

Decoding the Role of O-GlcNAcylation in Hepatocellular Carcinoma

Abstract
1. Introduction
2. O-GlcNAcylation
3. Role of O-GlcNAcylation in HCC
4. O-GlcNAcylation of Hsp27
5. O-GlcNAcylation of HDAC1
6. O-GlcNAcylation of TFRC
7. O-GlcNAcylation of YAP
8. O-GlcNAcylation of AMOT
9. O-GlcNAcylation of TRIB2
10. O-GlcNAcylation of HGS
11. O-GlcNAcylation of CHK2
12. O-GlcNAcylation of APA1
13. O-GlcNAcylation of eIF4E
14. O-GlcNAcylation of SIX1
15. O-GlcNAcylation of RACK1
16. O-GlcNAcylation of SLC7A11
17. O-GlcNAcylation of SPOP
18. O-GlcNAcylation of FOXA2
19. O-GlcNAcylation of Skp2
20. O-GlcNAcylation of RAB10
21. O-GlcNAcylation of YTHDF2
22. O-GlcNAcylation and Immunotherapy
23. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Martinello, M.; Solomon, S.S.; Terrault, N.A.; Dore, G.J. Hepatitis C. Lancet 2023, 402, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Kanwal, F.; Llovet, J.M. Global trends in hepatocellular carcinoma epidemiology: Implications for screening, prevention and therapy. Nat. Rev. Clin. Oncol. 2023, 20, 864–884. [Google Scholar] [CrossRef]
- Huang, D.Q.; Singal, A.G.; Kanwal, F.; Lampertico, P.; Buti, M.; Sirlin, C.B.; Nguyen, M.H.; Loomba, R. Hepatocellular carcinoma surveillance—Utilization, barriers and the impact of changing aetiology. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Willoughby, C.E.; Singal, A.G.; Greten, T.F.; Heikenwalder, M.; El-Serag, H.B.; Finn, R.S.; Friedman, S.L. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: Pathogenesis and treatment. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhao, Y.; He, C.; Qian, L.; Huang, P. Ultrasonography of Hepatocellular Carcinoma: From Diagnosis to Prognosis. J. Clin. Transl. Hepatol. 2024, 12, 516–524. [Google Scholar] [CrossRef]
- Candita, G.; Rossi, S.; Cwiklinska, K.; Fanni, S.C.; Cioni, D.; Lencioni, R.; Neri, E. Imaging Diagnosis of Hepatocellular Carcinoma: A State-of-the-Art Review. Diagnostics 2023, 13, 625. [Google Scholar] [CrossRef]
- Lu, Y.; Lin, B.; Li, M. The role of alpha-fetoprotein in the tumor microenvironment of hepatocellular carcinoma. Front. Oncol. 2024, 14, 1363695. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, H.; Zhang, L.; Zhu, A.X.; Bernards, R.; Qin, W.; Wang, C. Evolving therapeutic landscape of advanced hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 203–222. [Google Scholar] [CrossRef]
- Hamaya, S.; Oura, K.; Morishita, A.; Masaki, T. Cisplatin in Liver Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 10858. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.J. Targeting angiogenesis in cancer: Clinical development of bevacizumab. Nat. Clin. Pract. Oncol. 2004, 1, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Pinyol, R.; Yarchoan, M.; Singal, A.G.; Marron, T.U.; Schwartz, M.; Pikarsky, E.; Kudo, M.; Finn, R.S. Adjuvant and neoadjuvant immunotherapies in hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2024, 21, 294–311. [Google Scholar] [CrossRef]
- Ma, G.L.; Lin, W.F. Immune checkpoint inhibition mediated with liposomal nanomedicine for cancer therapy. Mil. Med. Res. 2023, 10, 20. [Google Scholar] [CrossRef]
- Xu, Y.; Song, G.; Xie, S.; Jiang, W.; Chen, X.; Chu, M.; Hu, X.; Wang, Z.W. The roles of PD-1/PD-L1 in the prognosis and immunotherapy of prostate cancer. Mol. Ther. 2021, 29, 1958–1969. [Google Scholar] [CrossRef]
- Hou, B.; Chen, T.; Zhang, H.; Li, J.; Wang, P.; Shang, G. The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels in tumor microenvironment to improve immunotherapy. Front. Immunol. 2023, 14, 1123244. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Pan, S.; Chen, X.; Wang, Z.W.; Zhu, X. The role of lncRNAs and circRNAs in the PD-1/PD-L1 pathway in cancer immunotherapy. Mol. Cancer 2021, 20, 116. [Google Scholar] [CrossRef]
- Lu, Y.; Gao, Y.; Yang, H.; Hu, Y.; Li, X. Nanomedicine-boosting icaritin-based immunotherapy of advanced hepatocellular carcinoma. Mil. Med. Res. 2022, 9, 69. [Google Scholar] [CrossRef]
- Kitamura, N.; Galligan, J.J. A global view of the human post-translational modification landscape. Biochem. J. 2023, 480, 1241–1265. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Chen, K.; Zhai, L.; Liu, M.; Liu, B.; Tan, M. Substrate and Functional Diversity of Protein Lysine Post-translational Modifications. Genom. Proteom. Bioinform. 2024, 22, qzae019. [Google Scholar] [CrossRef]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 59–70. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W.; Chen, Q.; Yuan, Y.; Wang, P. Targeting CSC-related transcription factors by E3 ubiquitin ligases for cancer therapy. Semin. Cancer Biol. 2022, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A.C.O. Signalling mechanisms and cellular functions of SUMO. Nat. Rev. Mol. Cell Biol. 2022, 23, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.; Umit Kaniskan, H.; Jin, J.; Gozani, O. Epigenetics and beyond: Targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. [Google Scholar] [CrossRef]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Gunal, S.; Hardman, R.; Kopriva, S.; Mueller, J.W. Sulfation pathways from red to green. J. Biol. Chem. 2019, 294, 12293–12312. [Google Scholar] [CrossRef] [PubMed]
- Guil-Luna, S.; Sanchez-Montero, M.T.; Rodriguez-Ariza, A. S-Nitrosylation at the intersection of metabolism and autophagy: Implications for cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 189012. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, X.; Liang, T.; Bai, X. O-GlcNAcylation: An important post-translational modification and a potential therapeutic target for cancer therapy. Mol. Med. 2022, 28, 115. [Google Scholar] [CrossRef]
- He, X.F.; Hu, X.; Wen, G.J.; Wang, Z.; Lin, W.J. O-GlcNAcylation in cancer development and immunotherapy. Cancer Lett. 2023, 566, 216258. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qu, S.; Jin, H.; Jia, Q.; Li, M. Role of O-GlcNAcylation in cancer biology. Pathol. Res. Pract. 2024, 253, 155001. [Google Scholar] [CrossRef]
- Liu, X.; Cai, Y.D.; Chiu, J.C. Regulation of protein O-GlcNAcylation by circadian, metabolic, and cellular signals. J. Biol. Chem. 2024, 300, 105616. [Google Scholar] [CrossRef]
- Le Minh, G.; Esquea, E.M.; Young, R.G.; Huang, J.; Reginato, M.J. On a sugar high: Role of O-GlcNAcylation in cancer. J. Biol. Chem. 2023, 299, 105344. [Google Scholar] [CrossRef]
- Maynard, J.C.; Burlingame, A.L.; Medzihradszky, K.F. Cysteine S-linked N-acetylglucosamine (S-GlcNAcylation), A New Post-translational Modification in Mammals. Mol. Cell Proteom. 2016, 15, 3405–3411. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Cui, J.; Huang, Q.; Qi, B.; Xia, Z. Roles of O-GlcNAcylation in Mitochondrial Homeostasis and Cardiovascular Diseases. Antioxidants 2024, 13, 571. [Google Scholar] [CrossRef]
- Ramakrishnan, P. O-GlcNAcylation and immune cell signaling: A review of known and a preview of unknown. J. Biol. Chem. 2024, 300, 107349. [Google Scholar] [CrossRef]
- Chen, L.; Hu, M.; Chen, L.; Peng, Y.; Zhang, C.; Wang, X.; Li, X.; Yao, Y.; Song, Q.; Li, J.; et al. Targeting O-GlcNAcylation in cancer therapeutic resistance: The sugar Saga continues. Cancer Lett. 2024, 588, 216742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y. Emerging roles of O-GlcNAcylation in protein trafficking and secretion. J. Biol. Chem. 2024, 300, 105677. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zheng, J.; Ren, W.; Li, S.; Yang, S.; Zhi, K.; Gao, L. O-GlcNAcylation: Roles and potential therapeutic target for bone pathophysiology. Cell Commun. Signal 2024, 22, 279. [Google Scholar] [CrossRef]
- Shi, R.R.; He, T.Q.; Lin, M.S.; Xu, J.; Gu, J.H.; Xu, H. O-GlcNAcylation in ischemic diseases. Front. Pharmacol. 2024, 15, 1377235. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Ji, S.; Xu, H.; Yu, S. O-GlcNAcylation: A pro-survival response to acute stress in the cardiovascular and central nervous systems. Eur. J. Med. Res. 2024, 29, 174. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Lian, X.; Holscher, C.; Xue, G. O-GlcNAcylation and Its Roles in Neurodegenerative Diseases. J. Alzheimers Dis. 2024, 97, 1051–1068. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Mu, J.; Gao, Z.; Huang, S.; Chen, L. Biological Functions and Potential Therapeutic Significance of O-GlcNAcylation in Hepatic Cellular Stress and Liver Diseases. Cells 2024, 13, 805. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Li, S.; Yin, X.; Sun, K.; Song, J.; Ren, W.; Gao, L.; Zhi, K. Protein O-GlcNAcylation regulates DNA damage response: A novel target for cancer therapy. Int. J. Biol. Macromol. 2024, 264, 130351. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zhou, L.; Yang, Z.; Lai, M.; Xie, H.; Wu, L.; Xing, C.; Zhang, F.; Zheng, S. O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med. Oncol. 2012, 29, 985–993. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Z.; Xu, M.; Zhang, D.; Ling, J.; Yu, P.; Shen, Y. O-GlycNacylation Remission Retards the Progression of Non-Alcoholic Fatty Liver Disease. Cells 2022, 11, 3637. [Google Scholar] [CrossRef]
- Zhang, J.; Xun, M.; Li, C.; Chen, Y. The O-GlcNAcylation and its promotion to hepatocellular carcinoma. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188806. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Gan, L.; Zhang, S.; Cui, F.J.; Cun, W.; Li, Y.; Kang, N.X.; Gao, M.D.; Liu, K.Y. Translocation of HSP27 into liver cancer cell nucleus may be associated with phosphorylation and O-GlcNAc glycosylation. Oncol. Rep. 2012, 28, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Tao, T.; Zhang, D.; Liu, X.; Qiu, H.; Han, L.; Xu, Z.; Xiao, Y.; Cheng, C.; Shen, A. O-GlcNAcylation of histone deacetylases 1 in hepatocellular carcinoma promotes cancer progression. Glycobiology 2016, 26, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, Y.; Li, X.; Zhou, J.; Yang, W.; Wang, X.; Jiao, S.; Zuo, W.; You, Z.; Ying, W.; et al. O-GlcNAcylation regulates the stability of transferrin receptor (TFRC) to control the ferroptosis in hepatocellular carcinoma cells. Redox Biol. 2024, 73, 103182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Murshed, A.; Li, H.; Ma, J.; Zhen, N.; Ding, M.; Zhu, J.; Mao, S.; Tang, X.; Liu, L.; et al. O-GlcNAcylation enhances sensitivity to RSL3-induced ferroptosis via the YAP/TFRC pathway in liver cancer. Cell Death Discov. 2021, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qiao, Y.; Wu, Q.; Chen, Y.; Zou, S.; Liu, X.; Zhu, G.; Zhao, Y.; Chen, Y.; Yu, Y.; et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 2017, 8, 15280. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, Z.; Shi, Y.; Sun, F. AMOT is required for YAP function in high glucose induced liver malignancy. Biochem. Biophys. Res. Commun. 2018, 495, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Xu, Y.; Wang, J.; Qiao, Y.; Zhang, Y.; Zhang, X.; Chen, Y.; Wu, Q.; Zhao, Y.; Zhu, G.; et al. Reciprocal regulation between O-GlcNAcylation and tribbles pseudokinase 2 (TRIB2) maintains transformative phenotypes in liver cancer cells. Cell Signal 2016, 28, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Cheng, Y.; Geng, D.; Fan, Z.; Lin, B.; Zhu, Q.; Li, J.; Qin, W.; Yi, W. O-GlcNAcylation regulates epidermal growth factor receptor intracellular trafficking and signaling. Proc. Natl. Acad. Sci. USA 2022, 119, e2107453119. [Google Scholar] [CrossRef]
- Xiang, J.; Chen, C.; Liu, R.; Gou, D.; Chang, L.; Deng, H.; Gao, Q.; Zhang, W.; Tuo, L.; Pan, X.; et al. Gluconeogenic enzyme PCK1 deficiency promotes CHK2 O-GlcNAcylation and hepatocellular carcinoma growth upon glucose deprivation. J. Clin. Investig. 2021, 131, e144703. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, R.; Chu, Z.; Le, B.; Zeng, H.; Zhang, X.; Wu, Q.; Zhu, G.; Chen, Y.; Liu, Y.; et al. High glucose stimulates proliferative capacity of liver cancer cells possibly via O-GlcNAcylation-dependent transcriptional regulation of GJC1. J. Cell Physiol. 2018, 234, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Duan, M.; Xing, Y.; Liu, C.; Yang, F.; Li, Y.; Yang, T.; Wei, Y.; Gao, Q.; Jiang, J. O-GlcNAc transferase activates stem-like cell potential in hepatocarcinoma through O-GlcNAcylation of eukaryotic initiation factor 4E. J. Cell Mol. Med. 2019, 23, 2384–2398. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Jiang, M.; Wu, N.; Xu, B.; Li, W.; Liu, H.; Su, S.; Shi, Y.; Liu, H.; Gao, X.; et al. O-GlcNAcylation of SIX1 enhances its stability and promotes Hepatocellular Carcinoma Proliferation. Theranostics 2020, 10, 9830–9842. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Wu, H.; Jia, D.; Wu, W.; Ren, S.; Wang, L.; Song, S.; Guo, X.; Liu, F.; Ruan, Y.; et al. O-GlcNAcylation of RACK1 promotes hepatocellular carcinogenesis. J. Hepatol. 2018, 68, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Long, G.; Hu, K.; Xiao, D.; Liu, S.; Xiao, L.; Zhou, L.; Tao, Y. Targeting USP8 Inhibits O-GlcNAcylation of SLC7A11 to Promote Ferroptosis of Hepatocellular Carcinoma via Stabilization of OGT. Adv. Sci. 2023, 10, e2302953. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Chang, W.Y.; Gong, D.A.; Huang, L.Y.; Liu, R.; Liu, Y.; Xia, J.; Wang, K.; Tang, N.; Huang, A.L. O-GlcNAcylation of SPOP promotes carcinogenesis in hepatocellular carcinoma. Oncogene 2023, 42, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wu, Q.; Guo, X.; Huang, T.; Xie, X.; Wang, L.; Liu, Y.; Shi, L.; Li, W.; Zhang, J.; et al. O-GlcNAcylation promotes the migratory ability of hepatocellular carcinoma cells via regulating FOXA2 stability and transcriptional activity. J. Cell Physiol. 2021, 236, 7491–7503. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Yin, J.; Zhang, Z.; Chen, Z.; Huang, L.; Tang, N.; Wang, K. O-GlcNAcylation of E3 ubiquitin ligase SKP2 promotes hepatocellular carcinoma proliferation. Oncogene 2024, 43, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Ma, G.; Zhong, Z.; Xie, X.; Li, B.; Long, D. O-GlcNAcylation of RAB10 promotes hepatocellular carcinoma progression. Carcinogenesis 2023, 44, 785–794. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, Y.; Yin, J.; Tang, N.; Wang, K.; Huang, L.; Hu, J.; Feng, Z.; Gao, Q.; Huang, A. O-GlcNAcylation of YTHDF2 promotes HBV-related hepatocellular carcinoma progression in an N(6)-methyladenosine-dependent manner. Signal Transduct. Target. Ther. 2023, 8, 63. [Google Scholar] [CrossRef]
- Shan, R.; Liu, N.; Yan, Y.; Liu, B. Apoptosis, autophagy and atherosclerosis: Relationships and the role of Hsp27. Pharmacol. Res. 2021, 166, 105169. [Google Scholar] [CrossRef] [PubMed]
- Lampros, M.; Vlachos, N.; Voulgaris, S.; Alexiou, G.A. The Role of Hsp27 in Chemotherapy Resistance. Biomedicines 2022, 10, 897. [Google Scholar] [CrossRef]
- Zheng, S.; Liang, Y.; Li, L.; Tan, Y.; Liu, Q.; Liu, T.; Lu, X. Revisiting the Old Data of Heat Shock Protein 27 Expression in Squamous Cell Carcinoma: Enigmatic HSP27, More Than Heat Shock. Cells 2022, 11, 1665. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.; Chi, J.G.; Lee, H. Expressions of HSP70 and HSP27 in hepatocellular carcinoma. J. Korean Med. Sci. 2005, 20, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Luk, J.M.; Lam, C.T.; Siu, A.F.; Lam, B.Y.; Ng, I.O.; Hu, M.Y.; Che, C.M.; Fan, S.T. Proteomic profiling of hepatocellular carcinoma in Chinese cohort reveals heat-shock proteins (Hsp27, Hsp70, GRP78) up-regulation and their associated prognostic values. Proteomics 2006, 6, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Kang, N.X.; Li, Y.; Sun, L.; Gan, L.; Cui, F.J.; Gao, M.D.; Liu, K.Y. Regulation of HSP27 on NF-kappaB pathway activation may be involved in metastatic hepatocellular carcinoma cells apoptosis. BMC Cancer 2009, 9, 100. [Google Scholar] [CrossRef]
- Cheng, J.; Lv, Z.; Weng, X.; Ye, S.; Shen, K.; Li, M.; Qin, Y.; Hu, C.; Zhang, C.; Wu, J.; et al. Hsp27 Acts as a Master Molecular Chaperone and Plays an Essential Role in Hepatocellular Carcinoma Progression. Digestion 2015, 92, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Huang, C.Y.; Pan, T.L.; Chen, W.Y.; Ho, C.T.; Liu, T.Z.; Chang, Y.J. Proteomic Characterization of Annexin l (ANX1) and Heat Shock Protein 27 (HSP27) as Biomarkers for Invasive Hepatocellular Carcinoma Cells. PLoS ONE 2015, 10, e0139232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tao, X.; Jin, G.; Jin, H.; Wang, N.; Hu, F.; Luo, Q.; Shu, H.; Zhao, F.; Yao, M.; et al. A Targetable Molecular Chaperone Hsp27 Confers Aggressiveness in Hepatocellular Carcinoma. Theranostics 2016, 6, 558–570. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, H.; Xing, X.; Ma, Y.; Ji, S.; Xu, X.; Zhao, X.; Wang, S.; Jiang, W.; Fang, C.; et al. CD13 Induces Autophagy to Promote Hepatocellular Carcinoma Cell Chemoresistance Through the P38/Hsp27/CREB/ATG7 Pathway. J. Pharmacol. Exp. Ther. 2020, 374, 512–520. [Google Scholar] [CrossRef]
- Sharma, A.; Upadhyay, A.K.; Bhat, M.K. Inhibition of Hsp27 and Hsp40 potentiates 5-fluorouracil and carboplatin mediated cell killing in hepatoma cells. Cancer Biol. Ther. 2009, 8, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wu, W.; Zhou, Y.; Xie, Q.; Liu, T.; Jin, J.; Liu, K. HSP27 expression levels are associated with the sensitivity of hepatocellular carcinoma cells to 17-allylamino-17-demethoxygeldanamycin. Future Oncol. 2013, 9, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Du, J.; Xu, J.; Meng, X.; Tian, J.; Yang, J.; Liang, H. SUMOylation of HSP27 by small ubiquitin-like modifier 2/3 promotes proliferation and invasion of hepatocellular carcinoma cells. Cancer Biol. Ther. 2017, 18, 552–559. [Google Scholar] [CrossRef]
- Farooq, M.; Hozzein, W.N.; Elsayed, E.A.; Taha, N.A.; Wadaan, M.A. Identification of histone deacetylase 1 protein complexes in liver cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Iakova, P.; Jiang, Y.; Lewis, K.; Sullivan, E.; Jawanmardi, N.; Donehower, L.; Timchenko, L.; Timchenko, N.A. Transcriptional and translational regulation of C/EBPbeta-HDAC1 protein complexes controls different levels of p53, SIRT1, and PGC1alpha proteins at the early and late stages of liver cancer. J. Biol. Chem. 2013, 288, 14451–14462. [Google Scholar] [CrossRef]
- Xie, H.J.; Noh, J.H.; Kim, J.K.; Jung, K.H.; Eun, J.W.; Bae, H.J.; Kim, M.G.; Chang, Y.G.; Lee, J.Y.; Park, H.; et al. HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS ONE 2012, 7, e34265. [Google Scholar] [CrossRef]
- Al-Yhya, N.; Khan, M.F.; Almeer, R.S.; Alshehri, M.M.; Aldughaim, M.S.; Wadaan, M.A. Pharmacological inhibition of HDAC1/3-interacting proteins induced morphological changes, and hindered the cell proliferation and migration of hepatocellular carcinoma cells. Environ. Sci. Pollut. Res. Int. 2021, 28, 49000–49013. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.W.; Yao, R.Q.; Fan, Y.W.; Zhang, D.J.; Zhou, Y.; Wang, M.J.; Zhang, L.Y.; Dong, Y.; Li, Z.X.; Wang, S.Y.; et al. Interferon-alpha stimulates DExH-box helicase 58 to prevent hepatocyte ferroptosis. Mil. Med. Res. 2024, 11, 22. [Google Scholar] [CrossRef]
- Muhammad, J.S.; Bajbouj, K.; Shafarin, J.; Hamad, M. Estrogen-induced epigenetic silencing of FTH1 and TFRC genes reduces liver cancer cell growth and survival. Epigenetics 2020, 15, 1302–1318. [Google Scholar] [CrossRef]
- Guo, S.; Chen, Y.; Xue, X.; Yang, Y.; Wang, Y.; Qiu, S.; Cui, J.; Zhang, X.; Ma, L.; Qiao, Y.; et al. TRIB2 desensitizes ferroptosis via betaTrCP-mediated TFRC ubiquitiantion in liver cancer cells. Cell Death Discov. 2021, 7, 196. [Google Scholar] [CrossRef]
- Sun, H.; Qian, X.; Yang, W.; Zhou, W.; Zhou, C.; Liu, S.; Shi, H.; Tian, W. Novel prognostic signature based on HRAS, MAPK3 and TFRC identified to be associated with ferroptosis and the immune microenvironment in hepatocellular carcinoma. Am. J. Transl. Res. 2022, 14, 6924–6940. [Google Scholar]
- Hiromatsu, M.; Toshida, K.; Itoh, S.; Harada, N.; Kohashi, K.; Oda, Y.; Yoshizumi, T. Transferrin Receptor is Associated with Sensitivity to Ferroptosis Inducers in Hepatocellular Carcinoma. Ann. Surg. Oncol. 2023, 30, 8675–8689. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, W.Q.; Zhang, W.Q.; Xu, R.C.; Sun, J.L.; Zhang, G.C.; Liu, Z.Y.; Qi, Z.R.; Dong, L.; Weng, S.Q.; et al. Transferrin receptor 1 promotes hepatocellular carcinoma progression and metastasis by activating the mTOR signaling pathway. Hepatol. Int. 2024, 18, 636–650. [Google Scholar] [CrossRef]
- Perra, A.; Kowalik, M.A.; Ghiso, E.; Ledda-Columbano, G.M.; Di Tommaso, L.; Angioni, M.M.; Raschioni, C.; Testore, E.; Roncalli, M.; Giordano, S.; et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J. Hepatol. 2014, 61, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.H.; Kim, S.J.; Hyun, J. MicroRNAs Regulating Hippo-YAP Signaling in Liver Cancer. Biomedicines 2021, 9, 347. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, D. Role of the transcriptional coactivators YAP/TAZ in liver cancer. Curr. Opin. Cell Biol. 2019, 61, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Weiler, S.M.E.; Lutz, T.; Bissinger, M.; Sticht, C.; Knaub, M.; Gretz, N.; Schirmacher, P.; Breuhahn, K. TAZ target gene ITGAV regulates invasion and feeds back positively on YAP and TAZ in liver cancer cells. Cancer Lett. 2020, 473, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Zhao, Q.; An, L.; Jiao, S.; Li, R.; Sang, Y.; Liao, J.; Nie, P.; Wen, F.; Ju, J.; et al. A TNFR2-hnRNPK Axis Promotes Primary Liver Cancer Development via Activation of YAP Signaling in Hepatic Progenitor Cells. Cancer Res. 2021, 81, 3036–3050. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Weiler, S.M.E.; Toth, M.; Sticht, C.; Lutz, T.; Thomann, S.; De La Torre, C.; Straub, B.; Merker, S.; Ruppert, T.; et al. YAP-dependent induction of UHMK1 supports nuclear enrichment of the oncogene MYBL2 and proliferation in liver cancer cells. Oncogene 2019, 38, 5541–5550. [Google Scholar] [CrossRef]
- Ren, H.; Chen, Y.; Ao, Z.; Cheng, Q.; Yang, X.; Tao, H.; Zhao, L.; Shen, A.; Li, P.; Fu, Q. PDE4D binds and interacts with YAP to cooperatively promote HCC progression. Cancer Lett. 2022, 541, 215749. [Google Scholar] [CrossRef]
- Zhang, C.; Niu, Y.; Wang, Z.; Xu, X.; Li, Y.; Ma, L.; Wang, J.; Yu, Y. Corosolic acid inhibits cancer progression by decreasing the level of CDK19-mediated O-GlcNAcylation in liver cancer cells. Cell Death Dis. 2021, 12, 889. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Shen, Y.; Yang, J.; Li, S.; Wang, B.; Chen, Z.; Li, P.; Liu, P.; Yang, J. Angiomotin Family Members: Oncogenes or Tumor Suppressors? Int. J. Biol. Sci. 2017, 13, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, F.X. Angiomotin family proteins in the Hippo signaling pathway. Bioessays 2024, 46, e2400076. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ye, M.; Jin, X. Role of angiomotin family members in human diseases. Exp. Ther. Med. 2024, 27, 258. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Shen, Z.; Stemmer-Rachamimov, A.; Dawany, N.; Troutman, S.; Showe, L.C.; Liu, Q.; Shimono, A.; Sudol, M.; Holmgren, L.; et al. The p130 isoform of angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Sci. Signal 2013, 6, ra77. [Google Scholar] [CrossRef]
- Zhu, G.; Chen, Y.; Zhang, X.; Wu, Q.; Zhao, Y.; Chen, Y.; Sun, F.; Qiao, Y.; Wang, J. 12-O-Tetradecanoylphorbol-13-acetate (TPA) is anti-tumorigenic in liver cancer cells via inhibiting YAP through AMOT. Sci. Rep. 2017, 7, 44940. [Google Scholar] [CrossRef] [PubMed]
- Salome, M.; Campos, J.; Keeshan, K. TRIB2 and the ubiquitin proteasome system in cancer. Biochem. Soc. Trans. 2015, 43, 1089–1094. [Google Scholar] [CrossRef]
- Fang, Y.; Zekiy, A.O.; Ghaedrahmati, F.; Timoshin, A.; Farzaneh, M.; Anbiyaiee, A.; Khoshnam, S.E. Tribbles homolog 2 (Trib2), a pseudo serine/threonine kinase in tumorigenesis and stem cell fate decisions. Cell Commun. Signal 2021, 19, 41. [Google Scholar] [CrossRef]
- Mayoral-Varo, V.; Jimenez, L.; Link, W. The Critical Role of TRIB2 in Cancer and Therapy Resistance. Cancers 2021, 13, 2701. [Google Scholar] [CrossRef]
- Wang, J.; Park, J.S.; Wei, Y.; Rajurkar, M.; Cotton, J.L.; Fan, Q.; Lewis, B.C.; Ji, H.; Mao, J. TRIB2 acts downstream of Wnt/TCF in liver cancer cells to regulate YAP and C/EBPalpha function. Mol. Cell 2013, 51, 211–225. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Weng, W.; Qiao, Y.; Ma, L.; Xiao, W.; Yu, Y.; Pan, Q.; Sun, F. Impaired phosphorylation and ubiquitination by p70 S6 kinase (p70S6K) and Smad ubiquitination regulatory factor 1 (Smurf1) promote tribbles homolog 2 (TRIB2) stability and carcinogenic property in liver cancer. J. Biol. Chem. 2013, 288, 33667–33681. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, Y.; Wang, J. Ubiquitin E3 ligase SCF(beta-TRCP) regulates TRIB2 stability in liver cancer cells. Biochem. Biophys. Res. Commun. 2013, 441, 555–559. [Google Scholar] [CrossRef]
- Xu, S.; Tong, M.; Huang, J.; Zhang, Y.; Qiao, Y.; Weng, W.; Liu, W.; Wang, J.; Sun, F. TRIB2 inhibits Wnt/beta-Catenin/TCF4 signaling through its associated ubiquitin E3 ligases, beta-TrCP, COP1 and Smurf1, in liver cancer cells. FEBS Lett. 2014, 588, 4334–4341. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Chen, Y.; Yang, Y.; Zhang, X.; Ma, L.; Xue, X.; Qiao, Y.; Wang, J. TRIB2 modulates proteasome function to reduce ubiquitin stability and protect liver cancer cells against oxidative stress. Cell Death Dis. 2021, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Komada, M.; Kitamura, N. The Hrs/STAM complex in the downregulation of receptor tyrosine kinases. J. Biochem. 2005, 137, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. Hrs and endocytic sorting of ubiquitinated membrane proteins. Cell Struct. Funct. 2002, 27, 403–408. [Google Scholar] [CrossRef] [PubMed]
- da Rocha, A.A.; Giorgi, R.R.; de Sa, S.V.; Correa-Giannella, M.L.; Fortes, M.A.; Cavaleiro, A.M.; Machado, M.C.; Cescato, V.A.; Bronstein, M.D.; Giannella-Neto, D. Hepatocyte growth factor-regulated tyrosine kinase substrate (HGS) and guanylate kinase 1 (GUK1) are differentially expressed in GH-secreting adenomas. Pituitary 2006, 9, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, W.; Guo, Z.; Ju, Q.; Zhu, L.; Gao, J.; Zhou, L.; Liu, F.; Xu, Y.; Zhan, Q.; et al. A novel TP53 pathway influences the HGS-mediated exosome formation in colorectal cancer. Sci. Rep. 2016, 6, 28083. [Google Scholar] [CrossRef] [PubMed]
- Matthew-Onabanjo, A.N.; Janusis, J.; Mercado-Matos, J.; Carlisle, A.E.; Kim, D.; Levine, F.; Cruz-Gordillo, P.; Richards, R.; Lee, M.J.; Shaw, L.M. Beclin 1 Promotes Endosome Recruitment of Hepatocyte Growth Factor Tyrosine Kinase Substrate to Suppress Tumor Proliferation. Cancer Res. 2020, 80, 249–262. [Google Scholar] [CrossRef]
- Xiao, B.L.; Wang, X.L.; Xia, H.F.; Zhang, L.Z.; Wang, K.M.; Chen, Z.K.; Zhong, Y.H.; Jiang, H.G.; Zhou, F.X.; Wang, W.; et al. HRS Regulates Small Extracellular Vesicle PD-L1 Secretion and Is Associated with Anti-PD-1 Treatment Efficacy. Cancer Immunol. Res. 2023, 11, 228–240. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, J.; Wang, B.; Lu, Y.; Yang, J.; Zhong, W.; Yu, Z.; Qin, Z.; Xiao, B.; Wang, K.; et al. HRS mediates tumor immune evasion by regulating proteostasis-associated interferon pathway activation. Cell Rep. 2023, 42, 113352. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Tanaka, N.; Aoki, J.; Tanaka, Y.; Murata, K.; Kyuuma, M.; Kobayashi, H.; Ishii, N.; Yaegashi, N.; Sugamura, K. Inhibition of tumor growth and metastasis by depletion of vesicular sorting protein Hrs: Its regulatory role on E-cadherin and beta-catenin. Cancer Res. 2007, 67, 5162–5171. [Google Scholar] [CrossRef] [PubMed]
- Canal, F.; Anthony, E.; Lescure, A.; Del Nery, E.; Camonis, J.; Perez, F.; Ragazzon, B.; Perret, C. A kinome siRNA screen identifies HGS as a potential target for liver cancers with oncogenic mutations in CTNNB1. BMC Cancer 2015, 15, 1020. [Google Scholar] [CrossRef] [PubMed]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Mustofa, M.K.; Tanoue, Y.; Tateishi, C.; Vaziri, C.; Tateishi, S. Roles of Chk2/CHEK2 in guarding against environmentally induced DNA damage and replication-stress. Environ. Mol. Mutagen. 2020, 61, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef] [PubMed]
- Perona, R.; Moncho-Amor, V.; Machado-Pinilla, R.; Belda-Iniesta, C.; Sanchez Perez, I. Role of CHK2 in cancer development. Clin. Transl. Oncol. 2008, 10, 538–542. [Google Scholar] [CrossRef]
- Lulli, M.; Del Coco, L.; Mello, T.; Sukowati, C.; Madiai, S.; Gragnani, L.; Forte, P.; Fanizzi, F.P.; Mazzocca, A.; Rombouts, K.; et al. DNA Damage Response Protein CHK2 Regulates Metabolism in Liver Cancer. Cancer Res. 2021, 81, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lin, N.; Xu, L.; Liu, B.; Feitelson, M.A. UCN-01 induces S and G2/M cell cycle arrest through the p53/p21 (waf1) or CHK2/CDC25C pathways and can suppress invasion in human hepatoma cell lines. BMC Cancer 2013, 13, 167. [Google Scholar] [CrossRef]
- Huang, H.; Hu, M.; Zhao, R.; Li, P.; Li, M. Dihydromyricetin suppresses the proliferation of hepatocellular carcinoma cells by inducing G2/M arrest through the Chk1/Chk2/Cdc25C pathway. Oncol. Rep. 2013, 30, 2467–2475. [Google Scholar] [CrossRef]
- Carloni, V.; Lulli, M.; Madiai, S.; Mello, T.; Hall, A.; Luong, T.V.; Pinzani, M.; Rombouts, K.; Galli, A. CHK2 overexpression and mislocalisation within mitotic structures enhances chromosomal instability and hepatocellular carcinoma progression. Gut 2018, 67, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Shi, Y.; Wu, W.; Wu, H.; Chang, L.; Peng, P.; Zhang, L.; Fan, J.; Gu, J.; Ruan, Y. Reticulon 3-mediated Chk2/p53 activation suppresses hepatocellular carcinogenesis and is blocked by hepatitis B virus. Gut 2021, 70, 2159–2171. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Kamimura, K.; Ogawa, K.; Oda, C.; Tanaka, Y.; Horigome, R.; Ohtsuka, M.; Miura, H.; Fujisawa, K.; Yamamoto, N.; et al. Cyclin D1 Binding Protein 1 Responds to DNA Damage through the ATM-CHK2 Pathway. J. Clin. Med. 2022, 11, 851. [Google Scholar] [CrossRef]
- Leslie, J.; Hunter, J.E.; Collins, A.; Rushton, A.; Russell, L.G.; Ramon-Gil, E.; Laszczewska, M.; McCain, M.; Zaki, M.Y.W.; Knox, A.; et al. c-Rel-dependent Chk2 signaling regulates the DNA damage response limiting hepatocarcinogenesis. Hepatology 2023, 78, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhuang, Y.; Wang, B.; Yuan, B.; Du, S.; Zeng, Z. The miR-34a-5p-c-MYC-CHK1/CHK2 Axis Counteracts Cancer Stem Cell-Like Properties and Enhances Radiosensitivity in Hepatocellular Cancer Through Repression of the DNA Damage Response. Radiat. Res. 2023, 199, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Desplantez, T. Cardiac Cx43, Cx40 and Cx45 co-assembling: Involvement of connexins epitopes in formation of hemichannels and Gap junction channels. BMC Cell Biol. 2017, 18, 3. [Google Scholar] [CrossRef]
- Xiang, Q.; Liu, X.L.; Chen, J.J.; Yang, L.; Liu, L.N.; Deng, J.; Tao, J.S.; Li, X.H. A Review of Gap Junction Protein and its Potential Role in Nervous System-Related Disease. Protein Pept. Lett. 2023, 30, 891–899. [Google Scholar] [CrossRef]
- Zlomuzica, A.; Plank, L.; Dere, E. A new path to mental disorders: Through gap junction channels and hemichannels. Neurosci. Biobehav. Rev. 2022, 142, 104877. [Google Scholar] [CrossRef]
- Beckmann, A.; Hainz, N.; Tschernig, T.; Meier, C. Facets of Communication: Gap Junction Ultrastructure and Function in Cancer Stem Cells and Tumor Cells. Cancers 2019, 11, 288. [Google Scholar] [CrossRef]
- Sirnes, S.; Honne, H.; Ahmed, D.; Danielsen, S.A.; Rognum, T.O.; Meling, G.I.; Leithe, E.; Rivedal, E.; Lothe, R.A.; Lind, G.E. DNA methylation analyses of the connexin gene family reveal silencing of GJC1 (Connexin45) by promoter hypermethylation in colorectal cancer. Epigenetics 2011, 6, 602–609. [Google Scholar] [CrossRef]
- Ahmed, D.; Lothe, R.A.; Rivedal, E.; Lind, G.E. Quantitative validation of GJC1 promoter hypermethylation in benign and malignant colorectal tumors. Endocr. Relat. Cancer 2011, 18, C31–C34. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Palacios-Prado, N.; Saez, J.C.; Lee, J. Identification of Cx45 as a Major Component of GJs in HeLa Cells. Biomolecules 2020, 10, 1389. [Google Scholar] [CrossRef]
- Chen, X.; An, Y.; Tan, M.; Xie, D.; Liu, L.; Xu, B. Biological functions and research progress of eIF4E. Front. Oncol. 2023, 13, 1076855. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, A.; D’Agostino, M.; Ardiccioni, C.; Maracci, C.; Motta, S.; La Teana, A.; Di Marino, D. Control of the eIF4E activity: Structural insights and pharmacological implications. Cell Mol. Life Sci. 2021, 78, 6869–6885. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhong, W.; Cao, R. Phosphorylation of the mRNA cap-binding protein eIF4E and cancer. Cell Signal 2020, 73, 109689. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.M.; Yu, X.N.; Huang, R.Z.; Zhu, H.R.; Chen, X.P.; Xiong, J.; Chen, Z.Y.; Huang, X.X.; Shen, X.Z.; Zhu, J.M. Prognostic significance of eukaryotic initiation factor 4E in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, L.; Su, X.; Guo, S. Inhibition of eukaryotic initiation factor 4E phosphorylation by cercosporamide selectively suppresses angiogenesis, growth and survival of human hepatocellular carcinoma. Biomed. Pharmacother. 2016, 84, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Huang, R.; Wu, Q.; Li, P.; Chen, J.; Li, B.; Liu, H. The role of Six1 in the genesis of muscle cell and skeletal muscle development. Int. J. Biol. Sci. 2014, 10, 983–989. [Google Scholar] [CrossRef]
- Rafiq, A.; Aashaq, S.; Jan, I.; Beigh, M.A. SIX1 transcription factor: A review of cellular functions and regulatory dynamics. Int. J. Biol. Macromol. 2021, 193, 1151–1164. [Google Scholar] [CrossRef]
- Blevins, M.A.; Towers, C.G.; Patrick, A.N.; Zhao, R.; Ford, H.L. The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Expert. Opin. Ther. Targets 2015, 19, 213–225. [Google Scholar] [CrossRef]
- Wu, W.; Ren, Z.; Li, P.; Yu, D.; Chen, J.; Huang, R.; Liu, H. Six1: A critical transcription factor in tumorigenesis. Int. J. Cancer 2015, 136, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.T.; Man, K.; Sun, C.K.; Lee, T.K.; Poon, R.T.; Lo, C.M.; Fan, S.T. Clinicopathological significance of homeoprotein Six1 in hepatocellular carcinoma. Br. J. Cancer 2006, 95, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.T.; Lee, T.K.; Cheng, Q.; Wo, J.Y.; Sun, C.K.; Guo, D.Y.; Lim, Z.X.; Lo, C.M.; Poon, R.T.; Fan, S.T.; et al. Suppression of tumorigenesis and metastasis of hepatocellular carcinoma by shRNA interference targeting on homeoprotein Six1. Int. J. Cancer 2010, 127, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.W.; Dong, L.D.; Shang, W.J.; Pang, X.L.; Li, J.F.; Liu, L.; Wang, Y. HDAC5 promotes cell proliferation in human hepatocellular carcinoma by up-regulating Six1 expression. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 811–816. [Google Scholar]
- Cheng, Q.; Ning, D.; Chen, J.; Li, X.; Chen, X.P.; Jiang, L. SIX1 and DACH1 influence the proliferation and apoptosis of hepatocellular carcinoma through regulating p53. Cancer Biol. Ther. 2018, 19, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Jiang, M.; Du, F.; Chen, D.; Ye, T.; Xu, B.; Li, X.; Wang, W.; Qiu, Z.; Liu, H.; et al. miR-204-5p suppresses hepatocellular cancer proliferation by regulating homeoprotein SIX1 expression. FEBS Open Bio. 2018, 8, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Zhao, L.; Peng, C.; Ran, K.; Mu, R.; Ao, Y. LncRNA CRNDE promotes hepatocellular carcinoma progression by upregulating SIX1 through modulating miR-337-3p. J. Cell Biochem. 2019, 120, 16128–16142. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Huang, J.; Mo, J.; Da, X.; Li, Q.; Fan, M.; Lu, H. Exosomal lncRNA TUG1 from cancer-associated fibroblasts promotes liver cancer cell migration, invasion, and glycolysis by regulating the miR-524-5p/SIX1 axis. Cell Mol. Biol. Lett. 2022, 27, 17. [Google Scholar] [CrossRef]
- Chen, K.; Wei, H.; Pan, J.; Chen, Z.; Pan, D.; Gao, T.; Huang, J.; Huang, M.; Ou, M.; Zhong, W. Six1 is negatively correlated with poor prognosis and reduces 5-fluorouracil sensitivity via attenuating the stemness of hepatocellular carcinoma cells. Eur. J. Pharmacol. 2019, 861, 172599. [Google Scholar] [CrossRef]
- Liu, Y.; Kong, W.Y.; Yu, C.F.; Shao, Z.L.; Lei, Q.C.; Deng, Y.F.; Cai, G.X.; Zhuang, X.F.; Sun, W.S.; Wu, S.G.; et al. SNS-023 sensitizes hepatocellular carcinoma to sorafenib by inducing degradation of cancer drivers SIX1 and RPS16. Acta Pharmacol. Sin. 2023, 44, 853–864. [Google Scholar] [CrossRef]
- Critelli, R.M.; Milosa, F.; Romanzi, A.; Lasagni, S.; Marcelli, G.; Di Marco, L.; Pivetti, A.; Schepis, F.; Romagnoli, D.; Mancarella, S.; et al. Upregulation of the oestrogen target gene SIX1 is associated with higher growth speed and decreased survival in HCV-positive women with hepatocellular carcinoma. Oncol. Lett. 2022, 24, 395. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Senft, D.; Topisirovic, I.; Ronai, Z.A. RACK1 Function in Cell Motility and Protein Synthesis. Genes Cancer 2013, 4, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Chiba, N. Roles of RACK1 in centrosome regulation and carcinogenesis. Cell Signal 2022, 90, 110207. [Google Scholar] [CrossRef] [PubMed]
- Kershner, L.; Welshhans, K. RACK1 regulates neural development. Neural Regen. Res. 2017, 12, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Duff, D.; Long, A. Roles for RACK1 in cancer cell migration and invasion. Cell Signal 2017, 35, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Xie, D. RACK1, a versatile hub in cancer. Oncogene 2015, 34, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Sun, L.; Hao, Y.; Wang, L.; Xu, J.; Zhang, W.; Xie, J.; Guo, L.; Zhou, L.; Yun, X.; et al. Ribosomal RACK1 promotes chemoresistance and growth in human hepatocellular carcinoma. J. Clin. Investig. 2012, 122, 2554–2566. [Google Scholar] [CrossRef]
- Zhou, T.; Lv, X.; Guo, X.; Ruan, B.; Liu, D.; Ding, R.; Gao, Y.; Ding, J.; Dou, K.; Chen, Y. RACK1 modulates apoptosis induced by sorafenib in HCC cells by interfering with the IRE1/XBP1 axis. Oncol. Rep. 2015, 33, 3006–3014. [Google Scholar] [CrossRef]
- Wang, W.D.; Wen, Z.; Ji, W.; Ma, Y. RACK1 expression contributes to JNK activity, but JNK activity does not enhance RACK1 expression in hepatocellular carcinoma SMMC-7721 cells. Oncol. Lett. 2015, 9, 2767–2770. [Google Scholar] [CrossRef]
- Zhou, S.; Cao, H.; Zhao, Y.; Li, X.; Zhang, J.; Hou, C.; Ma, Y.; Wang, Q. RACK1 promotes hepatocellular carcinoma cell survival via CBR1 by suppressing TNF-alpha-induced ROS generation. Oncol. Lett. 2016, 12, 5303–5308. [Google Scholar] [CrossRef]
- Zou, Y.H.; Li, X.D.; Zhang, Q.H.; Liu, D.Z. RACK1 Silencing Induces Cell Apoptosis and Inhibits Cell Proliferation in Hepatocellular Carcinoma MHCC97-H Cells. Pathol. Oncol. Res. 2018, 24, 101–107. [Google Scholar] [CrossRef]
- Cao, J.; Zhao, M.; Liu, J.; Zhang, X.; Pei, Y.; Wang, J.; Yang, X.; Shen, B.; Zhang, J. RACK1 Promotes Self-Renewal and Chemoresistance of Cancer Stem Cells in Human Hepatocellular Carcinoma through Stabilizing Nanog. Theranostics 2019, 9, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, Y.M.; Sun, B.; Zhong, F.J.; Yang, L.Y. GNA14′s interaction with RACK1 inhibits hepatocellular carcinoma progression through reducing MAPK/JNK and PI3K/AKT signaling pathway. Carcinogenesis 2021, 42, 1357–1369. [Google Scholar] [CrossRef]
- Li, T.; Yi, J.; Wu, H.; Wang, K.; Zhou, B. SLC7A11 in hepatocellular carcinoma: Potential mechanisms, regulation, and clinical significance. Am. J. Cancer Res. 2024, 14, 2326–2342. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, W.; Liu, H.; Liu, N.; Chen, D.; Tian, D.; Wang, J. Research progress on SLC7A11 in the regulation of cystine/cysteine metabolism in tumors. Oncol. Lett. 2022, 23, 47. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar] [PubMed]
- Li, S.; Lu, Z.; Sun, R.; Guo, S.; Gao, F.; Cao, B.; Aa, J. The Role of SLC7A11 in Cancer: Friend or Foe? Cancers 2022, 14, 3059. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Wang, W.; Liu, B.; Yang, G.; Xu, Z.; Li, J.; Liu, Z. RNA binding protein DAZAP1 promotes HCC progression and regulates ferroptosis by interacting with SLC7A11 mRNA. Exp. Cell Res. 2021, 399, 112453. [Google Scholar] [CrossRef]
- Lyu, N.; Zeng, Y.; Kong, Y.; Chen, Q.; Deng, H.; Ou, S.; Bai, Y.; Tang, H.; Wang, X.; Zhao, M. Ferroptosis is involved in the progression of hepatocellular carcinoma through the circ0097009/miR-1261/SLC7A11 axis. Ann. Transl. Med. 2021, 9, 675. [Google Scholar] [CrossRef]
- Fan, Z.; Yang, G.; Zhang, W.; Liu, Q.; Liu, G.; Liu, P.; Xu, L.; Wang, J.; Yan, Z.; Han, H.; et al. Hypoxia blocks ferroptosis of hepatocellular carcinoma via suppression of METTL14 triggered YTHDF2-dependent silencing of SLC7A11. J. Cell Mol. Med. 2021, 25, 10197–10212. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Qiu, Y.; Wu, Z.; Liu, S.; Zuo, D.; Huang, Z.; Li, B.; Yuan, Y.; Niu, Y.; Qiu, J. METTL9-SLC7A11 axis promotes hepatocellular carcinoma progression through ferroptosis inhibition. Cell Death Discov. 2023, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, K.; Lu, Y.; Zhang, D.; Cheng, Y.; Li, L.; Huang, W.; He, G.; Liao, H.; Cai, L.; et al. ABCC5 facilitates the acquired resistance of sorafenib through the inhibition of SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia 2021, 23, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Pan, J.; Zhang, Y.; Li, Y.; Jin, S.; Zhong, C.; Chen, P.; Ma, J.; Hu, W.; Fan, X.; et al. C8orf76 Modulates Ferroptosis in Liver Cancer via Transcriptionally Up-Regulating SLC7A11. Cancers 2022, 14, 3410. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zheng, W.; Guan, J.; Liu, H.; Dan, Y.; Zhu, L.; Song, Y.; Zhou, Y.; Zhao, X.; Zhang, Y.; et al. SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ. 2023, 30, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Bao, W.; Zhang, S.; Chen, B.; Zhou, X.; Zhao, J.; Shi, Z.; Zhang, T.; Chen, Z.; Wang, L.; et al. LncRNA HEPFAL accelerates ferroptosis in hepatocellular carcinoma by regulating SLC7A11 ubiquitination. Cell Death Dis. 2022, 13, 734. [Google Scholar] [CrossRef]
- Shi, Z.; Li, Z.; Jin, B.; Ye, W.; Wang, L.; Zhang, S.; Zheng, J.; Lin, Z.; Chen, B.; Liu, F.; et al. Loss of LncRNA DUXAP8 synergistically enhanced sorafenib induced ferroptosis in hepatocellular carcinoma via SLC7A11 de-palmitoylation. Clin. Transl. Med. 2023, 13, e1300. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, L. Long noncoding RNA CASC11 suppresses sorafenib-triggered ferroptosis via stabilizing SLC7A11 mRNA in hepatocellular carcinoma cells. Discov. Oncol. 2023, 14, 145. [Google Scholar] [CrossRef]
- Jin, D.; Hui, Y.; Liu, D.; Li, N.; Leng, J.; Wang, G.; Wang, Q.; Lu, Z. LINC00942 inhibits ferroptosis and induces the immunosuppression of regulatory T cells by recruiting IGF2BP3/SLC7A11 in hepatocellular carcinoma. Funct. Integr. Genom. 2024, 24, 29. [Google Scholar] [CrossRef]
- Zong, K.; Lin, C.; Luo, K.; Deng, Y.; Wang, H.; Hu, J.; Chen, S.; Li, R. Ferroptosis-related lncRNA NRAV affects the prognosis of hepatocellular carcinoma via the miR-375-3P/SLC7A11 axis. BMC Cancer 2024, 24, 496. [Google Scholar] [CrossRef]
- Wang, Y.F.; Feng, J.Y.; Zhao, L.N.; Zhao, M.; Wei, X.F.; Geng, Y.; Yuan, H.F.; Hou, C.Y.; Zhang, H.H.; Wang, G.W.; et al. Aspirin triggers ferroptosis in hepatocellular carcinoma cells through restricting NF-kappaB p65-activated SLC7A11 transcription. Acta Pharmacol. Sin. 2023, 44, 1712–1724. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, J.; Tian, W.; Ge, C.; Su, Y.; Li, J.; Tian, H. AKR1C3 suppresses ferroptosis in hepatocellular carcinoma through regulation of YAP/SLC7A11 signaling pathway. Mol. Carcinog. 2023, 62, 833–844. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zhang, P.; Liu, J.; Wang, R.; Kaufman, R.J.; Yaden, B.C.; Karin, M. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J. Hepatol. 2023, 79, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, J.; Xiang, X.; Xie, C.; Lu, X.; Guo, H.; Sun, Y.; Shi, Z.; Song, H.; Qiu, N.; et al. An Esterase-Responsive SLC7A11 shRNA Delivery System Induced Ferroptosis and Suppressed Hepatocellular Carcinoma Progression. Pharmaceutics 2024, 16, 249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jin, X.; Huang, H. Deregulation of SPOP in Cancer. Cancer Res. 2023, 83, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Song, Y.; Ye, M.; Dai, X.; Zhu, X.; Wei, W. The diverse roles of SPOP in prostate cancer and kidney cancer. Nat. Rev. Urol. 2020, 17, 339–350. [Google Scholar] [CrossRef]
- Wang, L.; Lin, M.; Chu, M.; Liu, Y.; Ma, J.; He, Y.; Wang, Z.W. SPOP promotes ubiquitination and degradation of LATS1 to enhance kidney cancer progression. EBioMedicine 2020, 56, 102795. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, Y.; Pan, C.; Yan, L.; Wang, Z.W.; Zhu, X. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol. Cancer 2020, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, J.; Lyu, H.; Yang, J.; Wei, W.; Wang, Y.; Luo, H.; Zhang, Y.; Jiang, X.; Yi, H.; et al. CSN6-SPOP-HMGCS1 Axis Promotes Hepatocellular Carcinoma Progression via YAP1 Activation. Adv Sci 2024, 11, e2306827. [Google Scholar] [CrossRef]
- Yu, Z.; Wu, X.; Zhu, J.; Yan, H.; Li, Y.; Zhang, H.; Zhong, Y.; Lin, M.; Ye, G.; Li, X.; et al. BCLAF1 binds SPOP to stabilize PD-L1 and promotes the development and immune escape of hepatocellular carcinoma. Cell Mol. Life Sci. 2024, 81, 82. [Google Scholar] [CrossRef]
- Deng, Y.; Ding, W.; Ma, K.; Zhan, M.; Sun, L.; Zhou, Z.; Lu, L. SPOP point mutations regulate substrate preference and affect its function. Cell Death Dis. 2024, 15, 172. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.A.; Zhou, P.; Chang, W.Y.; Yang, J.Y.; Zhang, Y.L.; Huang, A.L.; Tang, N.; Wang, K. SPOP promotes CREB5 ubiquitination to inhibit MET signaling in liver cancer. Biochim. Biophys. Acta Mol. Cell Res. 2024, 1871, 119642. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, A.; Xue, M.; Zhu, X.; Liu, Y.; Chen, M. FOXA1 and FOXA2: The regulatory mechanisms and therapeutic implications in cancer. Cell Death Discov. 2024, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Ma, D.N.; Chen, Q.D.; Zhang, J.J.; Tian, Y.R.; Wang, Z.C.; Cai, H.; Lin, Y.; Sun, H.C. MicroRNA-200a inhibits cell growth and metastasis by targeting Foxa2 in hepatocellular carcinoma. J. Cancer 2017, 8, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xiang, L.; Hu, Z.; Ou, H.; Liu, X.; Yu, L.; Chen, W.; Jiang, L.; Yu, Q.; Fang, Y.; et al. Epigenetically silenced linc00261 contributes to the metastasis of hepatocellular carcinoma via inducing the deficiency of FOXA2 transcription. Am. J. Cancer Res. 2021, 11, 277–296. [Google Scholar] [PubMed]
- Wei, X.; Li, X.; Yan, W.; Zhang, X.; Sun, Y.; Zhang, F. SKP2 Promotes Hepatocellular Carcinoma Progression Through Nuclear AMPK-SKP2-CARM1 Signaling Transcriptionally Regulating Nutrient-Deprived Autophagy Induction. Cell Physiol. Biochem. 2018, 47, 2484–2497. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Dong, X.; Lin, P.; Jiang, J. Regulation of Akt/FoxO3a/Skp2 Axis Is Critically Involved in Berberine-Induced Cell Cycle Arrest in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2018, 19, 327. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bian, Y.; Takizawa, Y.; Hashimoto, T.; Ikoma, T.; Tanaka, J.; Kitamura, N.; Inagaki, Y.; Komada, M.; Tanaka, T. ERK-dependent downregulation of Skp2 reduces Myc activity with HGF, leading to inhibition of cell proliferation through a decrease in Id1 expression. Mol. Cancer Res. 2013, 11, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xuan, Z.; Song, W.; Han, W.; Chen, H.; Du, Y.; Xie, H.; Zhao, Y.; Zheng, S.; Song, P. EAG1 enhances hepatocellular carcinoma proliferation by modulating SKP2 and metastasis through pseudopod formation. Oncogene 2021, 40, 163–176. [Google Scholar] [CrossRef]
- Su, K.J.; Yu, Y.L. Downregulation of SHIP2 by Hepatitis B Virus X Promotes the Metastasis and Chemoresistance of Hepatocellular Carcinoma through SKP2. Cancers 2019, 11, 1065. [Google Scholar] [CrossRef]
- Du, Y.; Song, W.; Chen, J.; Chen, H.; Xuan, Z.; Zhao, L.; Chen, J.; Jin, C.; Zhou, M.; Tuo, B.; et al. The potassium channel KCa3.1 promotes cell proliferation by activating SKP2 and metastasis through the EMT pathway in hepatocellular carcinoma. Int. J. Cancer 2019, 145, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V. Rab3a and Rab10 are regulators of lysosome exocytosis and plasma membrane repair. Small GTPases 2018, 9, 349–351. [Google Scholar] [CrossRef]
- Yang, C.C.; Meng, G.X.; Dong, Z.R.; Li, T. Role of Rab GTPases in Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 1389–1397. [Google Scholar] [CrossRef]
- Wang, W.; Jia, W.D.; Hu, B.; Pan, Y.Y. RAB10 overexpression promotes tumor growth and indicates poor prognosis of hepatocellular carcinoma. Oncotarget 2017, 8, 26434–26447. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Pan, Q.; Yu, Y.; Zhong, X.P. microRNA-519d Induces Autophagy and Apoptosis of Human Hepatocellular Carcinoma Cells Through Activation of the AMPK Signaling Pathway via Rab10. Cancer Manag. Res. 2020, 12, 2589–2602. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wu, C.; Xu, H.; Zou, R.; Li, T.; Ye, S. miR-557 inhibits hepatocellular carcinoma progression through Wnt/beta-catenin signaling pathway by targeting RAB10. Aging 2024, 16, 3716–3733. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, X.; Wang, X. m(6)A binding protein YTHDF2 in cancer. Exp. Hematol. Oncol. 2022, 11, 21. [Google Scholar] [CrossRef]
- Liu, R.; Jia, Y.; Kong, G.; He, A. Novel insights into roles of N6-methyladenosine reader YTHDF2 in cancer progression. J. Cancer Res. Clin. Oncol. 2022, 148, 2215–2230. [Google Scholar] [CrossRef]
- Wang, H.; Cai, H.; Li, L. Comprehensive analysis of m6A reader YTHDF2 prognosis, immune infiltration, and related regulatory networks in hepatocellular carcinoma. Heliyon 2024, 10, e23204. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Zhong, L.; Liao, D.; Zhang, M.; Zeng, C.; Li, X.; Zhang, R.; Ma, H.; Kang, T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019, 442, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhang, H.; Liu, J.; Zhao, Z.; Wang, J.; Lu, Z.; Hu, B.; Zhou, J.; Zhao, Z.; Feng, M.; et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol. Cancer 2019, 18, 163. [Google Scholar] [CrossRef]
- Zhang, C.; Huang, S.; Zhuang, H.; Ruan, S.; Zhou, Z.; Huang, K.; Ji, F.; Ma, Z.; Hou, B.; He, X. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene 2020, 39, 4507–4518. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, Y.; Yu, C.; Lei, Q.; Cheng, J.; Kong, W.; Yu, Y.; Zhuang, X.; Sun, W.; Yin, S.; et al. HSP90beta Impedes STUB1-Induced Ubiquitination of YTHDF2 to Drive Sorafenib Resistance in Hepatocellular Carcinoma. Adv. Sci. 2023, 10, e2302025. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Xue, L.; Wei, Y.; Liang, J.; Jia, W.; Yong, T.; Chu, L.; Li, H.; Han, S.; Liao, J.; et al. YTHDF2 Is a Therapeutic Target for HCC by Suppressing Immune Evasion and Angiogenesis Through ETV5/PD-L1/VEGFA Axis. Adv. Sci. 2024, 11, e2307242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wang, H.; Chai, S.; Xu, L.; Lin, B.; Yi, W.; Wu, L. O-GlcNAcylation promotes tumor immune evasion by inhibiting PD-L1 lysosomal degradation. Proc. Natl. Acad. Sci. USA 2023, 120, e2216796120. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gong, W.; Wang, H.; Li, T.; Attri, K.S.; Lewis, R.E.; Kalil, A.C.; Bhinderwala, F.; Powers, R.; Yin, G.; et al. O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity 2019, 50, 576–590. [Google Scholar] [CrossRef]
- Shang, M.; Yang, H.; Yang, R.; Chen, T.; Fu, Y.; Li, Y.; Fang, X.; Zhang, K.; Zhang, J.; Li, H.; et al. The folate cycle enzyme MTHFD2 induces cancer immune evasion through PD-L1 up-regulation. Nat. Commun. 2021, 12, 1940. [Google Scholar] [CrossRef]
- Rodrigues Mantuano, N.; Stanczak, M.A.; Oliveira, I.A.; Kirchhammer, N.; Filardy, A.A.; Monaco, G.; Santos, R.C.; Fonseca, A.C.; Fontes, M.; Bastos, C.S., Jr.; et al. Hyperglycemia Enhances Cancer Immune Evasion by Inducing Alternative Macrophage Polarization through Increased O-GlcNAcylation. Cancer Immunol. Res. 2020, 8, 1262–1272. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, L.; Ge, D.; Tan, L.; Cao, B.; Fan, H.; Xue, L. Exosomal O-GlcNAc transferase from esophageal carcinoma stem cell promotes cancer immunosuppression through up-regulation of PD-1 in CD8+ T cells. Cancer Lett. 2021, 500, 98–106. [Google Scholar] [CrossRef]
- Shan, X.; Jiang, R.; Gou, D.; Xiang, J.; Zhou, P.; Xia, J.; Wang, K.; Huang, A.; Tang, N.; Huang, L. Identification of a diketopiperazine-based O-GlcNAc transferase inhibitor sensitizing hepatocellular carcinoma to CDK9 inhibition. FEBS J. 2023, 290, 4543–4561. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Duan, F.; Pan, Z.; Liu, X.; Lu, W.; Liang, C.; Fang, Z.; Peng, P.; Jia, D. Astragalus Polysaccharide Promotes Doxorubicin-Induced Apoptosis by Reducing O-GlcNAcylation in Hepatocellular Carcinoma. Cells 2023, 12, 866. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Y.; Zhang, C.; Chen, X.; Huang, H.; Li, W.; Zhang, J.; Liu, Y. Upregulation of OGT by Caveolin-1 promotes hepatocellular carcinoma cell migration and invasion. Cell Biol. Int. 2021, 45, 2251–2263. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
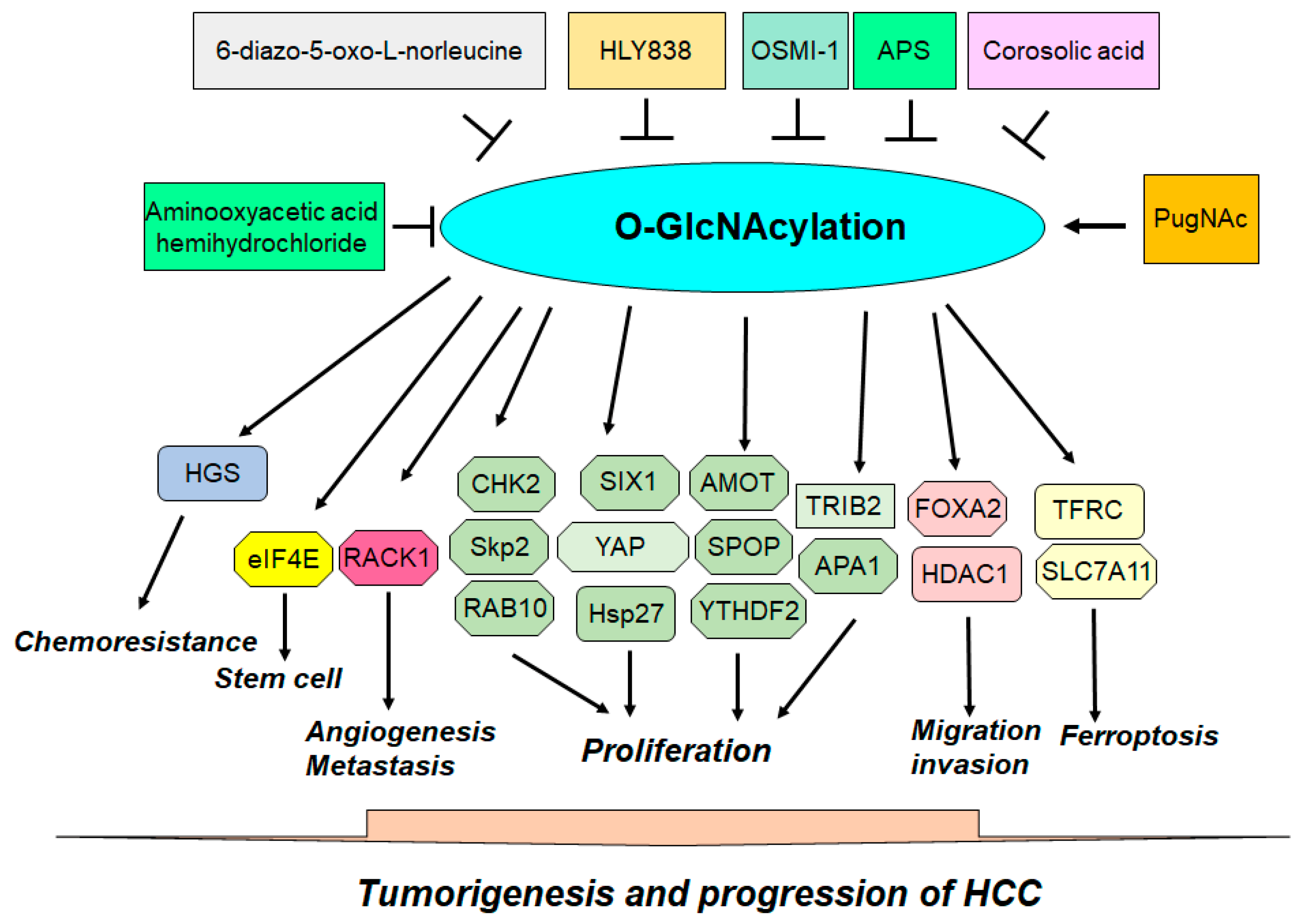
| Target | Mechanism | Function | Ref. |
|---|---|---|---|
| Hsp27 | The O-GlcNAcylation of Hsp27 regulates its nuclear translocation. | The O-GlcNAcylation and phosphorylation of Hsp27 influence biological activities in HCC. | [52] |
| HDAC1 | The O-GlcNAcylation enhances the phosphorylation and enzymatic activity of HDAC1 and influences the transcriptional regulation of p21 by altering histone acetylation levels. | O-GlcNAc-modified HDAC1 mutants impact HCC cell proliferation and cell invasion and migration capabilities in HCC. | [53] |
| TFRC | Erastin promotes the removal of O-GlcNAcylation from TFRC and reduces its interaction with MARCH8, leading to decreased polyubiquitination. | TFRC is modified by O-GlcNAcylation, affecting its sensitivity to Erastin-induced ferroptosis in HCC cells. | [54] |
| TFRC | The O-GlcNAcylation heightens the susceptibility of HCC cells to ferroptosis through the action of YAP. | O-GlcNAcylation promotes ferroptosis sensitivity through TFRC in HCC cells. | [55] |
| YAP | The O-GlcNAcylation increases the expression, stability, and functionality of YAP in liver cancer by regulating its phosphorylation. | The O-GlcNAcylation of YAP is necessary for liver cancer development induced by high glucose levels. | [56] |
| AMOT | High glucose levels increase O-GlcNAcylation of AMOT and facilitate the nuclear accumulation of YAP through AMOT. | Targeting AMOT O-GlcNAcylation offers an effective therapeutic strategy for treating liver cancer with diabetes. | [57] |
| TRIB2 | The O-GlcNAcylation of TRIB2 increases its protein stability. TRIB2 stabilizes GUCY1A3 by interacting with it and reducing its ubiquitination. | The O-GlcNAcylation of TRIB2 promotes transformative characteristics in liver cancer cells. | [58] |
| HGS | The O-GlcNAcylation of HGS reduces its interaction with STAM, disrupts the ESCRT-0 complex, and promotes HGS ubiquitination and EGFR accumulation. | The O-GlcNAcylation of HGS enhances tumor growth in mice and increases chemoresistance in liver cancer cells. | [59] |
| CHK2 | Reduced PCK1 expression promotes the O-GlcNAcylation of CHK2 and increases CHK2-dependent Rb phosphorylation. | The O-GlcNAcylation of CHK2 accelerates cell proliferation. | [60] |
| Target | Mechanism | Function | Ref. |
|---|---|---|---|
| APA1 | APA1 undergoes O-GlcNAcylation, which is critical for the HG-induced binding of APA1 to the GJC1 promoter. | Reducing O-GlcNAcylation eliminates the HG-driven increase in cell proliferation. | [61] |
| eIF4E | The O-GlcNAcylation of eIF4E is located at Thr168 and THr177, resulting in the protection of eIF4E from degradation. | High glucose augments stem-like cell functions by promoting the O-GlcNAcylation of eIF4E. | [62] |
| SIX1 | The O-GlcNAcylation of SIX1 prevents its breakdown through the ubiquitination pathway. | O-GlcNAcylation stabilizes SIX1 and enhances HCC cell proliferation. | [63] |
| RACK1 | RACK1 O-GlcNAcylation stabilizes the RACK1 protein and enhances its association with ribosomes and PRKCB, leading to the elevated phosphorylation of eIF4E. | The O-GlcNAcylation of RACK1 increases tumor growth, angiogenesis, and metastasis. | [64] |
| SLC7A11 | USP8 increases OGT stabilization by inhibiting OGT ubiquitination. OGT leads to SLC7A11 O-GlcNAcylation in HCC cells. | Promotes cystine importation from the extracellular environment and regulates ferroptosis. | [65] |
| SPOP | O-GlcNAcylation alters SPOP localization, moving it predominantly into the nucleus, reducing Nogo-B degradation. | This relocation enhances the progression of HCC. | [66] |
| FOXA2 | The O-GlcNAcylation of FOXA2 leads to the ubiquitin-dependent degradation of FOXA2 in metastatic HCC cell lines. | The O-GlcNAcylation of FOXA2 is critical for HCC metastasis, invasion, and migration through E-cadherin inhibition. | [67]. |
| Skp2 | Skp2 O-GlcNAcylation causes its stabilization by decreasing its degradation by the APC-CDH1 complex and enhances its interaction with Skp1, improving its function. | Allows Skp2 to effectively facilitate the cell cycle G1–S phase transition by targeting p27 and p21 for degradation and induces HCC cell proliferation. | [68] |
| RAB10 | RAB10 directly binds with OGT, and this O-GlcNAcylation increases the stability of the RAB10 protein. | Increased RAB10 levels promote the aggressive characteristics of HCC. | [69] |
| YTHDF2 | HBC infection increases YTHDF2 O-GlcNAcylation, leading to increased YTHDF2 stability due to the suppression of its ubiquitination. | YTHDF2 enhances the stability of MCM2 and MCM5 transcripts and accelerates cell cycle progression and HCC tumorigenesis. | [70] |
| Compound | Target | Function | Ref. |
|---|---|---|---|
| OSMI-1 | Inhibitor of OGT | Inhibits tumor progression in HCC. | [70] |
| Aminooxyacetic acid hemihydrochloride | HBP-mediated O-GlcNAcylation | Suppresses tumor growth in liver-specific Pck1-knockout mice. | [60] |
| 6-diazo-5-oxo-L-norleucine | HBP-induced O-GlcNAcylation | Inhibits tumor growth in liver-specific Pck1-knockout mice. | [60] |
| Corosolic acid | OGT and HBP | Inhibits HBP activation and OGT expression and represses YAP and O-GlcNAcylation through CDK19 suppression. | [101] |
| HLY838 | A new diketopiperazine-derived OGT inhibitor | Triggers a global reduction in cellular O-GlcNAc levels and increases CDK9 inhibitor-mediated anticancer activity through the inhibition of c-Myc and E2F1 in HCC. | [231] |
| APS | Reduces the stability of OGT and induces OGA expression | Induces doxorubicin-mediated apoptosis and elevated endoplasmic reticulum stress by reducing O-GlcNAcylation. | [232] |
| PugNAc | OGA inhibitor | Abrogates cell apoptosis and endoplasmic reticulum stress induced by doxorubicin and APS. | [232] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Hang, S.; Wang, Q.; Xu, L.; Wang, P. Decoding the Role of O-GlcNAcylation in Hepatocellular Carcinoma. Biomolecules 2024, 14, 908. https://doi.org/10.3390/biom14080908
Zhou X, Hang S, Wang Q, Xu L, Wang P. Decoding the Role of O-GlcNAcylation in Hepatocellular Carcinoma. Biomolecules. 2024; 14(8):908. https://doi.org/10.3390/biom14080908
Chicago/Turabian StyleZhou, Xinyu, Sirui Hang, Qingqing Wang, Liu Xu, and Peter Wang. 2024. "Decoding the Role of O-GlcNAcylation in Hepatocellular Carcinoma" Biomolecules 14, no. 8: 908. https://doi.org/10.3390/biom14080908
APA StyleZhou, X., Hang, S., Wang, Q., Xu, L., & Wang, P. (2024). Decoding the Role of O-GlcNAcylation in Hepatocellular Carcinoma. Biomolecules, 14(8), 908. https://doi.org/10.3390/biom14080908