
Clinal Variation in Short Tandem Repeats Linked to Gene Expression in Sunflower (Helianthus annuus L.)


Abstract
:1. Introduction
2. Materials and Methods
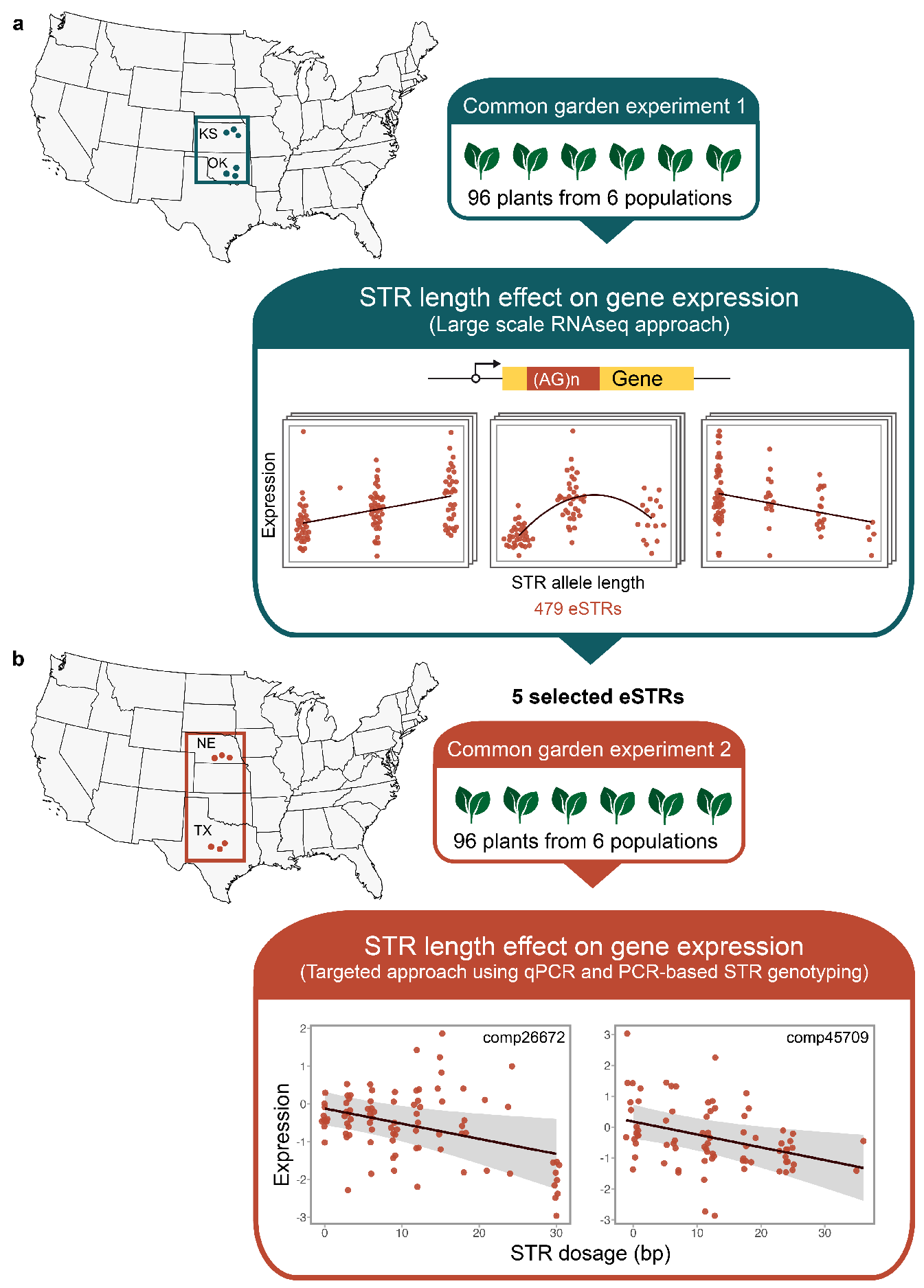
2.1. Sample Collection and Common Garden Experiment
2.2. RNA Extraction
2.3. Gene Expression Quantification
2.4. DNA Extraction and STR Genotyping
2.5. Effect of eSTR Length on Gene Expression
2.6. Latitudinal Variation in eSTR Length
3. Results
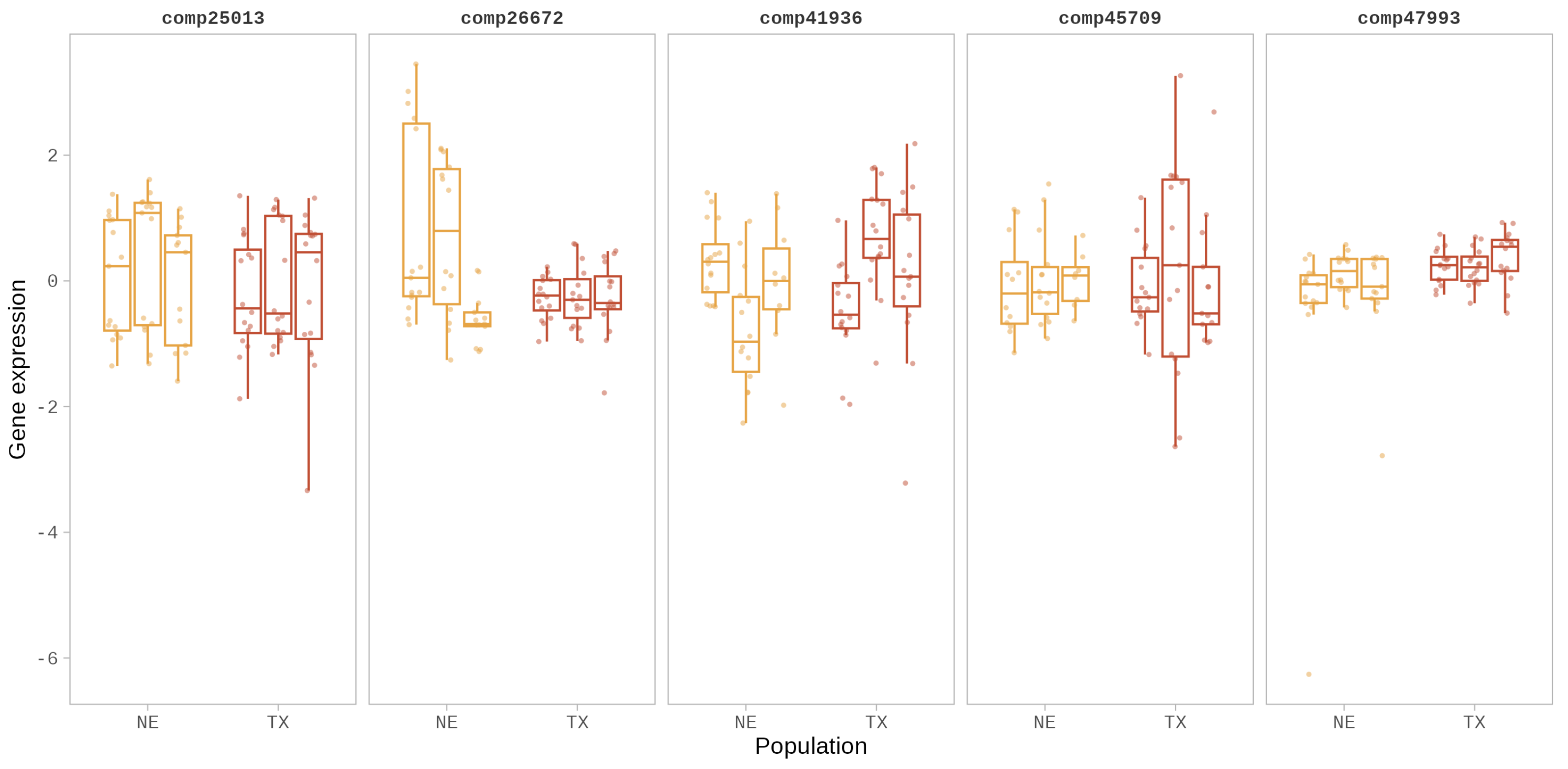
3.1. Gene Expression Variation in eSTR-Containing Genes
3.2. The Relationship between the eSTR Length and Gene Expression
3.3. Clinal Variation in eSTR Length
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shapiro, J.A.; von Sternberg, R. Why repetitive DNA is essential to genome function. Biol. Rev. 2005, 80, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, R.E.; Handsaker, R.E.; Sherman, M.A.; Barton, A.R.; Zheng, Y.; McCarroll, S.A.; Loh, P.R. Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science 2021, 373, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Gymrek, M.; Goren, A. Missing heritability may be hiding in repeats. Science 2021, 373, 1440–1441. [Google Scholar] [CrossRef] [PubMed]
- Kashi, Y.; King, D.; Soller, M. Simple sequence repeats as a source of quantitative genetic variation. Trends Genet. 1997, 13, 74–78. [Google Scholar] [CrossRef] [PubMed]
- King, D.G.; Soller, M.; Kashi, Y. Evolutionary tuning knobs. Endeavour 1997, 21, 36–40. [Google Scholar] [CrossRef]
- Trifonov, E.N. Tuning function of tandemly repeating sequences: A molecular device for fast adaptation. In Evolutionary Theory and Processes: Modern Horizons; Springer: Berlin/Heidelberg, Germany, 2004; pp. 115–138. [Google Scholar]
- Kashi, Y.; King, D.G. Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 2006, 22, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Gemayel, R.; Vinces, M.D.; Legendre, M.; Verstrepen, K.J. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 2010, 44, 445–477. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within genes: Structure, function, and evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef]
- Fotsing, S.F.; Margoliash, J.; Wang, C.; Saini, S.; Yanicky, R.; Shleizer-Burko, S.; Goren, A.; Gymrek, M. The impact of short tandem repeat variation on gene expression. Nat. Genet. 2019, 51, 1652–1659. [Google Scholar] [CrossRef]
- Reinar, W.B.; Lalun, V.O.; Reitan, T.; Jakobsen, K.S.; Butenko, M.A. Length variation in short tandem repeats affects gene expression in natural populations of Arabidopsis thaliana. Plant Cell 2021, 33, 2221–2234. [Google Scholar] [CrossRef] [PubMed]
- Reinar, W.B.; Greulich, A.; Stø, I.M.; Knutsen, J.B.; Reitan, T.; Tørresen, O.K.; Jentoft, S.; Butenko, M.A.; Jakobsen, K.S. Adaptive protein evolution through length variation in short tandem repeats. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ranathunge, C.; Wheeler, G.L.; Chimahusky, M.E.; Perkins, A.D.; Pramod, S.; Welch, M.E. Transcribed microsatellite allele lengths are often correlated with gene expression in natural sunflower populations. Mol. Ecol. 2020, 29, 1704–1716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Andersen, E.C. Interplay between polymorphic short tandem repeats and gene expression variation in Caenorhabditis elegans. Mol. Biol. Evol. 2023, 40, msad067. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Koskela, E.; Mappes, T.; Mokkonen, M.; Sims, A.M.; Watts, P.C. Balancing selection maintains polymorphisms at neurogenetic loci in field experiments. Proc. Natl. Acad. Sci. USA 2017, 114, 3690–3695. [Google Scholar] [CrossRef] [PubMed]
- Ohadi, M.; Valipour, E.; Ghadimi-Haddadan, S.; Namdar-Aligoodarzi, P.; Bagheri, A.; Kowsari, A.; Rezazadeh, M.; Darvish, H.; Kazeminasab, S. Core promoter short tandem repeats as evolutionary switch codes for primate speciation. Am. J. Primatol. 2015, 77, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Rival, P.; Press, M.O.; Bale, J.; Grancharova, T.; Undurraga, S.F.; Queitsch, C. The conserved PFT1 tandem repeat is crucial for proper flowering in Arabidopsis thaliana. Genetics 2014, 198, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Rosas, U.; Mei, Y.; Xie, Q.; Banta, J.A.; Zhou, R.W.; Seufferheld, G.; Gerard, S.; Chou, L.; Bhambhra, N.; Parks, J.D.; et al. Variation in Arabidopsis flowering time associated with cis-regulatory variation in CONSTANS. Nat. Commun. 2014, 5, 3651. [Google Scholar] [CrossRef] [PubMed]
- Press, M.O.; McCoy, R.C.; Hall, A.N.; Akey, J.M.; Queitsch, C. Massive variation of short tandem repeats with functional consequences across strains of Arabidopsis thaliana. Genome Res. 2018, 28, 1169–1178. [Google Scholar] [CrossRef]
- Steinmeyer, C.; Mueller, J.C.; Kempenaers, B. Search for informative polymorphisms in candidate genes: Clock genes and circadian behaviour in blue tits. Genetica 2009, 136, 109–117. [Google Scholar] [CrossRef]
- Sawyer, L.A.; Hennessy, J.M.; Peixoto, A.A.; Rosato, E.; Parkinson, H.; Costa, R.; Kyriacou, C.P. Natural variation in a Drosophila clock gene and temperature compensation. Science 1997, 278, 2117–2120. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, A.; Fidler, A.E.; Kuhn, S.; Carter, K.L.; Hoffmann, A.; Barr, I.; Biard, C.; Charmantier, A.; Eens, M.; Korsten, P.; et al. Avian Clock gene polymorphism: Evidence for a latitudinal cline in allele frequencies. Mol. Ecol. 2007, 16, 4867–4880. [Google Scholar] [CrossRef] [PubMed]
- Lemay, M.A.; Russello, M.A. Latitudinal cline in allele length provides evidence for selection in a circadian rhythm gene. Biol. J. Linn. Soc. 2014, 111, 869–877. [Google Scholar] [CrossRef]
- O’Malley, K.G.; Banks, M.A. A latitudinal cline in the Chinook salmon (Oncorhynchus tshawytscha) Clock gene: Evidence for selection on PolyQ length variants. Proc. R. Soc. B Biol. Sci. 2008, 275, 2813–2821. [Google Scholar] [CrossRef] [PubMed]
- Ranathunge, C.; Chimahusky, M.E.; Welch, M.E. A comparative study of population genetic structure reveals patterns consistent with selection at functional microsatellites in common sunflower. Mol. Genet. Genom. MGG 2022, 297, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Blackman, B.K.; Michaels, S.D.; Rieseberg, L.H. Connecting the sun to flowering in sunflower adaptation. Mol. Ecol. 2011, 2017, 3503–3512. [Google Scholar] [CrossRef] [PubMed]
- Mcassey, E.; Corbi, J.; Burke, J.M. Range-wide phenotypic and genetic differentiation in wild sunflower. BMC Plant Biol. 2016, 16, 249. [Google Scholar] [CrossRef]
- Ranathunge, C.; Wheeler, G.L.; Chimahusky, M.E.; Kennedy, M.M.; Morrison, J.I.; Baldwin, B.S.; Perkins, A.D.; Welch, M.E. Transcriptome profiles of sunflower reveal the potential role of microsatellites in gene expression divergence. Mol. Ecol. 2018, 275, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Pramod, S.; Downs, K.E.; Welch, M.E. Gene expression assays for actin, ubiquitin, and three microsatellite-encoding genes in Helianthus annuus (Asteraceae). Am. J. Bot. 2012, 99, e350–e352. [Google Scholar] [CrossRef]
- Don, R.; Cox, P.T.; Wainwright, B.; Baker, K.; Mattick, J.S. ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 1991, 19, 4008. [Google Scholar] [CrossRef]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef] [PubMed]
- Gymrek, M.; Willems, T.; Guilmatre, A.; Zeng, H.; Markus, B.; Georgiev, S.; Daly, M.J.; Price, A.L.; Pritchard, J.K.; Sharp, A.J.; et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 2016, 48, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Vinces, M.D.; Legendre, M.; Caldara, M.; Hagihara, M.; Verstrepen, K.J. Unstable tandem repeats in promoters confer transcriptional evolvability. Science 2009, 324, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Fox, J.; Weisberg, S.; Adler, D.; Bates, D.; Baud-Bovy, G.; Ellison, S.; Firth, D.; Friendly, M.; Gorjanc, G.; Graves, S.; et al. Package “Car”; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Breheny, P.; Burchett, W. Visualization of regression models using visreg. R J. 2017, 9, 56. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Makowski, D.; Ben-Shachar, M.; Patil, I.; Lüdecke, D. Automated Results Reporting as a Practical Tool to Improve Reproducibility and Methodological Best Practices Adoption. CRAN 2020. Available online: https://easystats.github.io/report/ (accessed on 1 August 2022).
- Liang, Y.; Hao, J.; Wang, J.; Zhang, G.; Su, Y.; Liu, Z.J.; Wang, T. Statistical Genomics Analysis of Simple Sequence Repeats from the Paphiopedilum Malipoense Transcriptome Reveals Control Knob Motifs Modulating Gene Expression. Adv. Sci. 2024, 11, 2304848. [Google Scholar] [CrossRef]
- Oikawa, K.; Kasahara, M.; Kiyosue, T.; Kagawa, T.; Suetsugu, N.; Takahashi, F.; Kanegae, T.; Niwa, Y.; Kadota, A.; Wada, M. Chloroplast unusual positioning1 is essential for proper chloroplast positioning. Plant Cell 2003, 15, 2805–2815. [Google Scholar] [CrossRef]
- Kasahara, M.; Kagawa, T.; Oikawa, K.; Suetsugu, N.; Miyao, M.; Wada, M. Chloroplast avoidance movement reduces photodamage in plants. Nature 2002, 420, 829–832. [Google Scholar] [CrossRef]
- Kumar, S.; Bhatia, S. A polymorphic (GA/CT) n-SSR influences promoter activity of Tryptophan decarboxylase gene in Catharanthus roseus L. Don. Sci. Rep. 2016, 6, 33280. [Google Scholar] [CrossRef]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated regions of mRNAs. Genome Biol. 2002, 3, reviews0004.1. [Google Scholar] [CrossRef] [PubMed]
- Sandman, K.; Reeve, J.N. Archaeal nucleosome positioning by CTG repeats. J. Bacteriol. 1999, 181, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H. Chromatin structure of repeating CTG/CAG and CGG/CCG sequences in human disease. Front. Biosci. J. Virtual Libr. 2007, 12, 4731–4741. [Google Scholar] [CrossRef] [PubMed]
- Feder, M.; Walser, J. The biological limitations of transcriptomics in elucidating stress and stress responses. J. Evol. Biol. 2005, 18, 901–910. [Google Scholar] [CrossRef]
- Evans, T.G.; Podrabsky, J.E.; Stillman, J.H.; Tomanek, L. Considerations for the use of transcriptomics in identifying the ‘genes that matter’ for environmental adaptation. J. Exp. Biol. 2015, 218, 1925–1935. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| eSTR-Containing Gene | Putative Function | Repeat Motif | Region |
|---|---|---|---|
| comp26672 | Protein CHUP1, chloroplastic-like | CCTTCT | coding |
| comp25013 | ATP-dependent Clp protease proteolytic subunit 5, chloroplastic-like | GACGGT | 5′UTR |
| comp41936 | ATP synthase delta chain, chloroplastic-like | TCATT | 5′UTR |
| comp45709 | cytochrome P450 86A22-like (CYP86A22) | GTGTTT | 5′UTR |
| comp47993 | Putative dual specificity protein phosphatase DSP8 | TTCAA | 5′UTR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranathunge, C.; Welch, M.E. Clinal Variation in Short Tandem Repeats Linked to Gene Expression in Sunflower (Helianthus annuus L.). Biomolecules 2024, 14, 944. https://doi.org/10.3390/biom14080944
Ranathunge C, Welch ME. Clinal Variation in Short Tandem Repeats Linked to Gene Expression in Sunflower (Helianthus annuus L.). Biomolecules. 2024; 14(8):944. https://doi.org/10.3390/biom14080944
Chicago/Turabian StyleRanathunge, Chathurani, and Mark E. Welch. 2024. "Clinal Variation in Short Tandem Repeats Linked to Gene Expression in Sunflower (Helianthus annuus L.)" Biomolecules 14, no. 8: 944. https://doi.org/10.3390/biom14080944
APA StyleRanathunge, C., & Welch, M. E. (2024). Clinal Variation in Short Tandem Repeats Linked to Gene Expression in Sunflower (Helianthus annuus L.). Biomolecules, 14(8), 944. https://doi.org/10.3390/biom14080944