
Morphological Structure Identification, Comparative Mitochondrial Genomics and Population Genetic Analysis toward Exploring Interspecific Variations and Phylogenetic Implications of Malus baccata ‘ZA’ and Other Species


Abstract
1. Introduction
2. Materials and Methods
2.1. Material Collection, Sample Extraction, and DNA Sequencing
2.2. Sequencing Data Processing and Mitochondrial Genome Assembly
2.3. Annotation of Coding Sequence, Transfer RNA, and Ribosomal RNA in Mitogenome
2.4. Mitogenome Composition, Codon Usage Bias, and Collinearity Analysis
2.5. Identification of Mitochondrial Plastid DNAs and Exchange of Organelle Fragments
2.6. Population Evolution Based on Mitochondrial Genome
2.7. Phylogenetic Relationship and Interspecific Variation of Rosaceae
3. Results
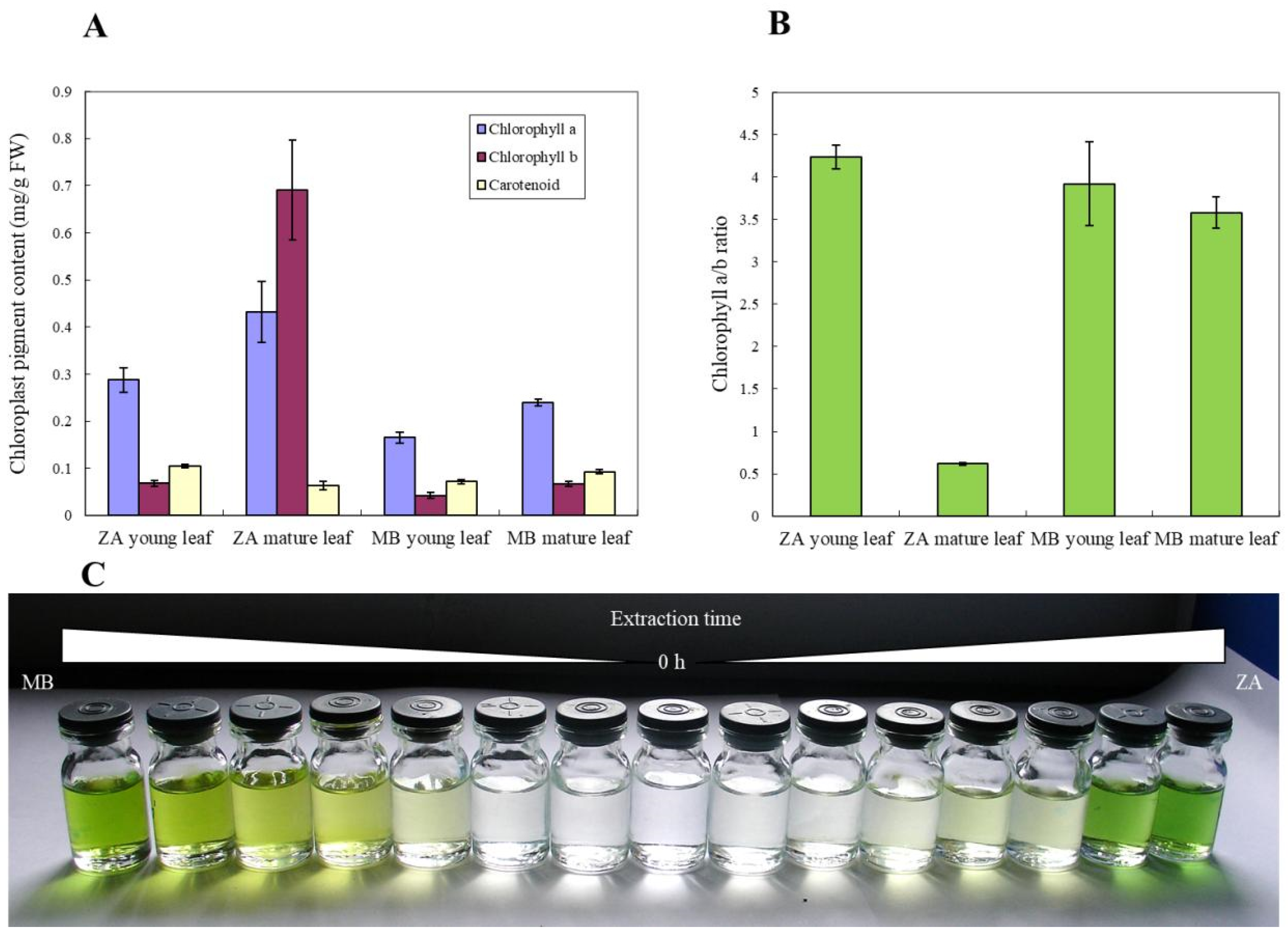
3.1. Morphological and Physiological Characteristics of M. baccata ‘ZA’
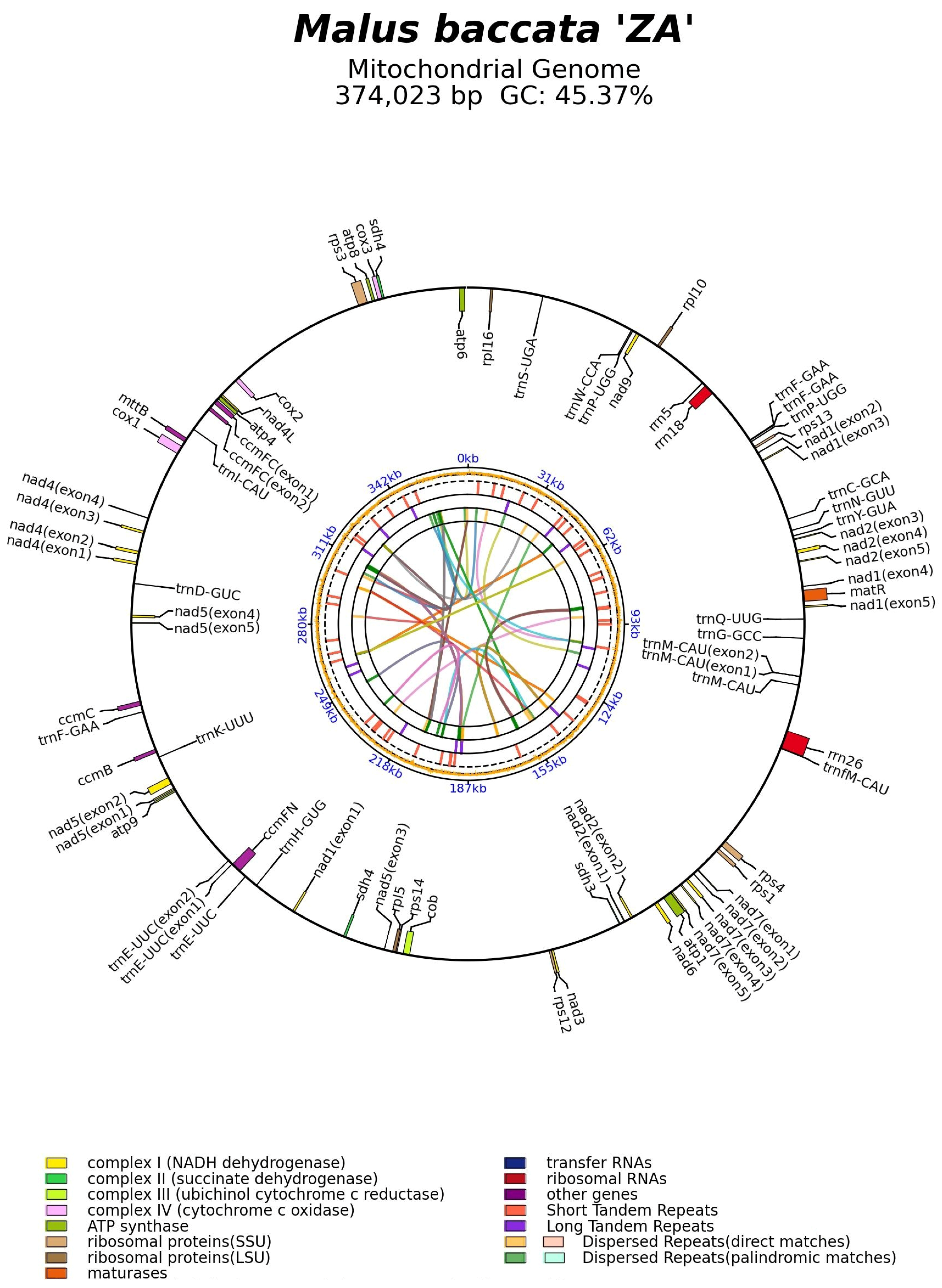
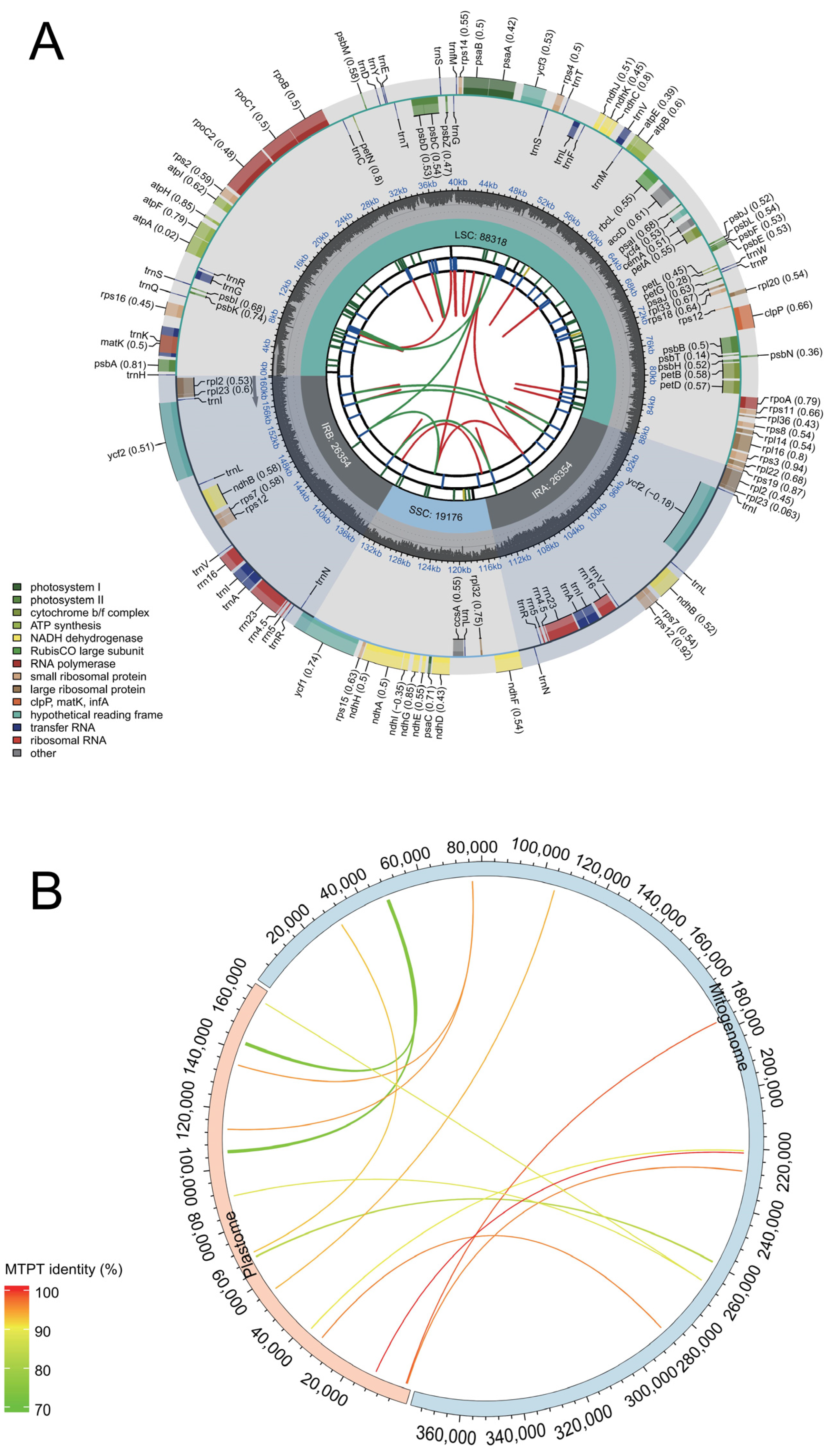
3.2. Basic Characteristics and Annotations of Malus baccata ‘ZA’ Mitogenome
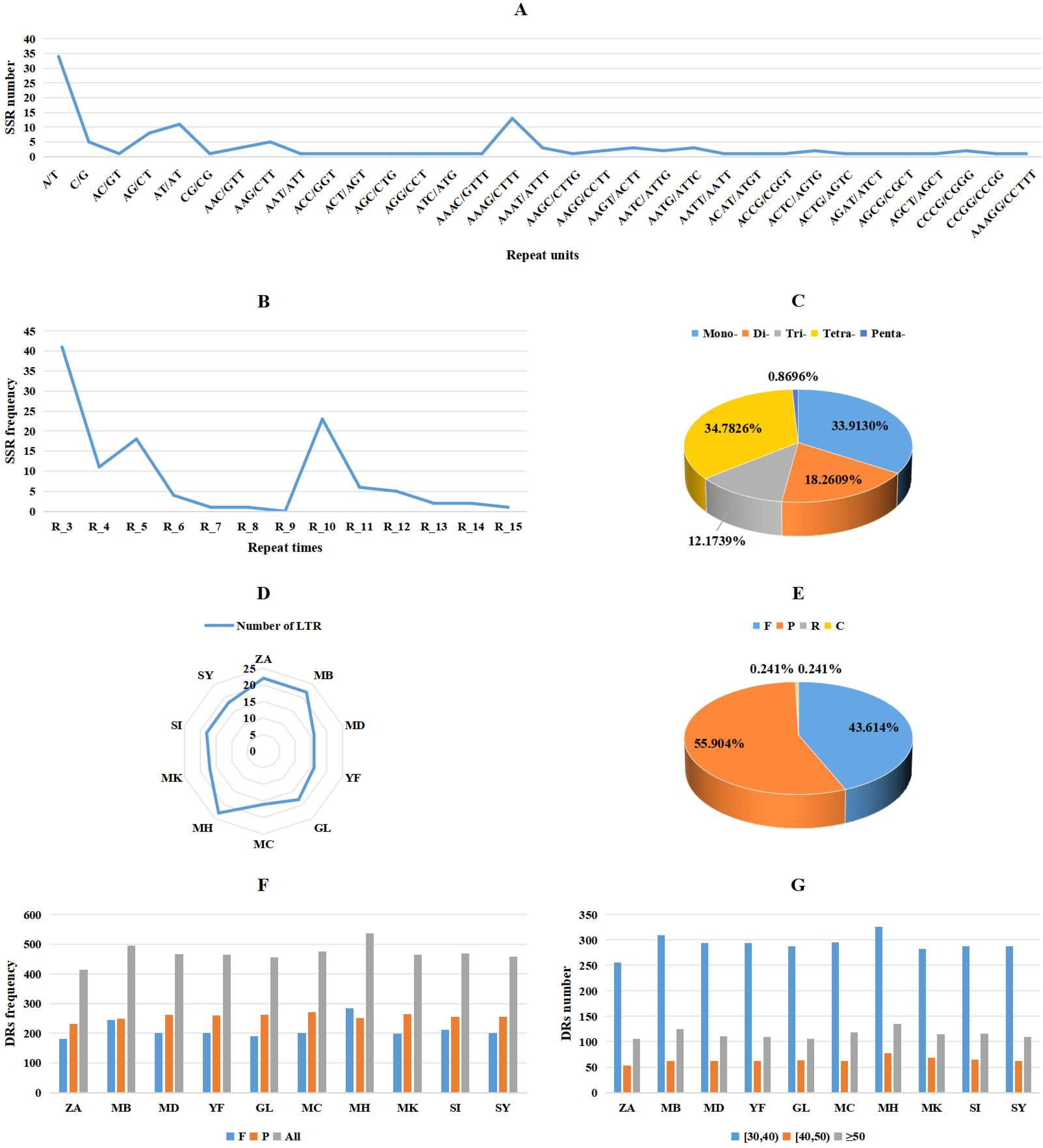
3.3. Repeat Sequences in Mitochondrial Genomes of M. baccata ‘ZA’ and Other Malus Species
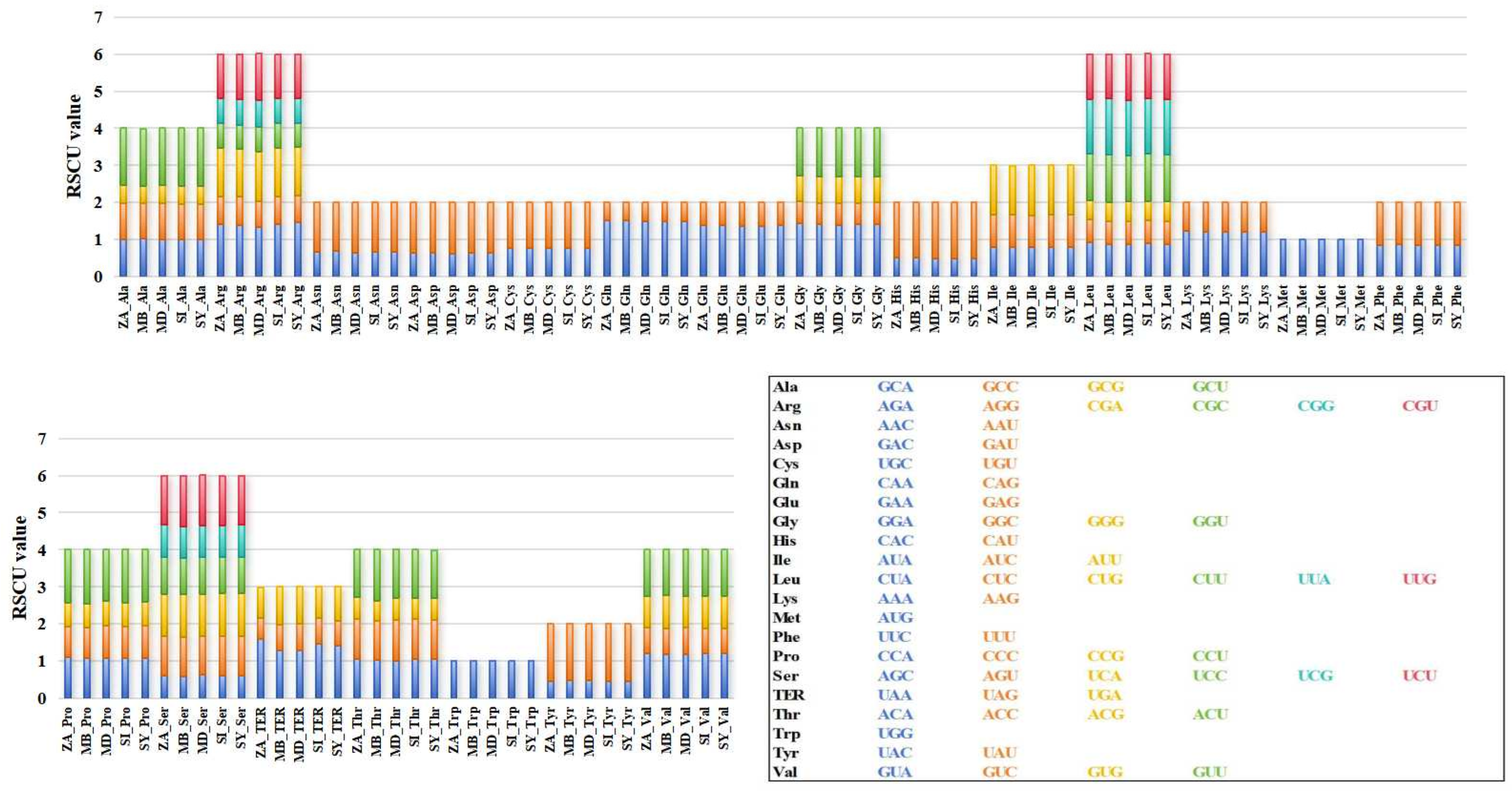
3.4. Codon Preference Analysis of Mitochondrial Coding Genes in M. baccata ‘ZA’
3.5. Interspecific and Intraspecific Collinearity of Mitogenomes
3.6. Assembly of Plastid Genome in M. baccata ‘ZA’ and Identification of MTPTs
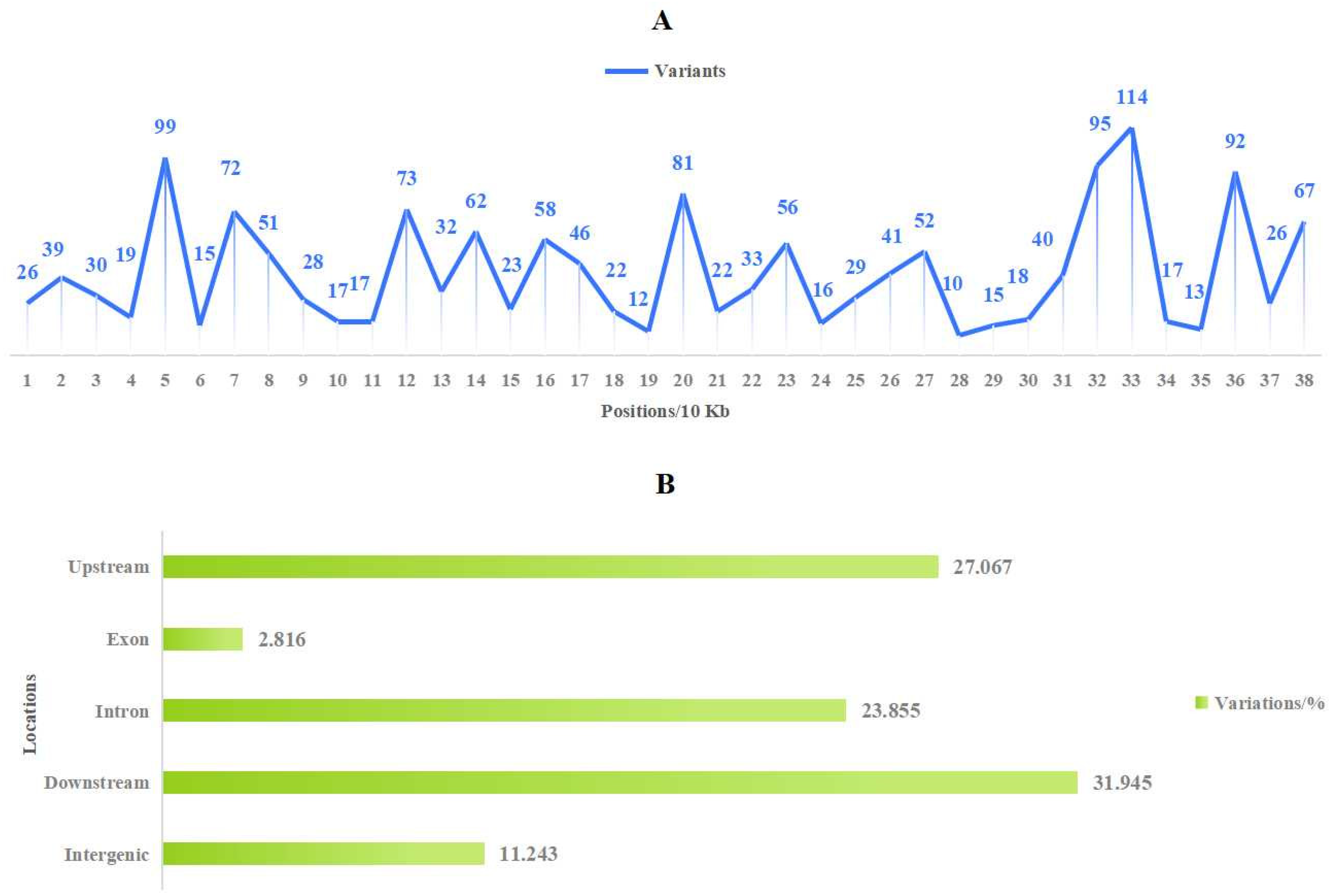
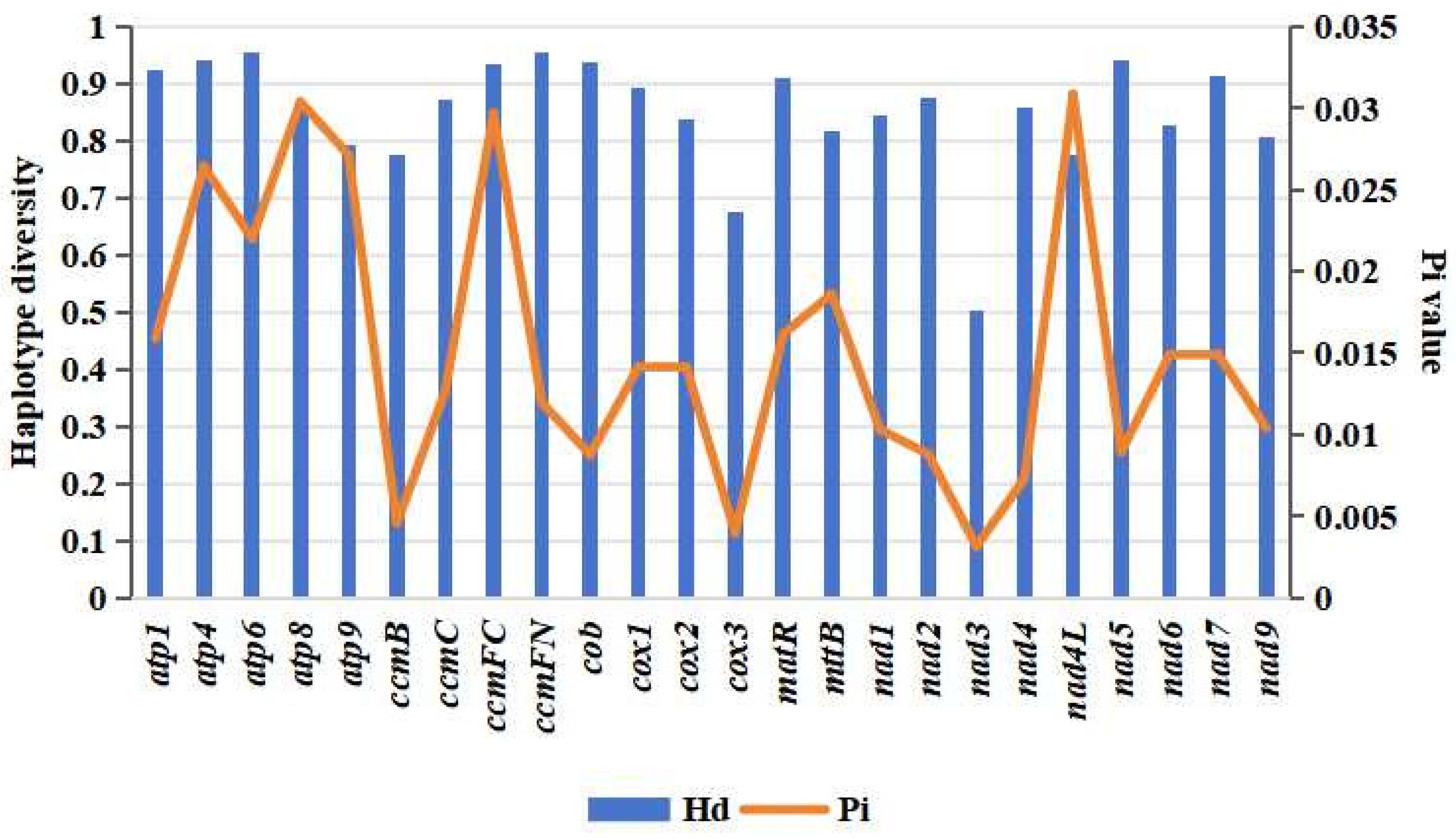
3.7. Population Evolution Analysis Based on Mitochondrial Genome Polymorphisms in Malus
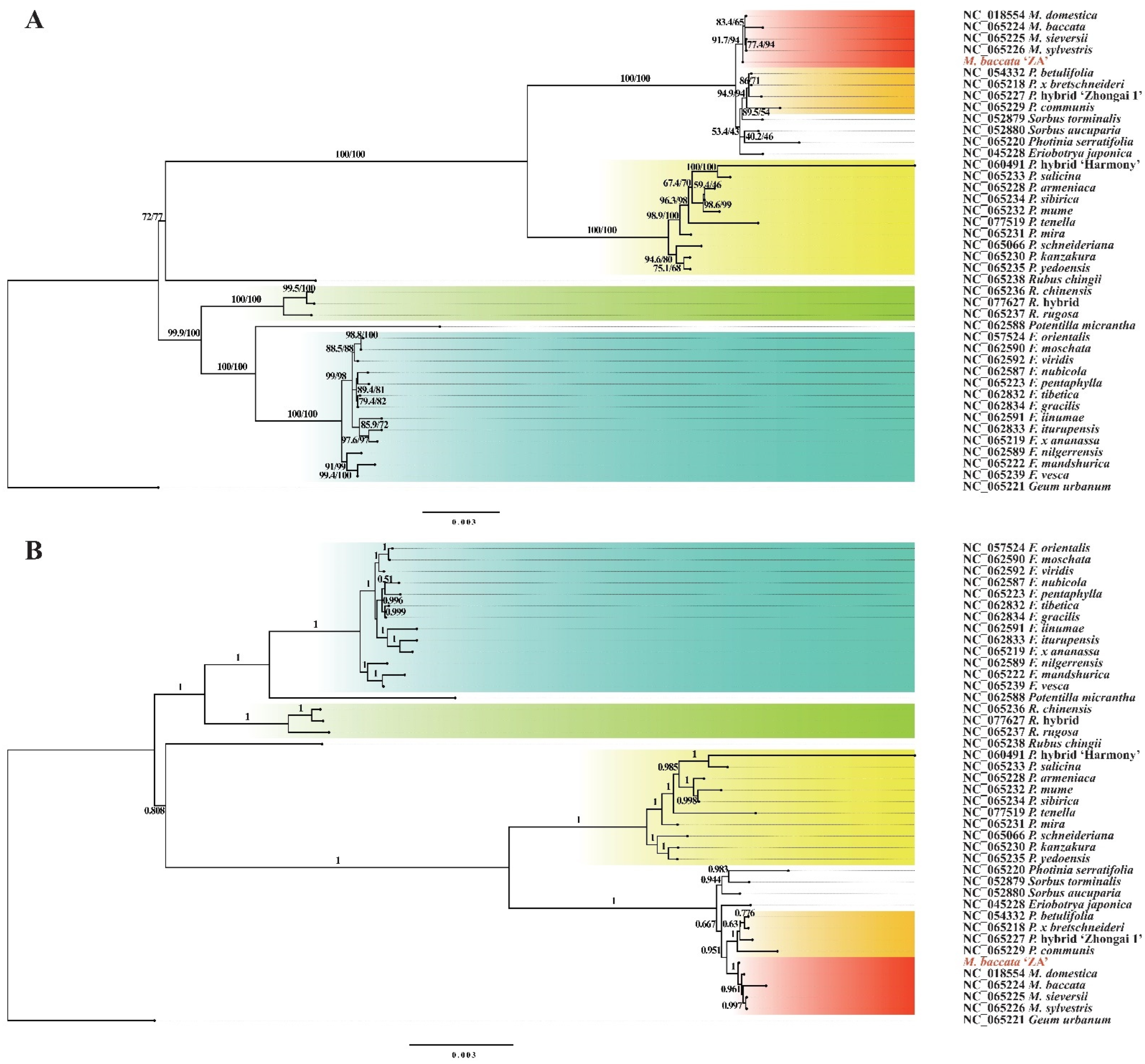
3.8. Phylogenetic Relationship between M. baccata ‘ZA’ and Other Species of Rosaceae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gao, Y.; Wang, D.-J.; Wang, K.; Cong, P.-H.; Li, L.-W.; Piao, J.-C. Analysis of genetic diversity and structure across a wide range of germplasm reveals genetic relationships among seventeen species of Malus Mill. native to China. J. Integr. Agric. 2021, 20, 3186–3198. [Google Scholar] [CrossRef]
- Wang, D.; Gao, Y.; Sun, S.; Lu, X.; Li, Q.; Li, L.; Wang, K.; Liu, J. Effects of salt stress on the antioxidant activity and malondialdehyde, solution protein, proline, and chlorophyll contents of three Malus species. Life 2022, 12, 1929. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, R.; Wang, D.; Yang, C.; Zhang, Y.; Sui, M.; Quan, J.; Sun, Y.; You, C.; Shen, X. Molecular structure and variation characteristics of the plastomes from six Malus baccata (L.) Borkh. individuals and comparative genomic analysis with other Malus species. Biomolecules 2023, 13, 962. [Google Scholar] [CrossRef] [PubMed]
- Stavitskaya, Z.; Dudareva, L.; Rudikovskii, A.; Garkava-Gustavsson, L.; Shabanova, E.; Levchuk, A.; Rudikovskaya, E. Evaluation of the carbohydrate composition of crabapple fruit tissues native to Northern Asia. Plants 2023, 12, 3472. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wang, Q.; Gao, J.; Li, C.; Du, X.; Ding, B.; Yang, T. Construction of a high-density genetic linkage map and QTL analysis of morphological traits in an F1 Malus domestica × Malus baccata hybrid. Physiol. Mol. Biol. Plants 2021, 27, 1997–2007. [Google Scholar] [CrossRef]
- Wang, X.; Wang, D.; Gao, N.; Han, Y.; Wang, X.; Shen, X.; You, C. Identification of the complete chloroplast genome of Malus zhaojiaoensis Jiang and its comparison and evolutionary analysis with other Malus species. Genes 2022, 13, 560. [Google Scholar] [CrossRef]
- Chen, X.; Cornille, A.; An, N.; Xing, L.; Ma, J.; Zhao, C.; Wang, Y.; Han, M.; Zhang, D. The East Asian wild apples, Malus baccata (L.) Borkh and Malus hupehensis (Pamp.) Rehder., are additional contributors to the genomes of cultivated European and Chinese varieties. Mol. Ecol. 2023, 32, 5125–5139. [Google Scholar] [CrossRef] [PubMed]
- Elansary, H.O.; Szopa, A.; Kubica, P.; O. El-Ansary, D.; Ekiert, H.; A. Al-Mana, F. Malus baccata var. gracilis and Malus toringoides bark polyphenol studies and antioxidant, antimicrobial and anticancer activities. Processes 2020, 8, 283. [Google Scholar] [CrossRef]
- Qin, X.; Hao, Q.; Wang, X.; Liu, Y.; Yang, C.; Sui, M.; Zhang, Y.; Hu, Y.; Chen, X.; Mao, Z.; et al. Complete chloroplast genome of the Malus baccata var. gracilis provides insights into the evolution and phylogeny of Malus species. Funct. Integr. Genom. 2024, 24, 13. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, X.; Ta, N.; Yuan, S.; Gong, Z.; Zhou, R. Zhaai shandingzi—Dwarf and cold resistant germplasm in Malus. China Fruits 1997, 3, 13–14. [Google Scholar]
- Wang, Y.; Shang, Y.; Wang, Y.; Wei, X.; Dong, W. Characteristics about embryo development of the hybridized offspring from pingyi tiancha [Malus hupehensis (Pamp.) Rehd. var. pingyiensis Jiang)] and zha’ai shandingzi [Malus baccata (L.) Borkh.]. Acta Hortic. Sin. 2008, 8, 1093–1100. [Google Scholar]
- Zhou, P.; Zhang, J.; Wang, Y.; Zhou, Y.; Wang, Y.; Zhou, H.; Dong, W. Ploidy identification and karyotype analysis of the hybrids crossed by pingyi tiancha [Malus hupehensis (Pamp.) Rehd] and zha’ai shangdingzi [Malus baccata (L.) Borkh.]. Chin. Agric. Sci. Bull. 2009, 25, 186–193. [Google Scholar]
- Wang, L.; Chen, J.; Xue, X.; Qin, G.; Gao, Y.; Li, K.; Zhang, Y.; Li, X.-J. Comparative analysis of mitogenomes among three species of grasshoppers (Orthoptera: Acridoidea: Gomphocerinae) and their phylogenetic implications. PeerJ 2023, 11, e16550. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bai, X.; Qian, H. Complete mitochondrial genome of Neuroctenus yunnanensis Hsiao, 1964 (Hemiptera: Aradidae: Mezirinae). Mitochondrial DNA B Resour. 2023, 8, 1373–1376. [Google Scholar] [CrossRef]
- Yang, F.; Long, L. Complete mitochondrial genome and phylogenetic analysis of the marine microalga Symbiochlorum hainanensis (Ulvophyceae, Chlorophyta). Mitochondrial DNA B Resour. 2023, 8, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Zhang, K.; Que, Y.; Li, Y. Assembly and analysis of the first complete mitochondrial genome of Punica granatum and the gene transfer from chloroplast genome. Front. Plant Sci. 2023, 14, 1132551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; He, W.; Kan, S.; Liao, X.; Jordan, D.R.; Mace, E.S.; Tao, Y.; Cruickshank, A.W.; Klein, R.; et al. Variation in mitogenome structural conformation in wild and cultivated lineages of sorghum corresponds with domestication history and plastome evolution. BMC Plant Biol. 2023, 23, 91. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Liu, F.; Jia, Q.; Du, H.; Chen, W.; Ruan, J.; Lei, J.; Li, D.-Z.; Mower, J.P.; Zhu, A. Fragaria mitogenomes evolve rapidly in structure but slowly in sequence and incur frequent multinucleotide mutations mediated by microinversions. New Phytol. 2022, 236, 745–759. [Google Scholar] [CrossRef]
- Lai, C.; Wang, J.; Kan, S.; Zhang, S.; Li, P.; Reeve, W.G.; Wu, Z.; Zhang, Y. Comparative analysis of mitochondrial genomes of Broussonetia spp. (Moraceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant Sci. 2022, 13, 1052151. [Google Scholar] [CrossRef]
- Duan, N.; Bai, Y.; Sun, H.; Wang, N.; Ma, Y.; Li, M.; Wang, X.; Jiao, C.; Legall, N.; Mao, L.; et al. Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat. Commun. 2017, 8, 249. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, C.; Schwaninger, H.; Chao, C.T.; Ma, Y.; Duan, N.; Khan, A.; Ban, S.; Xu, K.; Cheng, L.; et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 2020, 52, 1423–1432. [Google Scholar] [CrossRef]
- Chen, P.; Li, Z.; Zhang, D.; Shen, W.; Xie, Y.; Zhang, J.; Jiang, L.; Li, X.; Shen, X.; Geng, D.; et al. Insights into the effect of human civilization on Malus evolution and domestication. Plant Biotechnol. J. 2021, 19, 2206–2220. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhang, W.; Zhang, B.; Fang, T.; Wang, X.-F.; Cai, Y.; Ogutu, C.; Gao, L.; Chen, G.; Nie, X.; et al. Unraveling a genetic roadmap for improved taste in the domesticated apple. Mol. Plant 2021, 14, 1454–1471. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Chen, T.; Chen, X.; Zhang, S.; Zhu, J.; Tang, B.; Wang, A.; Dong, L.; Zhang, Z.; Yu, C.; Sun, Y.; et al. The Genome Sequence Archive family: Toward explosive data growth and diverse data types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar] [CrossRef]
- CNCB-NGDC Members and Partners. Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. The Sequence Read Archive: A decade more of explosive growth. Nucleic Acids Res. 2022, 50, D387–D390. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Carey, S.B.; Serrano, A.; Zhang, H.; Hargarten, H.; Hale, H.; Harkess, A.; Honaas, L. A phased, chromosome-scale genome of ‘Honeycrisp’ apple (Malus domestica). GigaByte 2022, 2022, gigabyte69. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Ni, Y.; Zhang, J.; Li, J.; Liu, C. Complete mitochondrial genome of Mentha spicata L. reveals multiple chromosomal configurations and RNA editing events. Int. J. Biol. Macromol. 2023, 251, 126257. [Google Scholar] [CrossRef]
- Yang, H.; Ni, Y.; Zhang, X.; Li, J.; Chen, H.; Liu, C. The mitochondrial genomes of Panax notoginseng reveal recombination mediated by repeats associated with DNA replication. Int. J. Biol. Macromol. 2023, 252, 126359. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, H.; Ni, Y.; Wu, B.; Li, J.; Burzyński, A.; Liu, C. Plant mitochondrial genome map (PMGmap): A software tool for the comprehensive visualization of coding, noncoding and genome features of plant mitochondrial genomes. Mol. Ecol. Resour. 2024, 24, e13952. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.-Y.; Graham, M.; Domselaar, G.V.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, R.; Peng, Y.; Chen, J.; Zhu, X.; Xie, K.; Ahmad, S.; Chen, J.; Wang, F.; Shen, M.; et al. The first mitochondrial genome of Melastoma dodecandrum resolved structure evolution in Melastomataceae and micro inversions from inner horizontal gene transfer. Ind. Crops Prod. 2023, 205, 117390. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Depamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, H.; Ni, Y.; Li, J.; Cai, Y.; Ma, B.; Yu, J.; Wang, J.; Liu, C. De novo hybrid assembly of the Salvia miltiorrhiza mitochondrial genome provides the first evidence of the multi-chromosomal mitochondrial DNA structure of Salvia species. Int. J. Mol. Sci. 2022, 23, 14267. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cheng, L.; Kim, K.W.; Park, Y.J. Evidence for selection events during domestication by extensive mitochondrial genome analysis between japonica and indica in cultivated rice. Sci. Rep. 2019, 9, 10846. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, M.; Chen, X.; Liu, Y.; Liu, B.; Li, J.; Wang, R.; Zhao, K.; Wu, J. Rearrangement and domestication as drivers of Rosaceae mitogenome plasticity. BMC Biol. 2022, 20, 181. [Google Scholar] [CrossRef] [PubMed]
- Goremykin, V.V.; Lockhart, P.J.; Viola, R.; Velasco, R. The mitochondrial genome of Malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 2012, 71, 615–626. [Google Scholar] [CrossRef]
- Ge, D.; Dong, J.; Guo, L.; Yan, M.; Zhao, X.; Yuan, Z. The complete mitochondrial genome sequence of cultivated apple (Malus domestica cv. ‘Yantai Fuji 8’). Mitochondrial DNA B Resour. 2020, 5, 1317–1318. [Google Scholar] [CrossRef]
- Zhai, X.; Wang, S.; Zheng, Y.; Yao, Y. Assembly and comparative analysis of four mitochondrial genomes of Malus. J. Beijing Univ. Agric. 2023, 38, 28–33. [Google Scholar]
- Duan, N.; Sun, H.; Wang, N.; Fei, Z.; Chen, X. The complete mitochondrial genome sequence of Malus hupehensis var. pinyiensis. Mitochondrial DNA B Resour. 2016, 27, 2905–2906. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gu, X.; Ma, J. Whole-genome assembly and evolutionary analysis of the Malus kansuensis (Rosaceae) mitochondrion. Mitochondrial DNA B Resour. 2021, 6, 3496–3497. [Google Scholar] [CrossRef] [PubMed]
- Kwasniak-Owczarek, M.; Janska, H. Experimental approaches to studying translation in plant semi-autonomous organelles. J. Exp. Bot. 2024, erae151. [Google Scholar] [CrossRef]
- Rozov, S.M.; Zagorskaya, A.A.; Konstantinov, Y.M.; Deineko, E.V. Three parts of the plant genome: On the way to success in the production of recombinant proteins. Plants 2022, 12, 38. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, N.; Li, S.; Grover, C.E.; Nie, H.; Wendel, J.F.; Hua, J. Plant mitochondrial genome evolution and cytoplasmic male sterility. Crit. Rev. Plant Sci. 2017, 36, 55–69. [Google Scholar] [CrossRef]
- Palmer, J.D.; Adams, K.L.; Cho, Y.; Parkinson, C.L.; Qiu, Y.L.; Song, K. Dynamic evolution of plant mitochondrial genomes: Mobile genes and introns and highly variable mutation rates. Proc. Natl. Acad. Sci. USA 2000, 97, 6960–6966. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.C. Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol. Evol. 2013, 5, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Newton, K.J. Plant mitochondrial genomes: Dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef]
- Tong, W.; He, Q.; Park, Y.J. Genetic variation architecture of mitochondrial genome reveals the differentiation in Korean landrace and weedy rice. Sci. Rep. 2017, 7, 43327. [Google Scholar] [CrossRef]
- Cao, S.; Zhang, H.; Liu, Y.; Sun, Y.; Chen, Z.J. Cytoplasmic genome contributions to domestication and improvement of modern maize. BMC Biol. 2024, 22, 64. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Gao, C.; Liu, J. Mitochondrial genome variation and intergenomic sequence transfers in Hevea species. Front. Plant Sci. 2024, 15, 1234643. [Google Scholar] [CrossRef]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024, 29, 754–769. [Google Scholar] [CrossRef]
- Liu, B.-B.; Ren, C.; Kwak, M.; Hodel, R.G.J.; Xu, C.; He, J.; Zhou, W.-B.; Huang, C.-H.; Ma, H.; Qian, G.-Z.; et al. Phylogenomic conflict analyses in the apple genus Malus s.l. reveal widespread hybridization and allopolyploidy driving diversification, with insights into the complex biogeographic history in the Northern Hemisphere. J. Integr. Plant Biol. 2022, 64, 1020–1043. [Google Scholar] [CrossRef]
- Wang, J.; Xue, L.; Zhang, X.; Hou, Y.; Zheng, K.; Fu, D.; Dong, W. A new function of MbIAA19 identified to modulate Malus plants dwarfing growth. Plants 2023, 12, 3097. [Google Scholar] [CrossRef]
- Li, J.; Tang, H.; Luo, H.; Tang, J.; Zhong, N.; Xiao, L. Complete mitochondrial genome assembly and comparison of Camellia sinensis var. Assamica cv. Duntsa. Front. Plant Sci. 2023, 14, 1117002. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zhang, Q.; Li, F.; Huang, J.; Zhang, M. Assembly and comparative analysis of the complete mitochondrial genome of Ilex metabaptista (Aquifoliaceae), a Chinese endemic species with a narrow distribution. BMC Plant Biol. 2023, 23, 393. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, S.; Zhang, D.; Han, M.; Jin, X.; Zhao, C.; Wang, S.; Xing, L.; Ma, J.; Ji, J.; et al. Sequencing of a wild apple (Malus baccata) genome unravels the differences between cultivated and wild apple species regarding disease resistance and cold tolerance. G3-Genes Genomes Genet. 2019, 9, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, F.; Wang, K.; Wang, D.; Gong, X.; Liu, L.; Richards, C.M.; Henk, A.D.; Volk, G.M. Genetic diversity of Malus cultivars and wild relatives in the Chinese National Repository of Apple Germplasm Resources. Tree Genet. Genomes 2015, 11, 106. [Google Scholar] [CrossRef]
- He, W.; Xiang, K.; Chen, C.; Wang, J.; Wu, Z. Master graph: An essential integrated assembly model for the plant mitogenome based on a graph-based framework. Brief. Bioinform. 2023, 24, bbac522. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Shen, F.; Han, F.; Qu, Y.; Hou, J.; Xu, K.; Xu, L.-A.; He, W.; Wu, Z.; Yin, T. PMAT: An efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Hortic. Res. 2024, 11, uhae023. [Google Scholar] [CrossRef]
- Zhou, W.; Armijos, C.E.; Lee, C.; Lu, R.; Wang, J.; Ruhlman, T.A.; Jansen, R.K.; Jones, A.M.; Jones, C.D. Plastid genome assembly using long-read data. Mol. Ecol. Resour. 2023, 23, 1442–1457. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family and Genus | Species | Reference | GenBank Accession | Sequence Length (bp) | Molecular Type | GC Content (%) | GC Skew |
|---|---|---|---|---|---|---|---|
| Rosaceae, Malus | M. baccata ‘ZA’ | This study | PP826182 | 374,023 | Circular DNA | 45.4 | −0.2695~0.2706 |
| M. baccata | [53] | NC_065224 1 | 400,769 | Circular DNA | 45.4 | −0.2739~0.2682 | |
| M. domestica | [54] | NC_018554 1 | 396,947 | Circular DNA | 45.4 | −0.2717~0.2695 | |
| M. domestica ‘Yantai fuji 8’ | [55] | MN964891 | 396,947 | Circular DNA | 45.4 | −0.2717~0.2695 | |
| M. domestica ‘Gala’ | [53] | ON478160 | 396,946 | Circular DNA | 45.4 | −0.2695~0.2717 | |
| M. domestica | * | OX352770 | 400,843 | Linear DNA | 45.4 | — | |
| M. domestica | * | OX352778 | 392,471 | Linear DNA | 45.4 | — | |
| M. domestica | * | OX352780 | 400,843 | Linear DNA | 45.4 | — | |
| M. domestica | * | OX352782 | 400,843 | Linear DNA | 45.4 | — | |
| M. domestica ‘Honeycrisp’ | This study; Data source: [32] | OR876282 | 396,949 | Circular DNA | 45.4 | −0.2717~0.2695 | |
| M. domestica ‘Fuji’ | [56] | — | 436,177 | — | 45.4 | — | |
| M. hupehensis var. mengshanensis | [57] | KR534606 | 422,555 | Circular DNA | 45.2 | −0.2682~0.2723 | |
| M. kansuensis | [58] | MW057419 | 385,436 | Circular DNA | 45.3 | −0.2717~0.2711 | |
| M. sieversii | [53] | NC_065225 1 | 385,869 | Circular DNA | 45.4 | −0.2711~0.2692 | |
| M. sylvestris | [53] | NC_065226 1 | 396,940 | Circular DNA | 45.4 | −0.2711~0.2692 | |
| M. sylvestris | * | OX352768 | 423,217 | Linear DNA | 45.5 | — | |
| M. × robusta | * | OY720342 | 385,872 | Linear DNA | 45.4 | — | |
| M. ‘SH6’ | [56] | — | 453,068 | — | 45.0 | — | |
| M. ‘Flame’ | [56] | — | 441,454 | — | 45.3 | — | |
| M. ‘Royalty’ | [56] | — | 397,430 | — | 45.3 | — |
| Gene Category | Gene Function | Gene Name |
|---|---|---|
| Core protein-coding genes | Subunit of NADH dehydrogenase (complex I) | nad1 c, nad2 c, nad3, nad4 b, nad4L, nad5 c, nad6, nad7 c, nad9 |
| Apocytochrome b (complex III) | cob | |
| Subunit of cytochrome c oxidase (complex IV) | cox1, cox2, cox3 | |
| Subunit of ATP synthase (complex V) | atp1, atp4, atp6, atp8, atp9 | |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFC a, ccmFN | |
| Maturase | matR | |
| Transport membrane protein | mttB | |
| Variable PCGs | Large subunit of ribosome | rpl5, rpl10, rpl16 |
| Small subunit of ribosome | rps1, rps3, rps4, rps12, rps13, rps14 | |
| Subunit of succinate dehydrogenase (complex II) | sdh3, sdh4 d | |
| tRNA genes | Transfer RNA | trnC-GCA, trnD-GUC, trnE-UUC a,d, trnF-GAA e, trnG-GCC, trnH-GUG, trnI-CAU, trnK-UUU, trnM-CAU a,d, trnfM-CAU, trnN-GUU, trnP-UGG d, trnQ-UUG, trnS-UGA, trnW-CCA, trnY-GUA |
| rRNA genes | Ribosomal RNA | rrn5, rrn18, rrn26 |
| MTPT Transfer Fragment | MTDNA Locations | CPDNA Locations | Identity (%) | Alignment Length (bp) | Mismatches | Gap Openings | Expected Value | Bit Score | Sequence Annotation |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 47,398…48,244 | 106,040…105,189 | 74.032 | 878 | 171 | 44 | 5.21 × 10−82 | 305 | Partial rrn16 |
| 2 | 47,386…48,244 | 142,469…143,332 | 73.933 | 890 | 175 | 44 | 5.21 × 10−82 | 305 | Partial rrn16 |
| 3 | 259,462…259,783 | 68,689…68,375 | 83.333 | 324 | 43 | 5 | 5.25 × 10−77 | 289 | Partial psbE, Partial (psbE_petL) |
| 4 | 220,541…220,679 | 37,818…37,682 | 90 | 140 | 10 | 4 | 1.19 × 10−43 | 178 | Partial psbC |
| 5 | 30,188…30,295 | 70,201…70,310 | 92.727 | 110 | 6 | 2 | 1.55 × 10−37 | 158 | Partial (petG_trnW-CCA), complete (trnW-CCA), Partial (trnW-CCA_trnP-UGG) |
| 6 | 227,435…227,519 | 35…118 | 96.471 | 85 | 2 | 1 | 5.60 × 10−32 | 139 | Partial (rpl2_trnH-GUG), complete trnH-GUG, Partial (trnH-GUG_psbA) |
| 7 | 287,676…287,759 | 32,888…32,971 | 96.429 | 84 | 3 | 0 | 5.60 × 10−32 | 139 | Partial (psbM_trnD-GUC), complete trnD-GUC, Partial (trnD-GUC_trnY_GUA) |
| 8 | 76,742…76,826 | 113,280…113,196 | 95.349 | 86 | 2 | 2 | 7.24 × 10−31 | 135 | Partial (trnR-ACG_trnN-GUU), complete trnN-GUU, Partial (trnN-GUU_ndhF) |
| 9 | 76,742…76,826 | 135,241…135,325 | 95.349 | 86 | 2 | 2 | 7.24 × 10−31 | 135 | Partial (ycf1_trnN-GUU), complete trnN-GUU, Partial (trnN-GUU_trnR-ACG) |
| 10 | 104,344…104,422 | 55,944…55,866 | 93.671 | 79 | 5 | 0 | 7.29 × 10−26 | 119 | Partial (trnV-UAC_trnM-CAU), complete trnM-CAU, Partial (trnM-CAU_atpE) |
| 11 | 176,322…176,385 | 395…458 | 98.438 | 64 | 1 | 0 | 3.39 × 10−24 | 113 | Partial psbA |
| 12 | 266,723…266,801 | 90,558…90,484 | 88.608 | 79 | 5 | 3 | 4.42 × 10−18 | 93.5 | Partial (rpl23_trnI-CAU), complete trnI-CAU |
| 13 | 266,723…266,801 | 157,963…158,037 | 88.608 | 79 | 5 | 3 | 4.42 × 10−18 | 93.5 | Complete trnI-CAU, Partial (trnI-CAU_rpl23) |
| 14 | 221,409…221,439 | 11,357…11,387 | 100 | 31 | 0 | 0 | 1.61× 10−7 | 58.4 | Partial atpA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, D.; Zhang, R.; Qin, X.; Shen, X.; You, C. Morphological Structure Identification, Comparative Mitochondrial Genomics and Population Genetic Analysis toward Exploring Interspecific Variations and Phylogenetic Implications of Malus baccata ‘ZA’ and Other Species. Biomolecules 2024, 14, 912. https://doi.org/10.3390/biom14080912
Wang X, Wang D, Zhang R, Qin X, Shen X, You C. Morphological Structure Identification, Comparative Mitochondrial Genomics and Population Genetic Analysis toward Exploring Interspecific Variations and Phylogenetic Implications of Malus baccata ‘ZA’ and Other Species. Biomolecules. 2024; 14(8):912. https://doi.org/10.3390/biom14080912
Chicago/Turabian StyleWang, Xun, Daru Wang, Ruifen Zhang, Xin Qin, Xiang Shen, and Chunxiang You. 2024. "Morphological Structure Identification, Comparative Mitochondrial Genomics and Population Genetic Analysis toward Exploring Interspecific Variations and Phylogenetic Implications of Malus baccata ‘ZA’ and Other Species" Biomolecules 14, no. 8: 912. https://doi.org/10.3390/biom14080912
APA StyleWang, X., Wang, D., Zhang, R., Qin, X., Shen, X., & You, C. (2024). Morphological Structure Identification, Comparative Mitochondrial Genomics and Population Genetic Analysis toward Exploring Interspecific Variations and Phylogenetic Implications of Malus baccata ‘ZA’ and Other Species. Biomolecules, 14(8), 912. https://doi.org/10.3390/biom14080912