A.; Tornabene, P.; Tiberi, P.; Minopoli, R.; Manfredi, A.; Mutarelli, M.; Rossi, S.; Simonelli, F.; Naggert, J.K.; Cacchiarelli, D.; et al. Triple Vectors Expand AAV Transfer Capacity in the Retina. Mol. Ther. 2018, 26, 524–541. [Google Scholar] [CrossRef] [PubMed]
- McClements, M.E.; Barnard, A.R.; Singh, M.S.; Charbel Issa, P.; Jiang, Z.; Radu, R.A.; MacLaren, R.E. An AAV Dual Vector Strategy Ameliorates the Stargardt Phenotype in Adult Abca4(-/-) Mice. Hum. Gene Ther. 2019, 30, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, T.A.C.; Georgiou, M.; Bainbridge, J.W.B.; Michaelides, M. Gene therapy for neovascular age-related macular degeneration: Rationale, clinical trials and future directions. Br. J. Ophthalmol. 2021, 105, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenbergh, C.; Coppieters, F.; Roels, D.; De Jaegere, S.; Flipts, H.; De Zaeytijd, J.; Walraedt, S.; Claes, C.; Fransen, E.; Van Camp, G. Mutations in splicing factor genes are a major cause of autosomal dominant retinitis pigmentosa in Belgian families. PLoS ONE 2017, 12, e0170038. [Google Scholar] [CrossRef] [PubMed]
- Sp, S.; Mitra, R.N.; Zheng, M.; Chrispell, J.D.; Wang, K.; Kwon, Y.S.; Weiss, E.R.; Han, Z. Gene augmentation for autosomal dominant retinitis pigmentosa using rhodopsin genomic loci nanoparticles in the P23H+/− knock-in murine model. Gene Ther. 2023, 30, 628–640. [Google Scholar] [CrossRef]
- Vasudevan, S.; Senapati, S.; Pendergast, M.; Park, P.S. Aggregation of rhodopsin mutants in mouse models of autosomal dominant retinitis pigmentosa. Nat. Commun. 2024, 15, 1451. [Google Scholar] [CrossRef]
- Ueno, S.; Koyanagi, Y.; Kominami, T.; Ito, Y.; Kawano, K.; Nishiguchi, K.M.; Rivolta, C.; Nakazawa, T.; Sonoda, K.H.; Terasaki, H. Clinical characteristics and high resolution retinal imaging of retinitis pigmentosa caused by RP1 gene variants. Jpn. J. Ophthalmol. 2020, 64, 485–496. [Google Scholar] [CrossRef]
- Lázaro-Guevara, J.M.; Flores-Robles, B.J.; Garrido-Lopez, K.M.; McKeown, R.J.; Flores-Morán, A.E.; Labrador-Sánchez, E.; Pinillos-Aransay, V.; Trasahedo, E.A.; López-Martín, J.A.; Soberanis, L.S.R.; et al. Identification of RP1 as the genetic cause of retinitis pigmentosa in a multi-generational pedigree using Extremely Low-Coverage Whole Genome Sequencing (XLC-WGS). Gene 2023, 851, 146956. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, V.W.; Feng, Y.; Tian, X.; Li, F.Y.; Truong, C.; Wang, G.; Chiang, P.W.; Lewis, R.A.; Wong, L.J. Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6213–6223. [Google Scholar] [CrossRef] [PubMed]
- Lisbjerg, K.; Grønskov, K.; Bertelsen, M.; Møller, L.B.; Kessel, L. Genetic Modifiers of Non-Penetrance and RNA Expression Levels in PRPF31-Associated Retinitis Pigmentosa in a Danish Cohort. Genes 2023, 14, 435. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.; Slembrouck-Brec, A.; Nanteau, C.; Terray, A.; Tymoshenko, Y.; Zagar, Y.; Reichman, S.; Xi, Z.; Sahel, J.A.; Fouquet, S.; et al. Modeling PRPF31 retinitis pigmentosa using retinal pigment epithelium and organoids combined with gene augmentation rescue. NPJ Regen. Med. 2022, 7, 39. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Meshad, N.; Hunt, S.; Jackson, N.; Churchill, A. A Combined in silico, in vitro and Clinical Approach to Characterize Novel Pathogenic Missense Variants in PRPF31 in Retinitis Pigmentosa. Front. Genet. 2019, 10, 248. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Boulanger-Scemama, E.; Lancelot, M.E.; Antonio, A.; Démontant, V.; Condroyer, C.; Letexier, M.; Saraiva, J.P.; Mohand-Saïd, S.; Sahel, J.A.; et al. Targeted next generation sequencing identifies novel mutations in RP1 as a relatively common cause of autosomal recessive rod-cone dystrophy. Biomed. Res. Int. 2015, 2015, 485624. [Google Scholar] [CrossRef] [PubMed]
- Kruczek, K.; Qu, Z.; Gentry, J.; Fadl, B.R.; Gieser, L.; Hiriyanna, S.; Batz, Z.; Samant, M.; Samanta, A.; Chu, C.J.; et al. Gene Therapy of Dominant CRX-Leber Congenital Amaurosis using Patient Stem Cell-Derived Retinal Organoids. Stem Cell Rep. 2021, 16, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Talib, M.; van Schooneveld, M.J.; Thiadens, A.A.; Fiocco, M.; Wijnholds, J.; Florijn, R.J.; Schalij-Delfos, N.E.; van Genderen, M.M.; Putter, H.; Cremers, F.P.M.; et al. CLINICAL AND GENETIC CHARACTERISTICS OF MALE PATIENTS WITH RPGR-ASSOCIATED RETINAL DYSTROPHIES: A Long-Term Follow-up Study. Retina 2019, 39, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Sladen, P.E.; Naeem, A.; Adefila-Ideozu, T.; Vermeule, T.; Busson, S.L.; Michaelides, M.; Naylor, S.; Forbes, A.; Lane, A.; Georgiadis, A. AAV-RPGR Gene Therapy Rescues Opsin Mislocalisation in a Human Retinal Organoid Model of RPGR-Associated X-Linked Retinitis Pigmentosa. Int. J. Mol. Sci. 2024, 25, 1839. [Google Scholar] [CrossRef]
- Awadh Hashem, S.; Georgiou, M.; Ali, R.R.; Michaelides, M. RPGR-Related Retinopathy: Clinical Features, Molecular Genetics, and Gene Replacement Therapy. Cold Spring Harb. Perspect. Med. 2023, 13, a041280. [Google Scholar] [CrossRef]
- Appelbaum, T.; Aguirre, G.D.; Beltran, W.A. Identification of circular RNAs hosted by the RPGR ORF15 genomic locus. RNA Biol. 2023, 20, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Mukherjee, S.; Agather, A.R.; Blain, D.; Cunningham, D.; Mays, R.; Sun, X.; Li, T.; Hufnagel, R.B.; Brooks, B.P.; et al. RPGR: Deep Phenotyping and Genetic Characterization with Findings Specific to the 3′-end of ORF15. Investig. Ophthalmol. Vis. Sci. 2023, 64, 19. [Google Scholar] [CrossRef] [PubMed]
- Cehajic-Kapetanovic, J.; Xue, K.; Martinez-Fernandez de la Camara, C.; Nanda, A.; Davies, A.; Wood, L.J.; Salvetti, A.P.; Fischer, M.D.; Aylward, J.W.; Barnard, A.R.; et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 2020, 26, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Aoun, M.; Passerini, I.; Chiurazzi, P.; Karali, M.; De Rienzo, I.; Sartor, G.; Murro, V.; Filimonova, N.; Seri, M.; Banfi, S. Inherited Retinal Diseases Due to RPE65 Variants: From Genetic Diagnostic Management to Therapy. Int. J. Mol. Sci. 2021, 22, 7207. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, V. X-Linked Retinitis Pigmentosa Gene Therapy: Preclinical Aspects. Ophthalmol. Ther. 2023, 12, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Robson, A.G.; Jovanovic, K.; Guimarães, T.A.C.; Ali, N.; Pontikos, N.; Uwaydat, S.H.; Mahroo, O.A.; Cheetham, M.E.; Webster, A.R.; et al. RP2-Associated X-linked Retinopathy: Clinical Findings, Molecular Genetics, and Natural History. Ophthalmology 2023, 130, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Robson, A.G.; Uwaydat, S.H.; Ji, M.H.; Shakarchi, A.F.; Pontikos, N.; Mahroo, O.A.; Cheetham, M.E.; Webster, A.R.; Hardcastle, A.J.; et al. RP2-Associated X-linked Retinopathy: Clinical Findings, Molecular Genetics, and Natural History in a Large Cohort of Female Carriers. Am. J. Ophthalmol. 2024, 261, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, V.K.L.; Takiuti, J.T.; Jauregui, R.; Lima, L.H.; Tsang, S.H. Structural disease progression in PDE6-associated autosomal recessive retinitis pigmentosa. Ophthalmic Genet. 2018, 39, 610–614. [Google Scholar] [CrossRef]
- Gopalakrishna, K.N.; Boyd, K.; Artemyev, N.O. Mechanisms of mutant PDE6 proteins underlying retinal diseases. Cell. Signal. 2017, 37, 74–80. [Google Scholar] [CrossRef]
- Yeo, J.H.; Jung, B.K.; Lee, H.; Baek, I.J.; Sung, Y.H.; Shin, H.S.; Kim, H.K.; Seo, K.Y.; Lee, J.Y. Development of a Pde6b Gene Knockout Rat Model for Studies of Degenerative Retinal Diseases. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1519–1526. [Google Scholar] [CrossRef]
- Hayashi, T.; Mizobuchi, K.; Kameya, S.; Yoshitake, K.; Iwata, T.; Nakano, T. A new PDE6A missense variant p.Arg544Gln in rod-cone dystrophy. Doc. Ophthalmol. 2021, 143, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Kuehlewein, L.; Zobor, D.; Andreasson, S.O.; Ayuso, C.; Banfi, S.; Bocquet, B.; Bernd, A.S.; Biskup, S.; Boon, C.J.F.; Downes, S.M.; et al. Clinical Phenotype and Course of PDE6A-Associated Retinitis Pigmentosa Disease, Characterized in Preparation for a Gene Supplementation Trial. JAMA Ophthalmol. 2020, 138, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Mowat, F.M.; Occelli, L.M.; Bartoe, J.T.; Gervais, K.J.; Bruewer, A.R.; Querubin, J.; Dinculescu, A.; Boye, S.L.; Hauswirth, W.W.; Petersen-Jones, S.M. Gene Therapy in a Large Animal Model of PDE6A-Retinitis Pigmentosa. Front. Neurosci. 2017, 11, 342. [Google Scholar] [CrossRef]
- Sothilingam, V.; Garcia Garrido, M.; Jiao, K.; Buena-Atienza, E.; Sahaboglu, A.; Trifunović, D.; Balendran, S.; Koepfli, T.; Mühlfriedel, R.; Schön, C.; et al. Retinitis pigmentosa: Impact of different Pde6a point mutations on the disease phenotype. Hum. Mol. Genet. 2015, 24, 5486–5499. [Google Scholar] [CrossRef] [PubMed]
- Han, I.C.; Wiley, L.A.; Ochoa, D.; Lang, M.J.; Harman, B.E.; Sheehan, K.M.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. Characterization of a novel Pde6b-deficient rat model of retinal degeneration and treatment with adeno-associated virus (AAV) gene therapy. Gene Ther. 2023, 30, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Toms, M.; Toualbi, L.; Almeida, P.V.; Harbottle, R.; Moosajee, M. Successful large gene augmentation of USH2A with non-viral episomal vectors. Mol. Ther. 2023, 31, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Fuster-García, C.; García-García, G.; González-Romero, E.; Jaijo, T.; Sequedo, M.D.; Ayuso, C.; Vázquez-Manrique, R.P.; Millán, J.M.; Aller, E. USH2A Gene Editing Using the CRISPR System. Mol. Ther. Nucleic Acids 2017, 8, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Mukherjee, R.; Holder, G.E.; Moore, A.T.; Webster, A.R. The clinical features of retinal disease due to a dominant mutation in RPE65. Mol. Vis. 2016, 22, 626–635. [Google Scholar] [PubMed]
- She, K.; Liu, Y.; Zhao, Q.; Jin, X.; Yang, Y.; Su, J.; Li, R.; Song, L.; Xiao, J.; Yao, S.; et al. Dual-AAV split prime editor corrects the mutation and phenotype in mice with inherited retinal degeneration. Signal Transduct. Target. Ther. 2023, 8, 57. [Google Scholar] [CrossRef]
- Wert, K.J.; Sancho-Pelluz, J.; Tsang, S.H. Mid-stage intervention achieves similar efficacy as conventional early-stage treatment using gene therapy in a pre-clinical model of retinitis pigmentosa. Hum. Mol. Genet. 2014, 23, 514–523. [Google Scholar] [CrossRef]
- Tatour, Y.; Tamaiev, J.; Shamaly, S.; Colombo, R.; Bril, E.; Rabinowitz, T.; Yaakobi, A.; Mezer, E.; Leibu, R.; Tiosano, B.; et al. A novel intronic mutation of PDE6B is a major cause of autosomal recessive retinitis pigmentosa among Caucasus Jews. Mol. Vis. 2019, 25, 155–164. [Google Scholar] [PubMed]
- Yang, J.M.; Kim, B.; Kwak, J.; Lee, M.K.; Kim, J.H.; Baek, I.J.; Sung, Y.H.; Lee, J.Y. Development of a novel knockout model of retinitis pigmentosa using Pde6b-knockout Long-Evans rats. Front. Med. 2022, 9, 909182. [Google Scholar] [CrossRef] [PubMed]
- Bitoque, D.B.; Silva, G.A. Molecular biology tools for the study and therapy of PDE6β mutations. J. Biotechnol. 2018, 284, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kuehlewein, L.; Zobor, D.; Stingl, K.; Kempf, M.; Nasser, F.; Bernd, A.; Biskup, S.; Cremers, F.P.M.; Khan, M.I.; Mazzola, P.; et al. Clinical Phenotype of PDE6B-Associated Retinitis Pigmentosa. Int. J. Mol. Sci. 2021, 22, 2374. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhang, W.; Zhang, S.; Feng, Y.; Xu, W.; Qi, J.; Zhang, Q.; Xu, C.; Liu, S.; Zhang, J.; et al. Vision rescue via unconstrained in vivo prime editing in degenerating neural retinas. J. Exp. Med. 2023, 220, e20220776. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Sun, Q.; Gu, M.; Qian, T.; Luo, D.; Liu, K.; Xu, X.; Yu, S. Multimodal imaging and genetic characteristics of Chinese patients with USH2A-associated nonsyndromic retinitis pigmentosa. Mol. Genet. Genom. Med. 2020, 8, e1479. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.J.; Wang, D.D.; Chen, F.; Sun, H.X.; Hu, F.Y.; Xu, P.; Li, J.; Liu, W.; Qi, Y.H.; Li, W.; et al. Prevalence and genetic-phenotypic characteristics of patients with USH2A mutations in a large cohort of Chinese patients with inherited retinal disease. Br. J. Ophthalmol. 2021, 105, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Toualbi, L.; Toms, M.; Moosajee, M. USH2A-retinopathy: From genetics to therapeutics. Exp. Eye Res. 2020, 201, 108330. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Mullins, R.F.; Streb, L.M.; Anfinson, K.; Eyestone, M.E.; Kaalberg, E.; Riker, M.J.; Drack, A.V.; Braun, T.A.; Stone, E.M. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. eLife 2013, 2, e00824. [Google Scholar] [CrossRef]
- Liu, X.; Bulgakov, O.V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4413–4418. [Google Scholar] [CrossRef]
- Yao, L.; Zhang, L.; Qi, L.S.; Liu, W.; An, J.; Wang, B.; Xue, J.H.; Zhang, Z.M. The Time Course of Deafness and Retinal Degeneration in a Kunming Mouse Model for Usher Syndrome. PLoS ONE 2016, 11, e0155619. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Erkilic, N.; Baux, D.; Mamaeva, D.; Hamel, C.P.; Meunier, I.; Roux, A.F.; Kalatzis, V. Genome Editing in Patient iPSCs Corrects the Most Prevalent USH2A Mutations and Reveals Intriguing Mutant mRNA Expression Profiles. Mol. Ther. Methods Clin. Dev. 2020, 17, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Conley, S.M.; Naash, M.I. RPE65: Role in the visual cycle, human retinal disease, and gene therapy. Ophthalmic Genet. 2009, 30, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Furhang, R.; Ray, A.; Duncan, T.; Soucy, J.; Mahdi, R.; Chaitankar, V.; Gieser, L.; Poliakov, E.; Qian, H.; et al. Aberrant RNA splicing is the major pathogenic effect in a knock-in mouse model of the dominantly inherited c.1430A>G human RPE65 mutation. Hum. Mutat. 2019, 40, 426–443. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Drack, A.V.; Simonelli, F.; Leroy, B.P.; Reape, K.Z.; High, K.A.; et al. Durability of Voretigene Neparvovec for Biallelic RPE65-Mediated Inherited Retinal Disease: Phase 3 Results at 3 and 4 Years. Ophthalmology 2021, 128, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhao, P.Y.; Branham, K.; Schlegel, D.; Fahim, A.T.; Jayasundera, T.K.; Khan, N.; Besirli, C.G. Real-world outcomes of voretigene neparvovec treatment in pediatric patients with RPE65-associated Leber congenital amaurosis. Graefes Arch. Clin. Exp. Ophthalmol. 2022, 260, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, K.L.; Cideciyan, A.V.; Swider, M.; Dufour, V.L.; Sumaroka, A.; Komáromy, A.M.; Hauswirth, W.W.; Iwabe, S.; Jacobson, S.G.; Beltran, W.A.; et al. Long-Term Structural Outcomes of Late-Stage RPE65 Gene Therapy. Mol. Ther. 2020, 28, 266–278. [Google Scholar] [CrossRef]
- Meng, D.; Ragi, S.D.; Tsang, S.H. Therapy in Rhodopsin-Mediated Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2020, 28, 2139–2149. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Fujita, K.; Ikeda, Y.; Kunikata, H.; Koyanagi, Y.; Akiyama, M.; Abe, T.; Wada, Y.; Sonoda, K.H.; Nakazawa, T. A founder Alu insertion in RP1 gene in Japanese patients with retinitis pigmentosa. Jpn. J. Ophthalmol. 2020, 64, 346–350. [Google Scholar] [CrossRef] [PubMed]
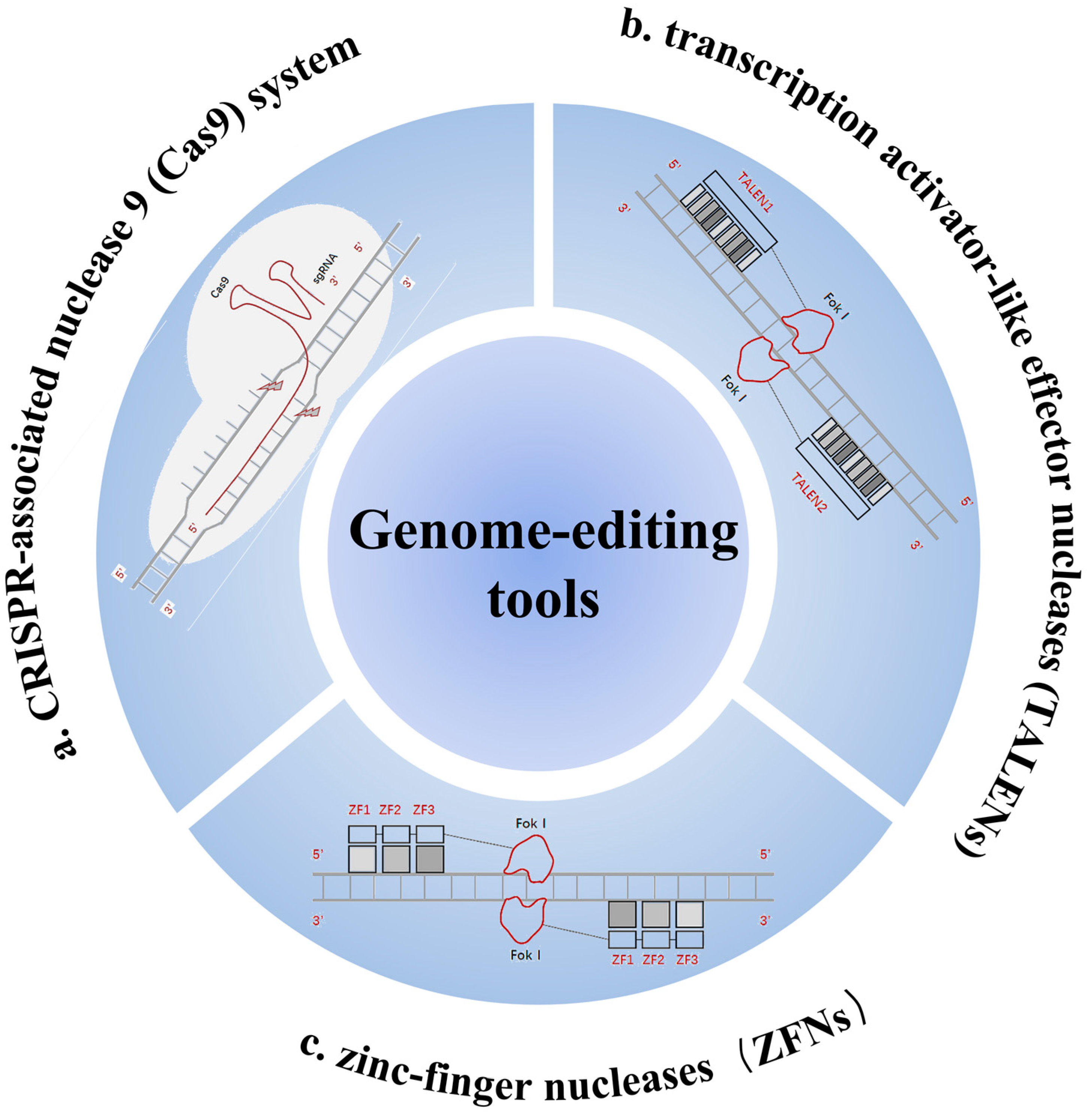
| Target Gene | Type of Method | Outcome | Sample Size | References |
|---|---|---|---|---|
| RhoP23H, RhoG188R | Western blotting | Beneficial effects in reducing retinal degeneration in mice expressing P23H rhodopsin mutants | (-) * | [59] |
| PRPF31 | Elucidated structure of the intact spliceosome to model the effect of a novel PRPF31 variant | Whole blood is not the optimal medium for monitoring PRPF31 mRNA expression | 176 | [60] |
| RP1 | XLC-WGS | Combining lineage information XLC-WGS is a cost-effective way to identify pathogenic variants | 17 | [61] |
| The Alu insertion in RP1 was screened with an optimized PCR-based method | The founder Alu insertion in RP1 is an important cause of autosomal recessive RP in Japanese patients and can be overlooked in standard targeted resequencing. | 26 | [62] |
| Target Gene | Type of Method | Outcome | Sample Size | References |
|---|---|---|---|---|
| RPGR | Optical coherence tomography (OCT) | Three-quarters of patients were male, exhibited cone-rod dystrophy | 66 | [74] |
| AAV8.coRPGR | data Data as vector concentrations decreased, subjects developed mild inflammation postoperatively but healed 6 months after hormone therapy | 18 | [71] | |
| RP2 | OCT and electrophysiology | Most carriers were asymptomatic, exhibiting subclinical characteristics, female carriers of RP2 variants can manifest RP, family history of affected females with RP does not exclude X-linked disease | 40 | [77] |
| Genotype | Target Gene | Genome-Editing Tool | Disease Application | Outcome | References |
|---|---|---|---|---|---|
| ADRP | RHO-P23H | hfCas9X-based sgRNAs | Toxic rhodopsin | Protective role of hfCas13X-mediated targeting in photoreceptor degeneration | [9] |
| P23H(+/−) | Rho and gRHO nanoparticle | Photoreceptor degeneration | Promising reduction of photoreceptor degeneration | [111] | |
| PRPF31 | CRISPR/Cas9 | Prevention of photoreceptor degeneration | Directly increasing PRPF31 expression in retinal organoids effectively prevents photoreceptor degeneration. K88N mutation can be alleviated by AAV-mediated CRX | [64] | |
| CRX-LCA, CRX-K88N | scRNA-seq, AAV | Determination of disease-associated changes in photoreceptor subtypes and evaluation of treatment effect following gene therapy | K88N mutation can be alleviated by AAV-mediated CRX | [112] | |
| X-RP | RPGR-KO | data CRISPR/Cas9, AAV-RPGR | Successful restoration of expression and localization of RPGR mRNAs and proteins connecting cilia in rod and cone photoreceptors | Photoreceptor degeneration | [69] |
| ARRP | RPE65 | AAV2-hRPE65v2 | Improvement and maintenance of functional vision after administration of voretigene neparvovec (VN; Luxturna [Spark Therapeutics, Inc.]) in patients with biallelic RPE65 mutation-associated inherited retinal disease | Significant improvement in visual function without serious adverse effects after one year, sustained over three to four years of follow-up | [105,106,107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Zong, X.; Cao, W.; Zhang, W.; Zhang, N.; Yang, N. Gene Therapy for Retinitis Pigmentosa: Current Challenges and New Progress. Biomolecules 2024, 14, 903. https://doi.org/10.3390/biom14080903
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene Therapy for Retinitis Pigmentosa: Current Challenges and New Progress. Biomolecules. 2024; 14(8):903. https://doi.org/10.3390/biom14080903
Chicago/Turabian StyleLiu, Yuchen, Xin Zong, Wenye Cao, Wenxi Zhang, Ningzhi Zhang, and Ning Yang. 2024. "Gene Therapy for Retinitis Pigmentosa: Current Challenges and New Progress" Biomolecules 14, no. 8: 903. https://doi.org/10.3390/biom14080903
APA StyleLiu, Y., Zong, X., Cao, W., Zhang, W., Zhang, N., & Yang, N. (2024). Gene Therapy for Retinitis Pigmentosa: Current Challenges and New Progress. Biomolecules, 14(8), 903. https://doi.org/10.3390/biom14080903