
Membrane Activity and Viroporin Assembly for the SARS-CoV-2 E Protein Are Regulated by Cholesterol
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Formation of Giant Unilamellar Vesicles (GUVs)
2.3. Confocal Fluorescence Microscopy
2.4. Calcein Leakage Assay
2.5. Formation of Planar Bilayer Lipid Membranes (BLMs)
2.6. Patch-Clamp Measurements
2.7. Formation of Small Liposomes
2.8. Atomic Force Microscopy
3. Results
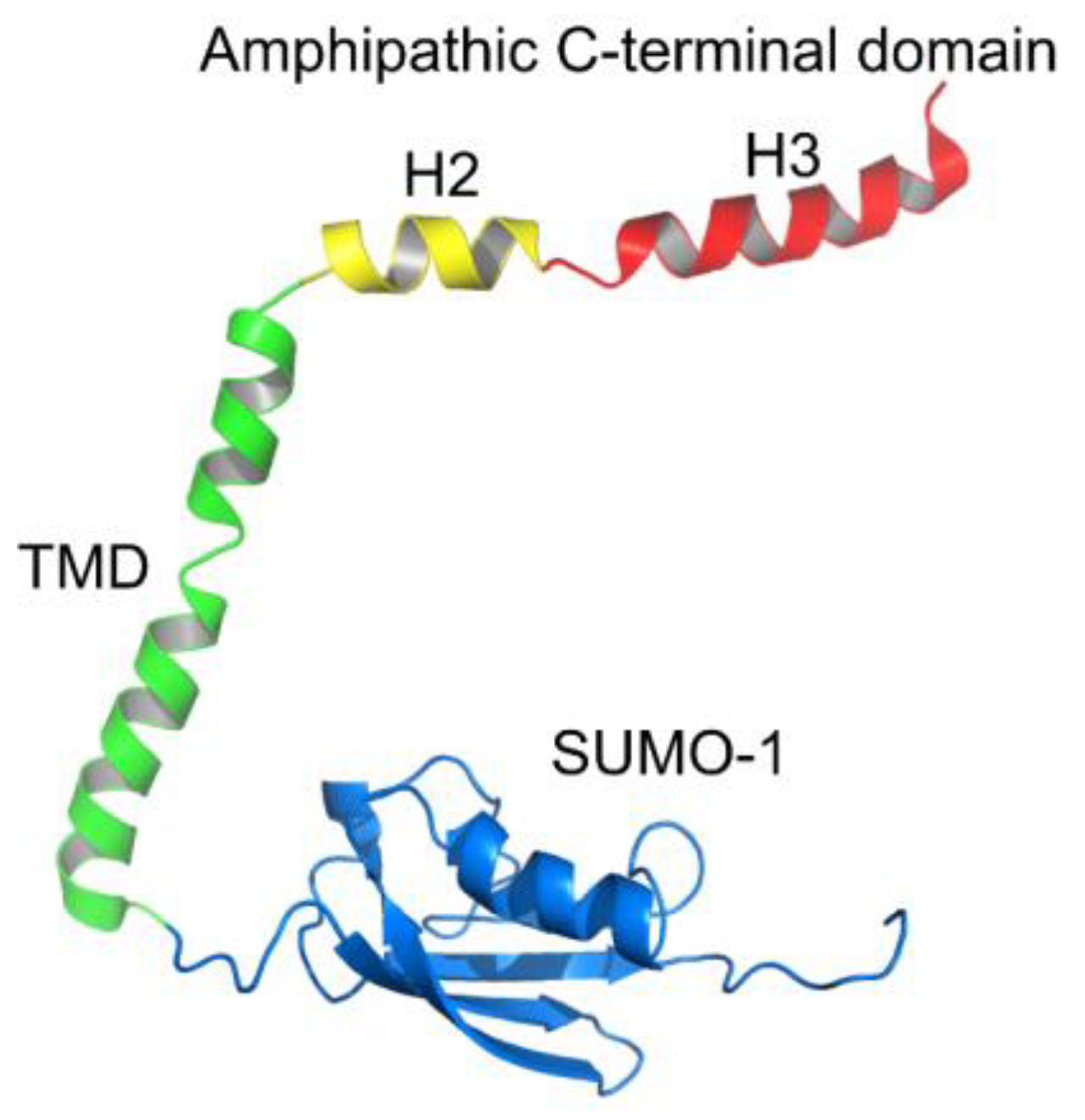
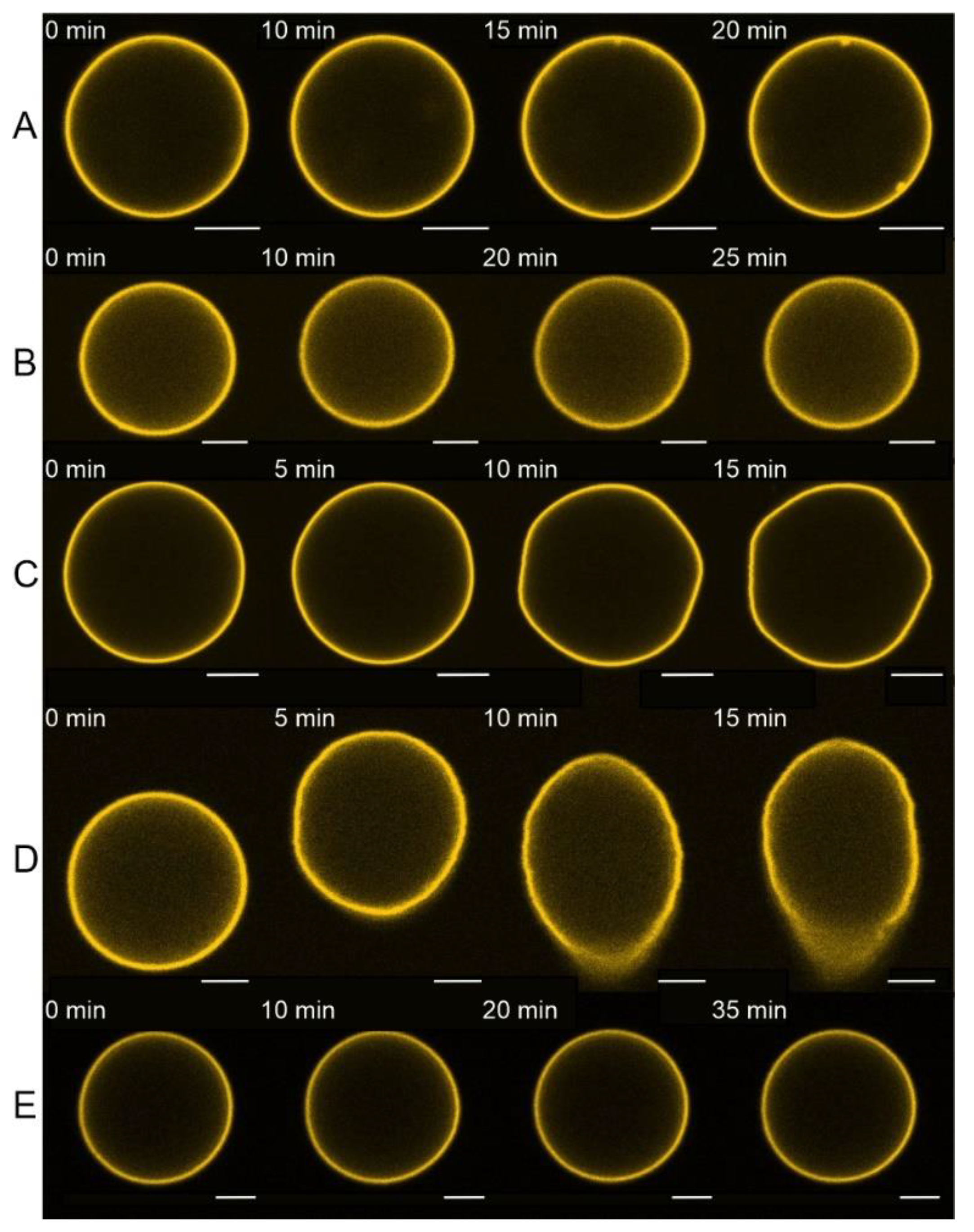
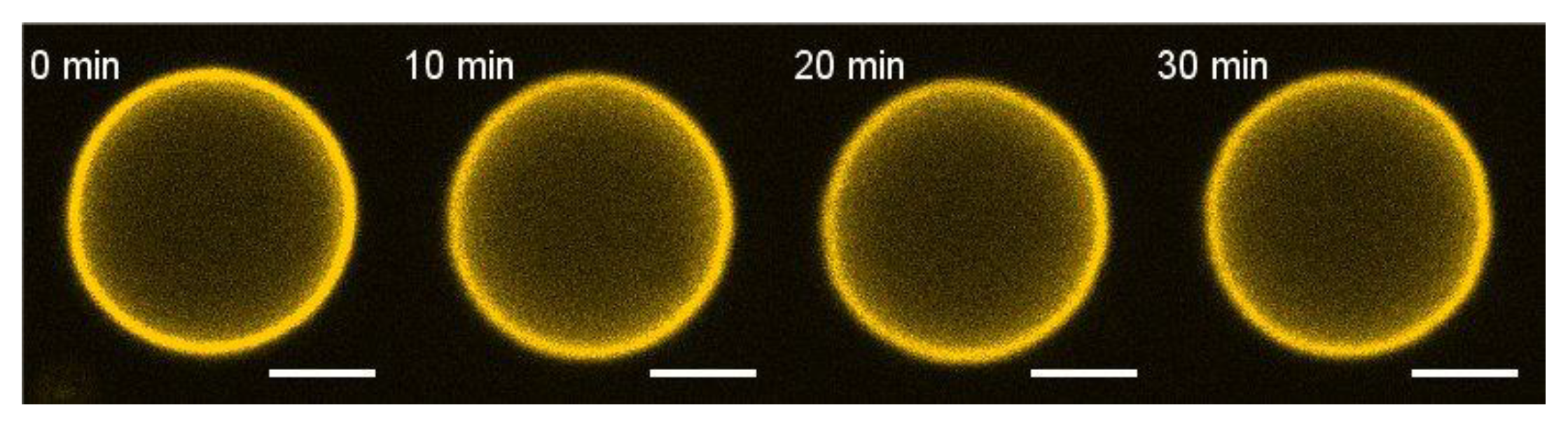
3.1. E Protein but Not Its H3 Peptide Deform GUVs with Charged Lipids and High Cholesterol Content
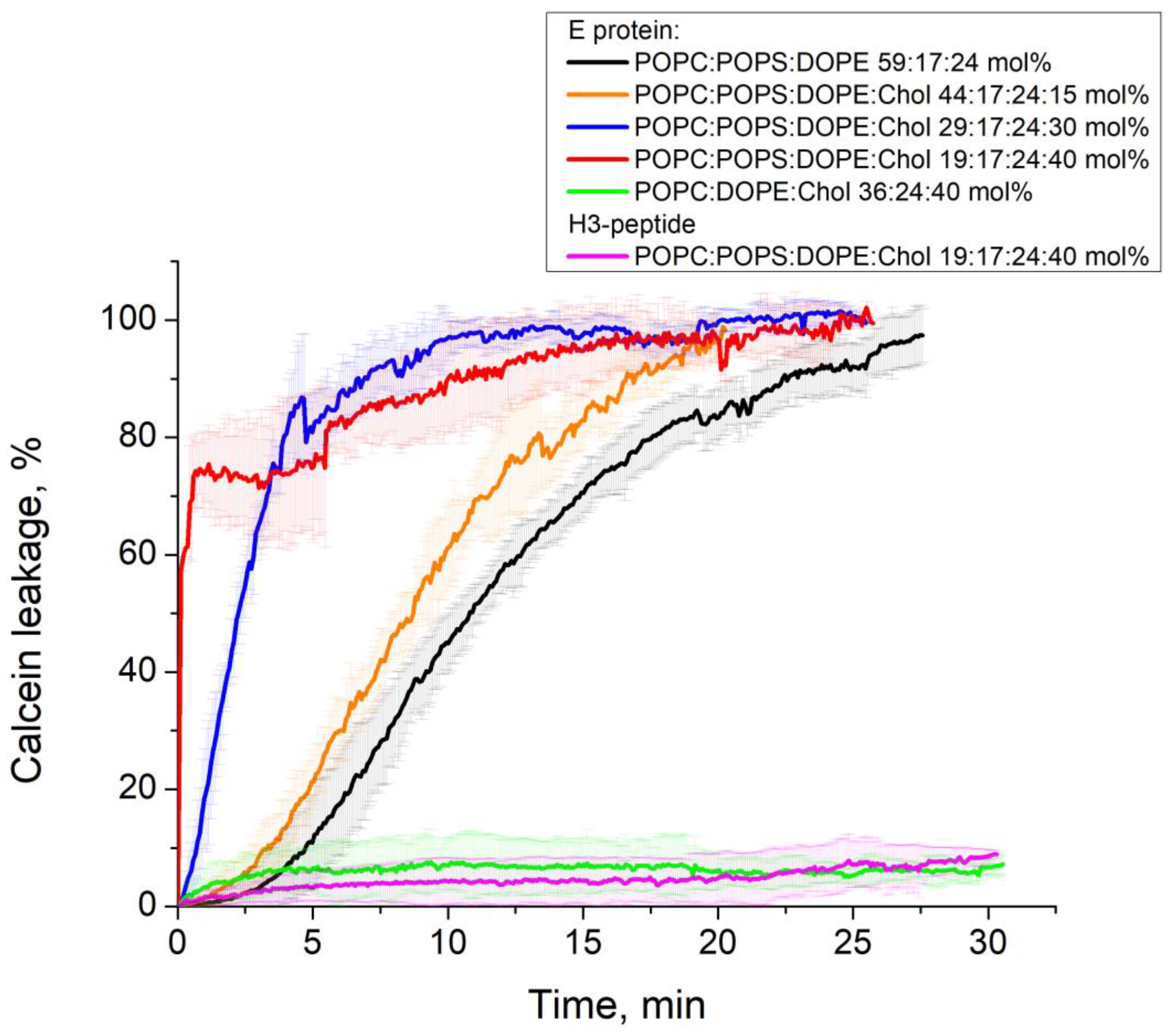
3.2. E Protein Induces Calcein Leakage from GUVs with Charged Lipids
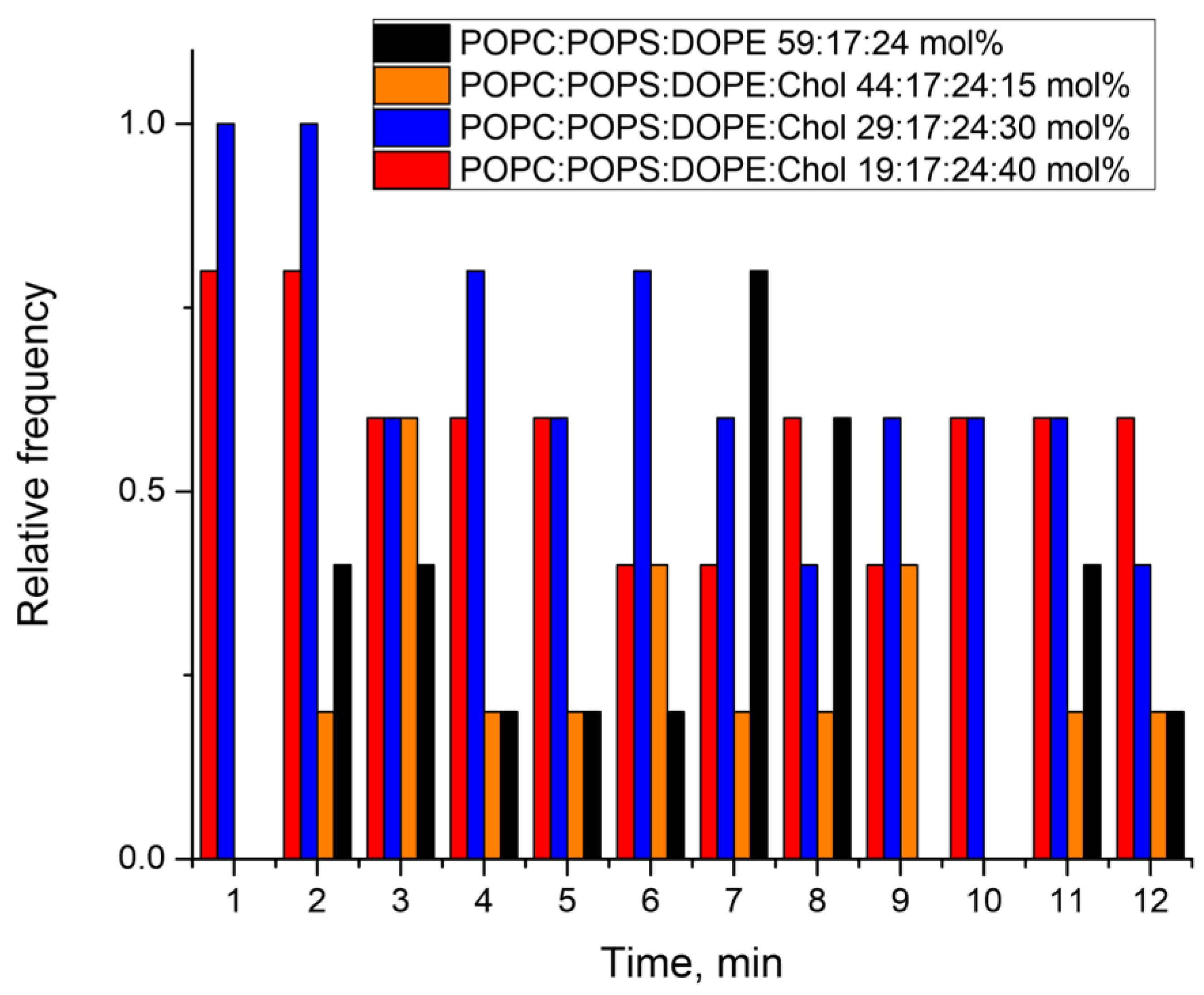
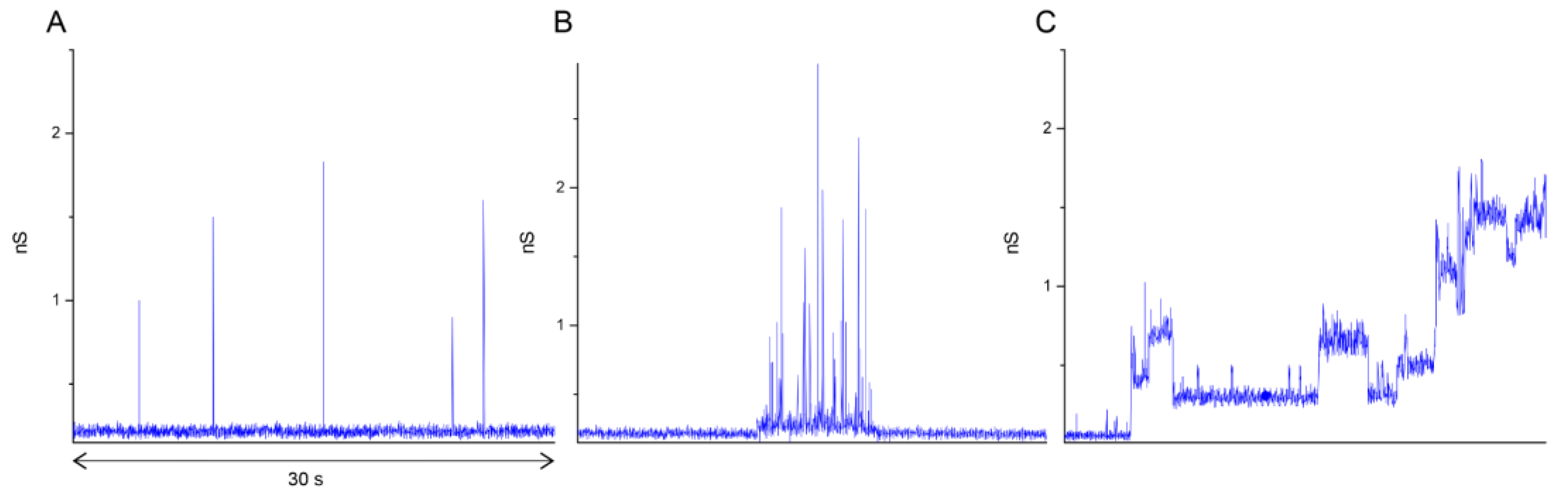
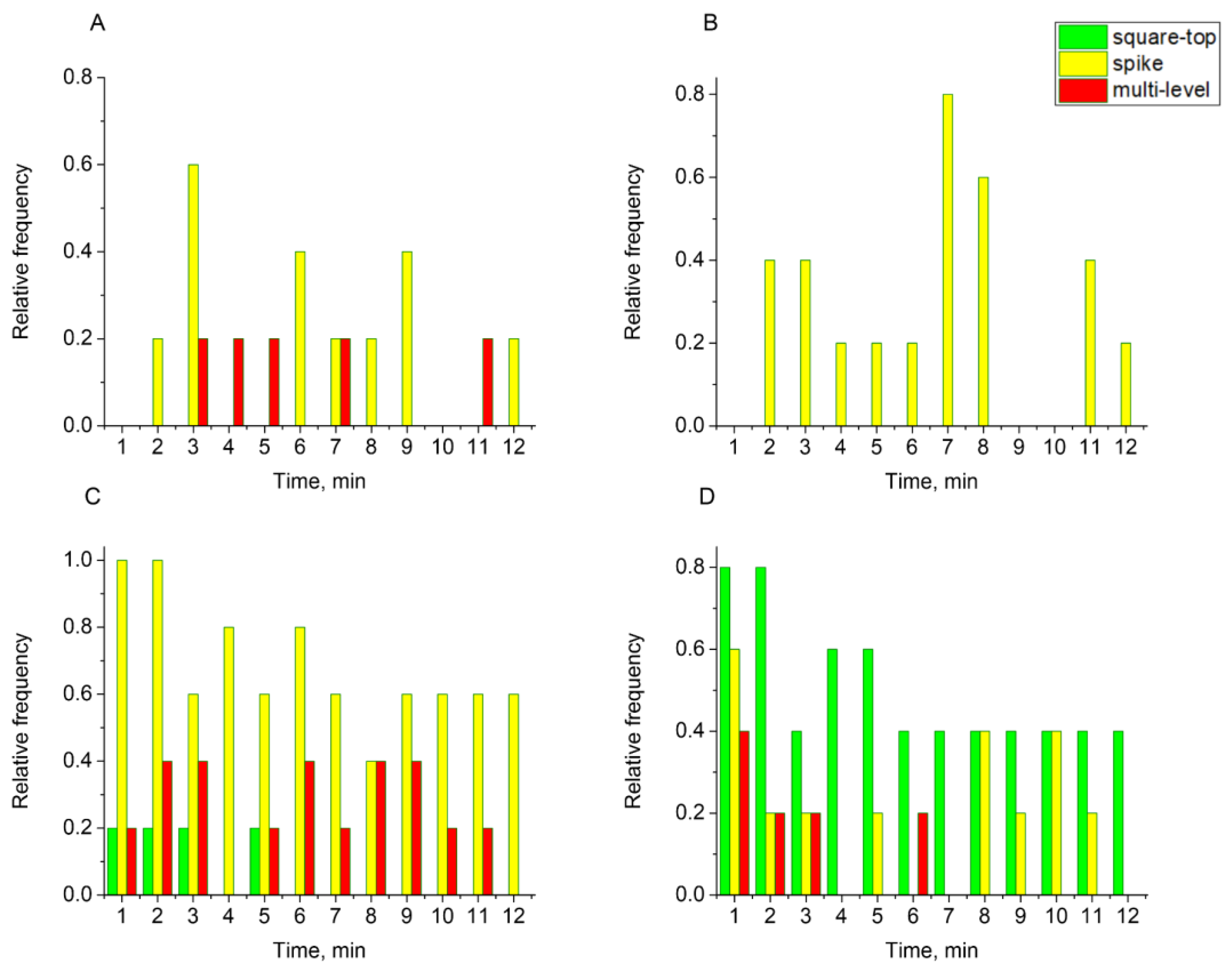
3.3. Patch-Clamp Experiments Reveal Different Types of E Protein-Induced Membrane Conductance
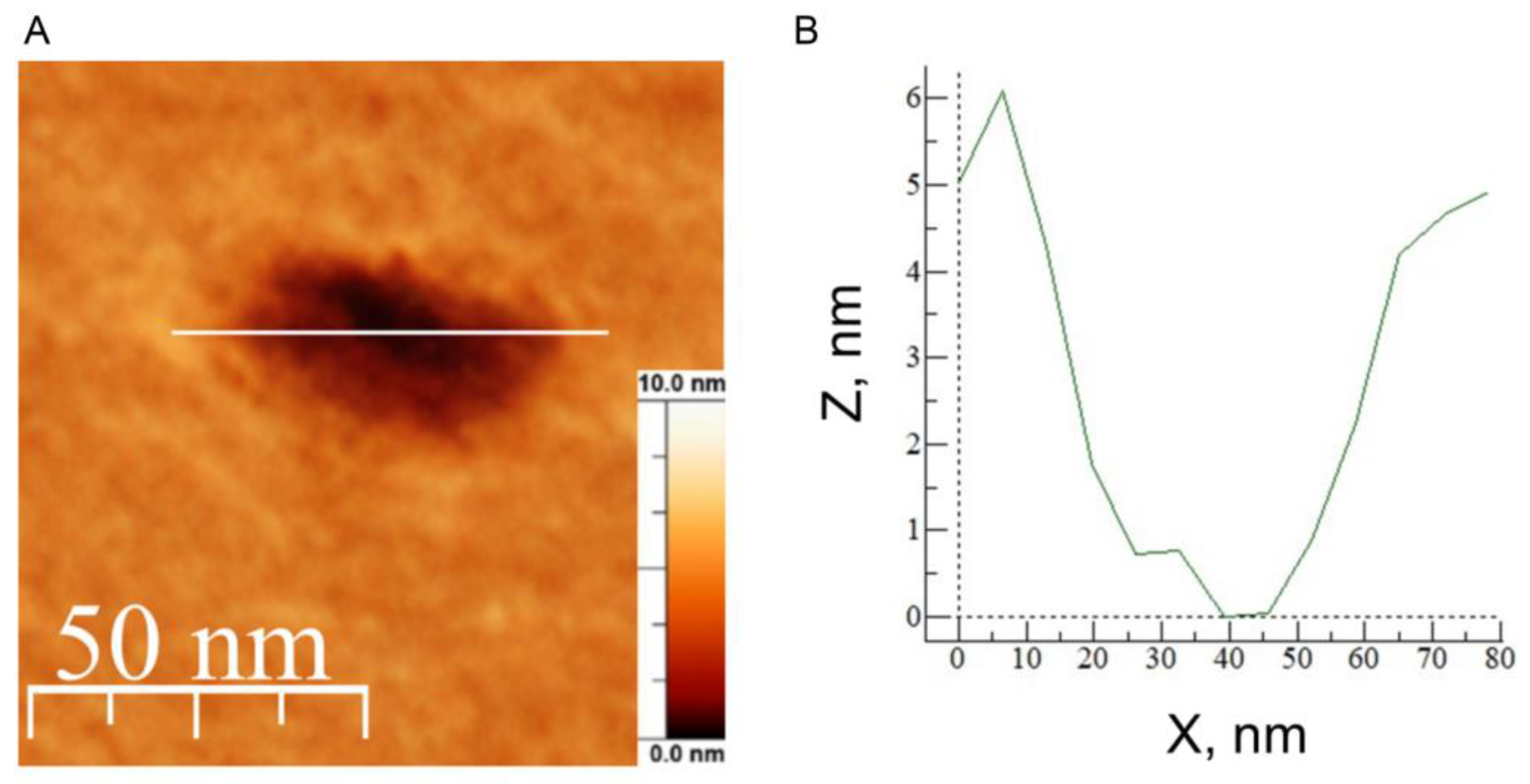
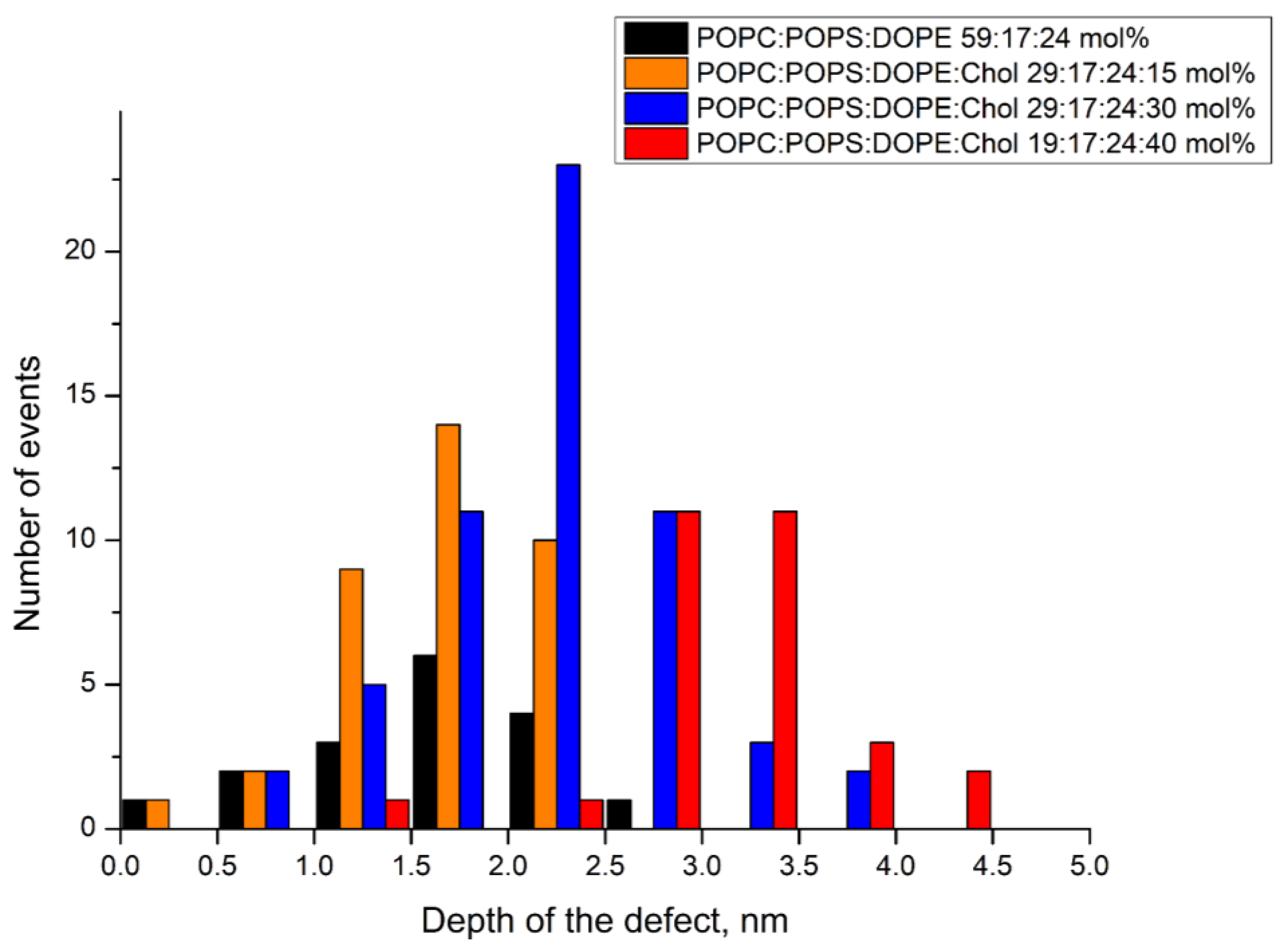
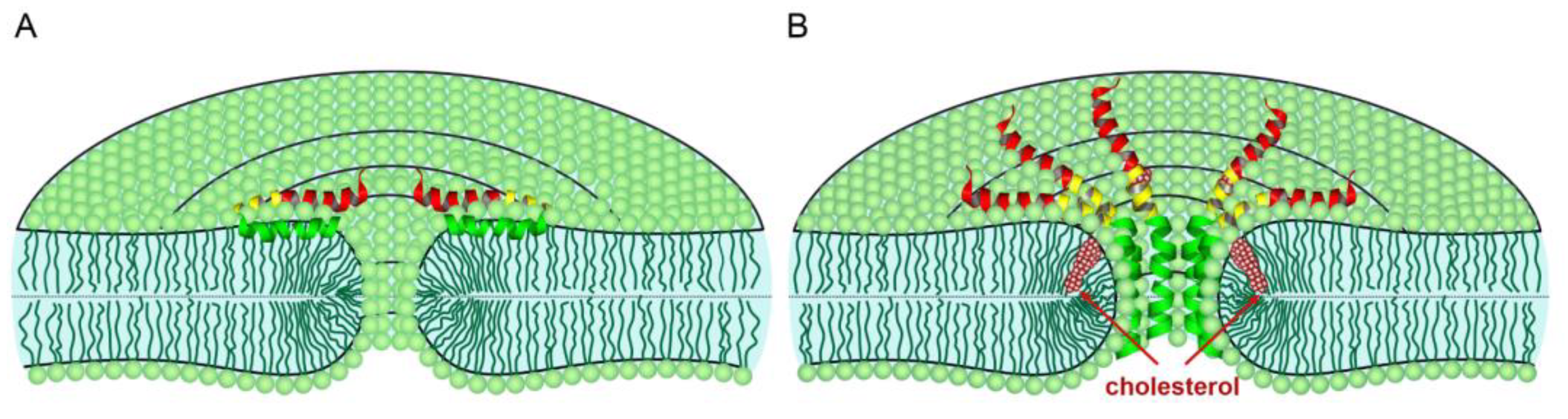
3.4. Atomic Force Microscopy Demonstrates the Ability of the E Protein to Form Semi-Transmembrane or Transmembrane Pores Depending on the Cholesterol Content of a Membrane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masters, P.S. The Molecular Biology of Coronaviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2006; Volume 66, pp. 193–292. ISBN 978-0-12-039869-0. [Google Scholar]
- Zhou, S.; Lv, P.; Li, M.; Chen, Z.; Xin, H.; Reilly, S.; Zhang, X. SARS-CoV-2 E Protein: Pathogenesis and Potential Therapeutic Development. Biomed. Pharmacother. 2023, 159, 114242. [Google Scholar] [CrossRef]
- Duart, G.; García-Murria, M.J.; Grau, B.; Acosta-Cáceres, J.M.; Martínez-Gil, L.; Mingarro, I. SARS-CoV-2 Envelope Protein Topology in Eukaryotic Membranes. Open Biol. 2020, 10, 200209. [Google Scholar] [CrossRef]
- Kuo, L.; Hurst, K.R.; Masters, P.S. Exceptional Flexibility in the Sequence Requirements for Coronavirus Small Envelope Protein Function. J. Virol. 2007, 81, 2249–2262. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Álvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.-J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A Severe Acute Respiratory Syndrome Coronavirus That Lacks the E Gene Is Attenuated In Vitro and In Vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef]
- Ortego, J.; Ceriani, J.E.; Patiño, C.; Plana, J.; Enjuanes, L. Absence of E Protein Arrests Transmissible Gastroenteritis Coronavirus Maturation in the Secretory Pathway. Virology 2007, 368, 296–308. [Google Scholar] [CrossRef]
- Verdiá-Báguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; DeDiego, M.L.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Coronavirus E Protein Forms Ion Channels with Functionally and Structurally-Involved Membrane Lipids. Virology 2012, 432, 485–494. [Google Scholar] [CrossRef]
- Xia, X.; Cheng, A.; Wang, M.; Ou, X.; Sun, D.; Mao, S.; Huang, J.; Yang, Q.; Wu, Y.; Chen, S.; et al. Functions of Viroporins in the Viral Life Cycle and Their Regulation of Host Cell Responses. Front. Immunol. 2022, 13, 890549. [Google Scholar] [CrossRef]
- Breitinger, U.; Farag, N.S.; Sticht, H.; Breitinger, H.-G. Viroporins: Structure, Function, and Their Role in the Life Cycle of SARS-CoV-2. Int. J. Biochem. Cell Biol. 2022, 145, 106185. [Google Scholar] [CrossRef]
- Farag, N.S.; Breitinger, U.; Breitinger, H.G.; El Azizi, M.A. Viroporins and Inflammasomes: A Key to Understand Virus-Induced Inflammation. Int. J. Biochem. Cell Biol. 2020, 122, 105738. [Google Scholar] [CrossRef]
- Alam, I.; Kamau, A.A.; Kulmanov, M.; Jaremko, Ł.; Arold, S.T.; Pain, A.; Gojobori, T.; Duarte, C.M. Functional Pangenome Analysis Shows Key Features of E Protein Are Preserved in SARS and SARS-CoV-2. Front. Cell. Infect. Microbiol. 2020, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. 2021, 30, 1114–1130. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. The Coronavirus E Protein: Assembly and Beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus Envelope Protein: Current Knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Grant, O.C.; Montgomery, D.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 Spike Protein Glycan Shield Reveals Implications for Immune Recognition. Sci. Rep. 2020, 10, 14991. [Google Scholar] [CrossRef]
- Balzar, M.; Bakker, H.A.M.; Briaire-de-Bruijn, I.H.; Fleuren, G.J.; Warnaar, S.O.; Litvinov, S.V. Cytoplasmic Tail Regulates the Intercellular Adhesion Function of the Epithelial Cell Adhesion Molecule. Mol. Cell. Biol. 1998, 18, 4833–4843. [Google Scholar] [CrossRef]
- Denieva, Z.G.; Kuzmin, P.I.; Galimzyanov, T.R.; Datta, S.A.K.; Rein, A.; Batishchev, O.V. Human Immunodeficiency Virus Type 1 Gag Polyprotein Modulates Membrane Physical Properties like a Surfactant: Potential Implications for Virus Assembly. ACS Infect. Dis. 2024, 10, 2870–2885. [Google Scholar] [CrossRef]
- Jimenez-Guardeño, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Enjuanes, L. The PDZ-Binding Motif of Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Is a Determinant of Viral Pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Proteins 3, 4, and 6 Induce Double-Membrane Vesicles. MBio 2013, 4, e00524-13. [Google Scholar] [CrossRef]
- Kuzmin, A.; Orekhov, P.; Astashkin, R.; Gordeliy, V.; Gushchin, I. Structure and Dynamics of the SARS-CoV-2 Envelope Protein Monomer. Proteins Struct. Funct. Bioinform. 2022, 90, 1102–1114. [Google Scholar] [CrossRef]
- Dregni, A.J.; McKay, M.J.; Surya, W.; Queralt-Martin, M.; Medeiros-Silva, J.; Wang, H.K.; Aguilella, V.; Torres, J.; Hong, M. The Cytoplasmic Domain of the SARS-CoV-2 Envelope Protein Assembles into a β-Sheet Bundle in Lipid Bilayers. J. Mol. Biol. 2023, 435, 167966. [Google Scholar] [CrossRef]
- Collins, L.T.; Elkholy, T.; Mubin, S.; Hill, D.; Williams, R.; Ezike, K.; Singhal, A. Elucidation of SARS-CoV-2 Budding Mechanisms through Molecular Dynamics Simulations of M and E Protein Complexes. J. Phys. Chem. Lett. 2021, 12, 12249–12255. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Is There a Link Between the Pathogenic Human Coronavirus Envelope Protein and Immunopathology? A Review of the Literature. Front. Microbiol. 2020, 11, 2086. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Xie, S.; Sun, B. Viral proteins function as ion channels. Biochim Biophys Acta 2011, 1808, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Wang, J.; Parthasarathy, K.; Liu, D.X. The Transmembrane Oligomers of Coronavirus Protein E. Biophys. J. 2005, 88, 1283–1290. [Google Scholar] [CrossRef]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and Drug Binding of the SARS-CoV-2 Envelope Protein Transmembrane Domain in Lipid Bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, K.; Lu, H.; Surya, W.; Vararattanavech, A.; Pervushin, K.; Torres, J. Expression and Purification of Coronavirus Envelope Proteins Using a Modified β-Barrel Construct. Protein Expr. Purif. 2012, 85, 133–141. [Google Scholar] [CrossRef]
- Li, Y.; Surya, W.; Claudine, S.; Torres, J. Structure of a Conserved Golgi Complex-Targeting Signal in Coronavirus Envelope Proteins. J. Biol. Chem. 2014, 289, 12535–12549. [Google Scholar] [CrossRef] [PubMed]
- Žerovnik, E. Viroporins vs. Other Pore-Forming Proteins: What Lessons Can We Take? Front. Chem. 2021, 9, 626059. [Google Scholar] [CrossRef]
- Asrani, P.; Seebohm, G.; Stoll, R. Potassium Viroporins as Model Systems for Understanding Eukaryotic Ion Channel Behaviour. Virus Res. 2022, 320, 198903. [Google Scholar] [CrossRef]
- Carrasco, L. Membrane Leakiness after Viral Infection and a New Approach to the Development of Antiviral Agents. Nature 1978, 272, 694–699. [Google Scholar] [CrossRef]
- Nieva, J.; Carrasco, L. Viroporins: Structures and Functions beyond Cell Membrane Permeabilization. Viruses 2015, 7, 5169–5171. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; Carrasco, L. Viroporins. FEBS Lett. 2003, 552, 28–34. [Google Scholar] [CrossRef]
- Cedillo-Barrón, L.; García-Cordero, J.; Visoso-Carvajal, G.; León-Juárez, M. Viroporins Manipulate Cellular Powerhouses and Modulate Innate Immunity. Viruses 2024, 16, 345. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and Biological Functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Balannik, V.; Pinto, L.H.; Lamb, R.A. Influenza Virus M2 Ion Channel Protein Is Necessary for Filamentous Virion Formation. J. Virol. 2010, 84, 5078–5088. [Google Scholar] [CrossRef]
- Suhy, D.A.; Giddings, T.H.; Kirkegaard, K. Remodeling the Endoplasmic Reticulum by Poliovirus Infection and by Individual Viral Proteins: An Autophagy-Like Origin for Virus-Induced Vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef]
- Barco, A.; Carrasco, L. A Human Virus Protein, Poliovirus Protein 2BC, Induces Membrane Proliferation and Blocks the Exocytic Pathway in the Yeast Saccharomyces Cerevisiae. EMBO J. 1995, 14, 3349–3364. [Google Scholar] [CrossRef]
- De Jong, A.S.; Visch, H.-J.; De Mattia, F.; Van Dommelen, M.M.; Swarts, H.G.; Luyten, T.; Callewaert, G.; Melchers, W.J.; Willems, P.H.; Van Kuppeveld, F.J. The Coxsackievirus 2B Protein Increases Efflux of Ions from the Endoplasmic Reticulum and Golgi, Thereby Inhibiting Protein Trafficking through the Golgi. J. Biol. Chem. 2006, 281, 14144–14150. [Google Scholar] [CrossRef]
- Kirkegaard, K. Subversion of the Cellular Autophagy Pathway by Viruses. In Autophagy in Infection and Immunity; Levine, B., Yoshimori, T., Deretic, V., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 335, pp. 323–333. ISBN 978-3-642-00301-1. [Google Scholar]
- Schnell, J.R.; Chou, J.J. Structure and Mechanism of the M2 Proton Channel of Influenza A Virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef]
- Martyna, A.; Bahsoun, B.; Badham, M.D.; Srinivasan, S.; Howard, M.J.; Rossman, J.S. Membrane Remodeling by the M2 Amphipathic Helix Drives Influenza Virus Membrane Scission. Sci. Rep. 2017, 7, 44695. [Google Scholar] [CrossRef]
- Wilson, L.; Mckinlay, C.; Gage, P.; Ewart, G. SARS Coronavirus E Protein Forms Cation-Selective Ion Channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, R.; Wang, W.; Lee, I.; Zhang, R.; Zhang, W.; Sun, J.; Xu, B.; Meng, X. Computational Study of the Ion and Water Permeation and Transport Mechanisms of the SARS-CoV-2 Pentameric E Protein Channel. Front. Mol. Biosci. 2020, 7, 565797. [Google Scholar] [CrossRef]
- Zhang, R.; Qin, H.; Prasad, R.; Fu, R.; Zhou, H.-X.; Cross, T.A. Dimeric Transmembrane Structure of the SARS-CoV-2 E Protein. Commun. Biol. 2023, 6, 1109. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.A.; Fapohunda, O.; Wang, Z.; Pham, H.; Taylor, M.T.; Kloss, B.; Park, S.H.; Opella, S.; Aspinwall, C.A.; Marty, M.T. Differences in Oligomerization of the SARS-CoV-2 Envelope Protein, Poliovirus VP4, and HIV Vpu. Biochemistry 2024, 63, 241–250. [Google Scholar] [CrossRef]
- Kondrashov, O.V.; Galimzyanov, T.R.; Jiménez-Munguía, I.; Batishchev, O.V.; Akimov, S.A. Membrane-Mediated Interaction of Amphipathic Peptides Can Be Described by a One-Dimensional Approach. Phys. Rev. E 2019, 99, 022401. [Google Scholar] [CrossRef]
- Monje-Galvan, V.; Voth, G.A. Molecular Interactions of the M and E Integral Membrane Proteins of SARS-CoV-2. Faraday Discuss. 2021, 232, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Caldas, L.A.; Carneiro, F.A.; Augusto, I.; Corrêa, I.A.; Da Costa, L.J.; Miranda, K.; Tanuri, A.; De Souza, W. SARS-CoV-2 Egress from Vero Cells: A Morphological Approach. Histochem. Cell Biol. 2024, 161, 59–67. [Google Scholar] [CrossRef]
- Somberg, N.H.; Wu, W.W.; Medeiros-Silva, J.; Dregni, A.J.; Jo, H.; DeGrado, W.F.; Hong, M. SARS-CoV-2 Envelope Protein Forms Clustered Pentamers in Lipid Bilayers. Biochemistry 2022, 61, 2280–2294. [Google Scholar] [CrossRef]
- Pinkwart, K.; Schneider, F.; Lukoseviciute, M.; Sauka-Spengler, T.; Lyman, E.; Eggeling, C.; Sezgin, E. Nanoscale Dynamics of Cholesterol in the Cell Membrane. J. Biol. Chem. 2019, 294, 12599–12609. [Google Scholar] [CrossRef] [PubMed]
- Dimova, R.; Riske, K.A. Electrodeformation, Electroporation, and Electrofusion of Giant Unilamellar Vesicles. In Handbook of Electroporation; Miklavcic, D., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–18. ISBN 978-3-319-26779-1. [Google Scholar]
- Islam, M.Z.; Alam, J.M.; Tamba, Y.; Karal, M.A.S.; Yamazaki, M. The Single GUV Method for Revealing the Functions of Antimicrobial, Pore-Forming Toxin, and Cell-Penetrating Peptides or Proteins. Phys. Chem. Chem. Phys. 2014, 16, 15752–15767. [Google Scholar] [CrossRef] [PubMed]
- Shnyrova, A.V.; Zimmerberg, J. Reconstitution of Membrane Budding with Unilamellar Vesicles. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 464, pp. 55–75. ISBN 978-0-12-374969-7. [Google Scholar]
- Mueller, P.; Rudin, D.O.; Ti Tien, H.; Wescott, W.C. Reconstitution of Cell Membrane Structure in Vitro and Its Transformation into an Excitable System. Nature 1962, 194, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Chui, J.K.W.; Fyles, T.M. Ionic Conductance of Synthetic Channels: Analysis, Lessons, and Recommendations. Chem. Soc. Rev. 2012, 41, 148–175. [Google Scholar] [CrossRef] [PubMed]
- Horcas, I.; Fernández, R.; Gómez-Rodríguez, J.M.; Colchero, J.; Gómez-Herrero, J.; Baro, A.M. WSXM: A Software for Scanning Probe Microscopy and a Tool for Nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705. [Google Scholar] [CrossRef] [PubMed]
- Loshkareva, A.S.; Popova, M.M.; Shilova, L.A.; Fedorova, N.V.; Timofeeva, T.A.; Galimzyanov, T.R.; Kuzmin, P.I.; Knyazev, D.G.; Batishchev, O.V. Influenza A Virus M1 Protein Non-Specifically Deforms Charged Lipid Membranes and Specifically Interacts with the Raft Boundary. Membranes 2023, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, Y.; Sakashita, S.; Shimizu, K.; Usui, K.; Kawano, R. Channel Current Analysis Estimates the Pore-Formation and the Penetration of Transmembrane Peptides. Analyst 2018, 143, 3540–3543. [Google Scholar] [CrossRef] [PubMed]
- Drabik, D.; Chodaczek, G.; Kraszewski, S.; Langner, M. Mechanical Properties Determination of DMPC, DPPC, DSPC, and HSPC Solid-Ordered Bilayers. Langmuir 2020, 36, 3826–3835. [Google Scholar] [CrossRef]
- Hammond, K.; Ryadnov, M.G.; Hoogenboom, B.W. Atomic Force Microscopy to Elucidate How Peptides Disrupt Membranes. Biochim. Biophys. Acta BBA Biomembr. 2021, 1863, 183447. [Google Scholar] [CrossRef]
- Aryal, C.M.; Bui, N.N.; Khadka, N.K.; Song, L.; Pan, J. The Helix 0 of Endophilin Modifies Membrane Material Properties and Induces Local Curvature. Biochim. Biophys. Acta BBA Biomembr. 2020, 1862, 183397. [Google Scholar] [CrossRef]
- Verdiá-Báguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; DeDiego, M.L.; Enjuanes, L.; Aguilella, V.M. Analysis of SARS-CoV E Protein Ion Channel Activity by Tuning the Protein and Lipid Charge. Biochim. Biophys. Acta BBA Biomembr. 2013, 1828, 2026–2031. [Google Scholar] [CrossRef]
- Pan, J.; Dalzini, A.; Khadka, N.K.; Aryal, C.M.; Song, L. Lipid Extraction by α-Synuclein Generates Semi-Transmembrane Defects and Lipoprotein Nanoparticles. ACS Omega 2018, 3, 9586–9597. [Google Scholar] [CrossRef]
- Khadka, N.K.; Hazen, P.; Haemmerle, D.; Mainali, L. Interaction of βL- and γ-Crystallin with Phospholipid Membrane Using Atomic Force Microscopy. Int. J. Mol. Sci. 2023, 24, 15720. [Google Scholar] [CrossRef]
- Chen, F.-Y.; Lee, M.-T.; Huang, H.W. Evidence for Membrane Thinning Effect as the Mechanism for Peptide-Induced Pore Formation. Biophys. J. 2003, 84, 3751–3758. [Google Scholar] [CrossRef] [PubMed]
- Krasnobaev, V.D.; Bershatsky, Y.V.; Bocharova, O.V.; Bocharov, E.V.; Batishchev, O.V. Amyloid Precursor Protein Changes Arrangement in a Membrane and Its Structure Depending on the Cholesterol Content. Membranes 2023, 13, 706. [Google Scholar] [CrossRef] [PubMed]
- Woolley, G.A. Channel-Forming Activity of Alamethicin: Effects of Covalent Tethering. Chem. Biodivers. 2007, 4, 1323–1337. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volovik, M.V.; Denieva, Z.G.; Gifer, P.K.; Rakitina, M.A.; Batishchev, O.V. Membrane Activity and Viroporin Assembly for the SARS-CoV-2 E Protein Are Regulated by Cholesterol. Biomolecules 2024, 14, 1061. https://doi.org/10.3390/biom14091061
Volovik MV, Denieva ZG, Gifer PK, Rakitina MA, Batishchev OV. Membrane Activity and Viroporin Assembly for the SARS-CoV-2 E Protein Are Regulated by Cholesterol. Biomolecules. 2024; 14(9):1061. https://doi.org/10.3390/biom14091061
Chicago/Turabian StyleVolovik, Marta V., Zaret G. Denieva, Polina K. Gifer, Maria A. Rakitina, and Oleg V. Batishchev. 2024. "Membrane Activity and Viroporin Assembly for the SARS-CoV-2 E Protein Are Regulated by Cholesterol" Biomolecules 14, no. 9: 1061. https://doi.org/10.3390/biom14091061
APA StyleVolovik, M. V., Denieva, Z. G., Gifer, P. K., Rakitina, M. A., & Batishchev, O. V. (2024). Membrane Activity and Viroporin Assembly for the SARS-CoV-2 E Protein Are Regulated by Cholesterol. Biomolecules, 14(9), 1061. https://doi.org/10.3390/biom14091061