
Mitochondrial Dysfunction in Glycogen Storage Disorders (GSDs)

Abstract
:1. Background
2. Glycogen Storage Disorders (GSDs) an Overview
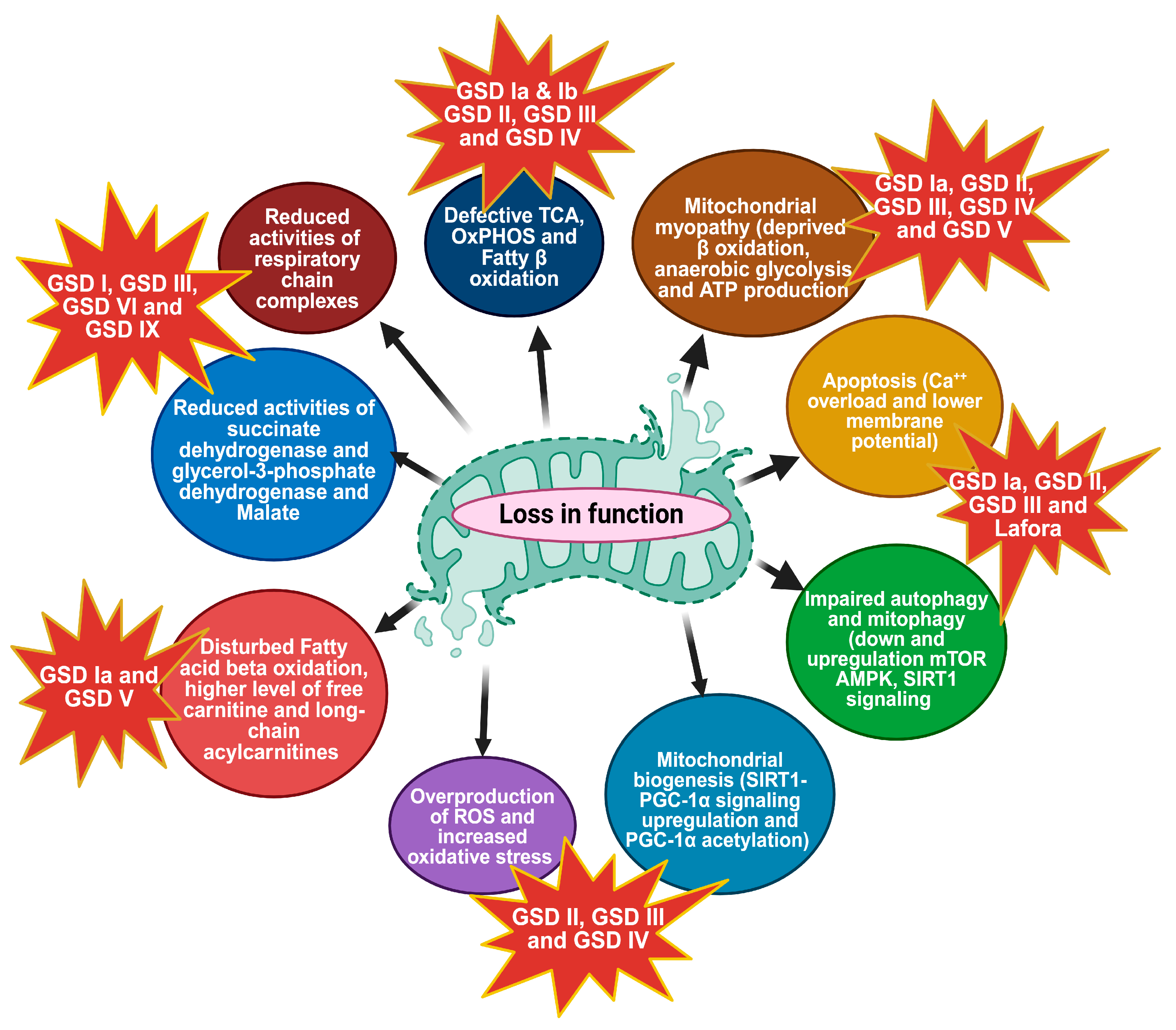
3. Mitochondrial Metabolic Dysfunction
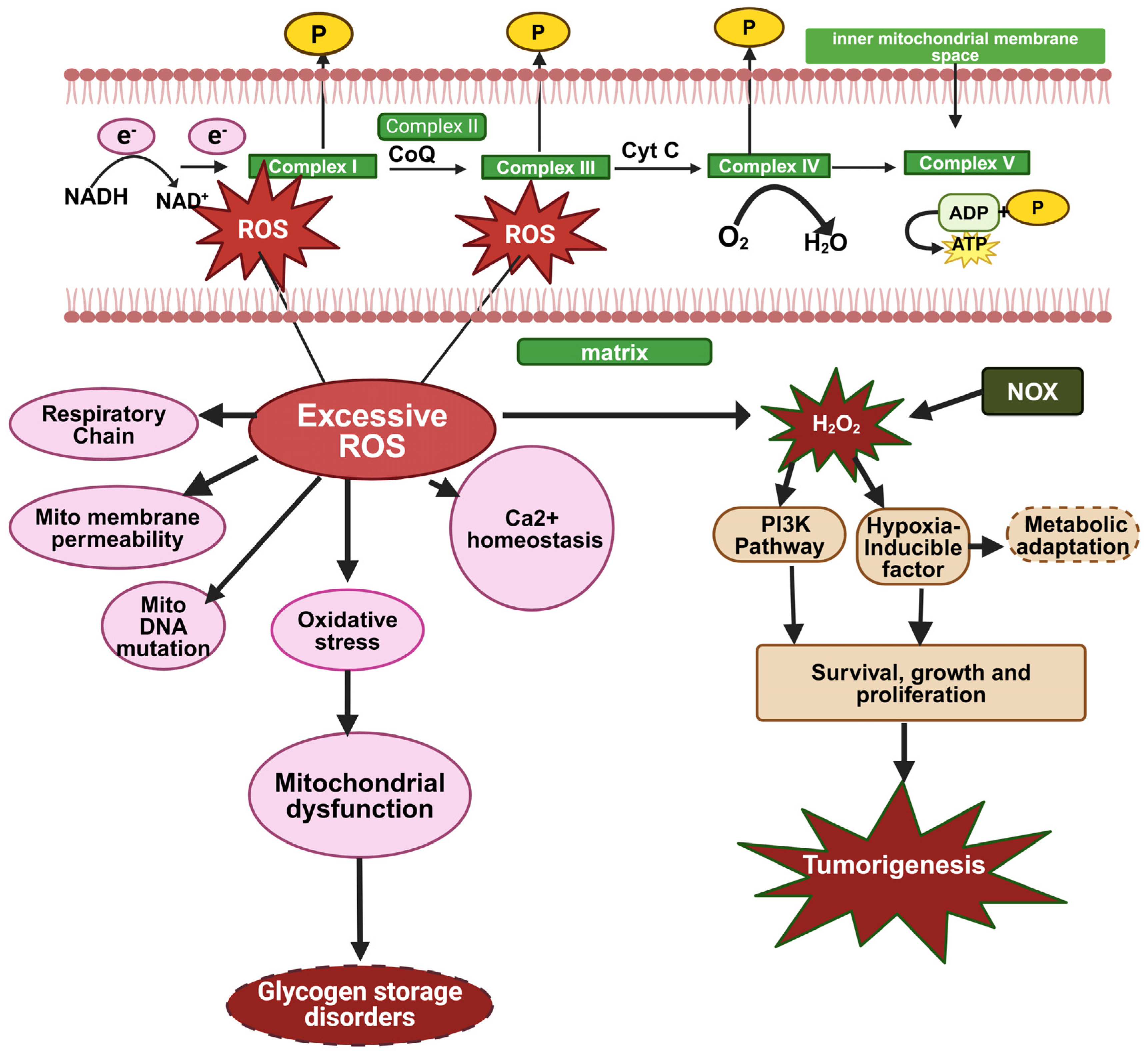
3.1. Overproduction of ROS and Oxidative Stress
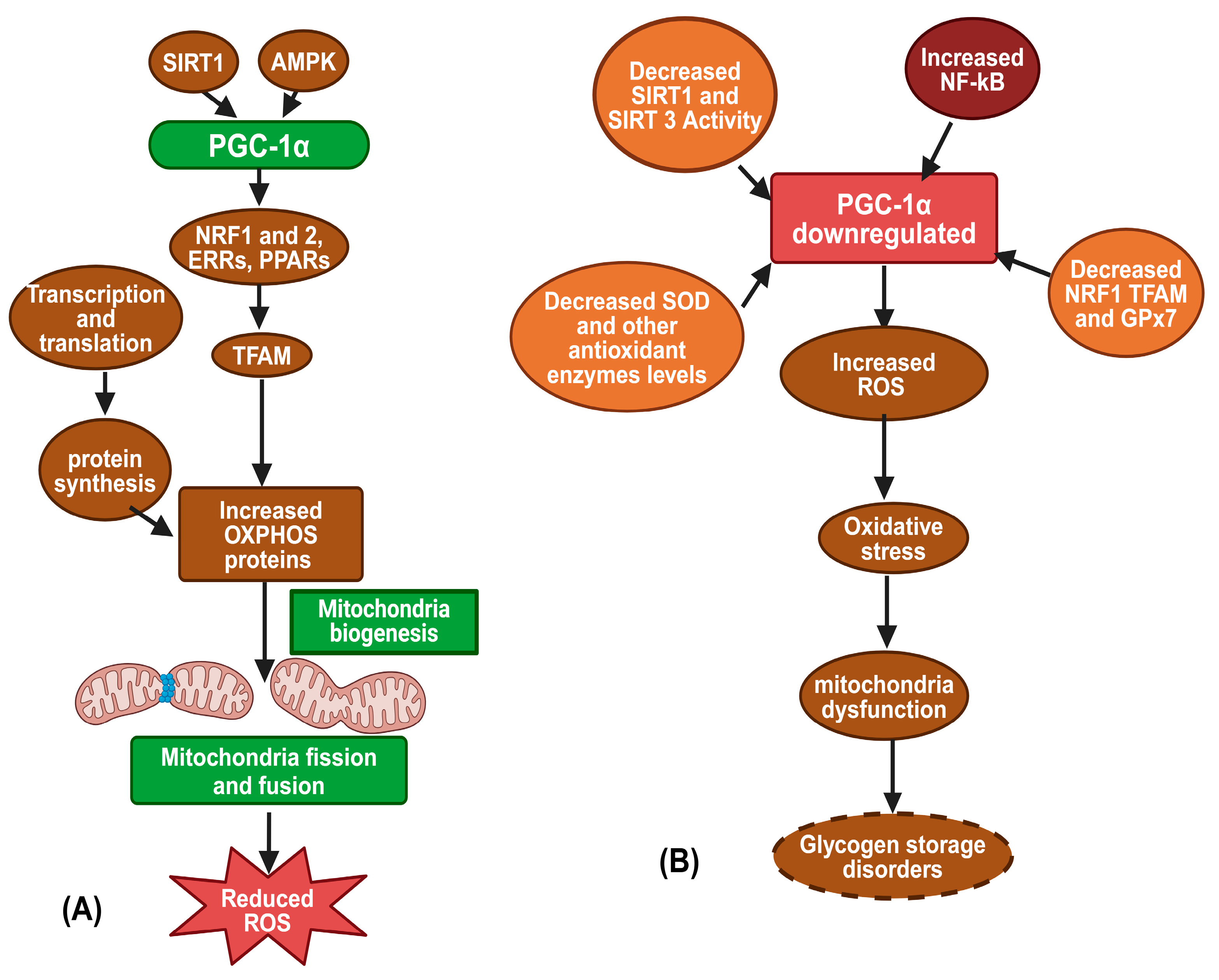
3.2. Mitochondrial Biogenesis
3.3. Autophagy and Mitophagy
3.4. Apoptosis and Mitochondrial Dysfunction
3.5. Mitochondrial Myopathy
4. Diagnosis and Therapeutic Strategies in GSDs
4.1. Diagnosis
4.2. Current Treatment Options and Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gümüş, E.; Özen, H. Glycogen storage diseases: An update. World J. Gastroenterol. 2023, 29, 3932–3963. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kanungo, S.; Wells, K.; Tribett, T.; El-Gharbawy, A. Glycogen metabolism and glycogen storage disorders. Ann. Transl. Med. 2018, 6, 474. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Massese, M.; Tagliaferri, F.; Dionisi-Vici, C.; Maiorana, A. Glycogen storage diseases with liver involvement: A literature review of GSD type 0, IV, VI, IX and XI. Orphanet J. Rare Dis. 2022, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- de Marchi, R.; Nalin, T.; Sperb-Ludwig, F.; Pinheiro, F.C.; Schwartz, I.V.D.; Steiner, C.E. Glycogen Storage Disease: Expert Opinion on Clinical Diagnosis Revisited after Molecular Testing. Genes 2023, 14, 2219. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- La Morgia, C.; Maresca, A.; Caporali, L.; Valentino, M.; Carelli, V. Mitochondrial diseases in adults. J. Intern. Med. 2020, 287, 592–608. [Google Scholar] [CrossRef]
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, e00313. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Oosterveer, M.H.; Mithieux, G.; Rajas, F. Mechanisms by which metabolic reprogramming in GSD1 liver generates a favorable tumorigenic environment. J. Inborn Errors Metab. Screen. 2019, 4, e160020. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, R.; Jiang, C.; Wang, J.; Zhou, Y.; Xu, X.; Wang, T.; Li, A.; Zhang, Y. Mitochondrial dysfunction is associated with hypertrophic cardiomyopathy in Pompe disease-specific induced pluripotent stem cell-derived cardiomyocytes. Cell Prolif. 2024, 57, e13573. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of mitochondrial dysfunction in lysosomal storage disorders: A review. J. Clin. Med. 2020, 9, 2596. [Google Scholar] [CrossRef]
- Sentner, C.P.; Hoogeveen, I.J.; Weinstein, D.A.; Santer, R.; Murphy, E.; McKiernan, P.J.; Steuerwald, U.; Beauchamp, N.J.; Taybert, J.; Laforêt, P. Glycogen storage disease type III: Diagnosis, genotype, management, clinical course and outcome. J. Inherit. Metab. Dis. 2016, 39, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Farah, B.L.; Sinha, R.A.; Wu, Y.; Singh, B.K.; Lim, A.; Hirayama, M.; Landau, D.J.; Bay, B.H.; Koeberl, D.D.; Yen, P.M. Hepatic mitochondrial dysfunction is a feature of glycogen storage disease type Ia (GSDIa). Sci. Rep. 2017, 7, 44408. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.; Sweetat, S.; Baraghithy, S.; Sprecher, U.; Marisat, M.; Bastu, S.; Glickstein, H.; Tam, J.; Rosenmann, H.; Weil, M.; et al. The Autophagic Activator GHF-201 Can Alleviate Pathology in a Mouse Model and in Patient Fibroblasts of Type III Glycogenosis. Biomolecules 2024, 14, 893. [Google Scholar] [CrossRef]
- Fiuza-Luces, C.; Andreu, A.; Rodríguez-Aguilera, J.; Martín, M.; Arenas, J.; Vissing, J.; Lucia, A.; Krag, T.; Pinós, T. Low aerobic capacity in McArdle disease: A role for mitochondrial network impairment? Mol. Metab. 2022, 66, 101648. [Google Scholar]
- Kamenets, E.A.; Gusarova, E.A.; Milovanova, N.V.; Itkis, Y.S.; Strokova, T.V.; Melikyan, M.A.; Garyaeva, I.V.; Rybkina, I.G.; Nikitina, N.V.; Zakharova, E.Y. Hepatic glycogen synthase (GYS2) deficiency: Seven novel patients and seven novel variants. JIMD Rep. 2020, 53, 39–44. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kasapkara, Ç.; Aycan, Z.; Açoğlu, E.; Senel, S.; Oguz, M.M.; Ceylaner, S. The variable clinical phenotype of three patients with hepatic glycogen synthase deficiency. J. Pediatr. Endocrinol. Metab. 2017, 30, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, O.; Pugliese, A.; Oteri, R.; Volta, S.; Ciranni, A.; Moggio, M.; Rodolico, C.; Toscano, A. A new phenotype of muscle glycogen synthase deficiency (GSD0B) characterized by an adult onset myopathy without cardiomyopathy. Neuromuscul. Disord. 2022, 32, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Kollberg, G.; Tulinius, M.; Gilljam, T.; Ostman-Smith, I.; Forsander, G.; Jotorp, P.; Oldfors, A.; Holme, E. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N. Engl. J. Med. 2007, 357, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Visser, G.; Rake, J.P.; Fernandes, J.; Labrune, P.; Leonard, J.V.; Moses, S.; Ullrich, K.; Smit, G.P. Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: Results of the European Study on Glycogen Storage Disease type I. J. Pediatr. 2000, 137, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Veiga-da-Cunha, M.; Wortmann, S.B.; Grünert, S.C.; Van Schaftingen, E. Treatment of the neutropenia associated with GSD1b and G6PC3 deficiency with SGLT2 inhibitors. Diagnostics 2023, 13, 1803. [Google Scholar] [CrossRef]
- Meena, N.K.; Raben, N. Pompe disease: New developments in an old lysosomal storage disorder. Biomolecules 2020, 10, 1339. [Google Scholar] [CrossRef]
- Szymańska, E.; Szymańska, S.; Truszkowska, G.; Ciara, E.; Pronicki, M.; Shin, Y.S.; Podskarbi, T.; Kępka, A.; Śpiewak, M.; Płoski, R. Variable clinical presentation of glycogen storage disease type IV: From severe hepatosplenomegaly to cardiac insufficiency. Some discrepancies in genetic and biochemical abnormalities. Arch. Med. Sci. 2018, 14, 237–247. [Google Scholar] [CrossRef]
- Kakhlon, O.; Vaknin, H.; Mishra, K.; D’Souza, J.; Marisat, M.; Sprecher, U.; Wald-Altman, S.; Dukhovny, A.; Raviv, Y.; Da’adoosh, B. Alleviation of a polyglucosan storage disorder by enhancement of autophagic glycogen catabolism. EMBO Mol. Med. 2021, 13, e14554. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.L.; Soler-Alfonso, C.; Kiely, B.T.; Asai, A.; Smith, A.L.; Bali, D.S.; Kang, P.B.; Landstrom, A.P.; Akman, H.O.; Burrow, T.A. Diagnosis and management of glycogen storage disease type IV, including adult polyglucosan body disease: A clinical practice resource. Mol. Genet. Metab. 2023, 138, 107525. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.D.; Pereira, Â.; Soares, A.R.; Guimas, A.; Rocha, S.; Cardoso, M.; Garrido, C.; Soares, C.A.; Nunes, I.S.; Fortuna, A.M. Diagnostic accuracy and the first genotype–phenotype correlation in glycogen storage disease type V. Pediatr. Res. 2023, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Labrador, E.; Weinstein, D.A. Glycogen storage disease type VI. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Aeppli, T.R.; Rymen, D.; Allegri, G.; Bode, P.K.; Häberle, J. Glycogen storage disease type VI: Clinical course and molecular background. Eur. J. Pediatr. 2020, 179, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, O.; Bruno, C.; Mongini, T.; Rodolico, C.; Aguennouz, M.h.; Barca, E.; Amati, A.; Cassandrini, D.; Serlenga, L.; Vita, G. Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscular Disord. 2012, 22, 325–330. [Google Scholar] [CrossRef]
- DiMauro, S.; Spiegel, R. Progress and problems in muscle glycogenoses. Acta Myol. 2011, 30, 96. [Google Scholar] [PubMed]
- Fernandes, S.A.; Cooper, G.E.; Gibson, R.A.; Kishnani, P.S. Benign or not benign? Deep phenotyping of liver Glycogen Storage Disease IX. Mol. Genet. Metab. 2020, 131, 299–305. [Google Scholar] [CrossRef]
- Roscher, A.; Patel, J.; Hewson, S.; Nagy, L.; Feigenbaum, A.; Kronick, J.; Raiman, J.; Schulze, A.; Siriwardena, K.; Mercimek-Mahmutoglu, S. The natural history of glycogen storage disease types VI and IX: Long-term outcome from the largest metabolic center in Canada. Mol. Genet. Metab. 2014, 113, 171–176. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Goldstein, J.; Austin, S.L.; Arn, P.; Bachrach, B.; Bali, D.S.; Chung, W.K.; El-Gharbawy, A.; Brown, L.M.; Kahler, S. Diagnosis and management of glycogen storage diseases type VI and IX: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2019, 21, 772–789. [Google Scholar] [CrossRef]
- Bali, D.S.; Goldstein, J.L.; Fredrickson, K.; Austin, S.; Pendyal, S.; Rehder, C.; Kishnani, P.S. Clinical and molecular variability in patients with PHKA2 variants and liver phosphorylase b kinase deficiency. JIMD Rep. 2017, 37, 63–72. [Google Scholar] [PubMed]
- Ellingwood, S.S.; Cheng, A. Biochemical and clinical aspects of glycogen storage diseases. J. Endocrinol. 2018, 238, R131–R141. [Google Scholar] [CrossRef]
- Kwong, A.K.Y.; Wong, S.S.N.; Rodenburg, R.J.; Smeitink, J.; Chan, G.C.F.; Fung, C.W. Human d-lactate dehydrogenase deficiency by LDHD mutation in a patient with neurological manifestations and mitochondrial complex IV deficiency. JIMD Rep. 2021, 60, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Lorenzo, P.; Rabasa, M.; Esteban, J.; Hidalgo Mayoral, I.; Domínguez-González, C.; Blanco-Echevarría, A.; Garrido-Moraga, R.; Lucia, A.; Blázquez, A.; Rubio, J.C. Clinical, biochemical, and molecular characterization of two families with novel mutations in the LDHA gene (GSD XI). Genes 2022, 13, 1835. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Chen, X.; Yang, J.; Ding, J. Lactate dehydrogenase D is a general dehydrogenase for D-2-hydroxyacids and is associated with D-lactic acidosis. Nat. Commun. 2023, 14, 6638. [Google Scholar] [CrossRef] [PubMed]
- Mamoune, A.; Bahuau, M.; Hamel, Y.; Serre, V.; Pelosi, M.; Habarou, F.; Nguyen Morel, M.-A.; Boisson, B.; Vergnaud, S.; Viou, M.T. A thermolabile aldolase A mutant causes fever-induced recurrent rhabdomyolysis without hemolytic anemia. PLoS Genet. 2014, 10, e1004711. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Svingou, M.; Kekou, K.; Vergnaud, S.; Xirou, S.; Niotakis, G.; Papadimas, G. Aldolase A deficiency: Report of new cases and literature review. Mol. Genet. Metab. Rep. 2021, 27, 100730. [Google Scholar] [CrossRef]
- Wigley, R.; Scalco, R.S.; Gardiner, A.R.; Godfrey, R.; Booth, S.; Kirk, R.; Hilton-Jones, D.; Houlden, H.; Heales, S.; Quinlivan, R. The need for biochemical testing in beta-enolase deficiency in the genomic era. JIMD Rep. 2019, 50, 40–43. [Google Scholar] [CrossRef]
- Buch, A.E.; Musumeci, O.; Wigley, R.; Stemmerik, M.P.G.; Eisum, A.S.V.; Madsen, K.L.; Preisler, N.; Hilton-Jones, D.; Quinlivan, R.; Toscano, A. Energy metabolism during exercise in patients with β-enolase deficiency (GSDXIII). JIMD Rep. 2021, 61, 60–66. [Google Scholar] [CrossRef]
- Malfatti, E.; Nilsson, J.; Hedberg-Oldfors, C.; Hernandez-Lain, A.; Michel, F.; Dominguez-Gonzalez, C.; Viennet, G.; Akman, H.O.; Kornblum, C.; Van den Bergh, P. A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Ann. Neurol. 2014, 76, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Visuttijai, K.; Hedberg-Oldfors, C.; Thomsen, C.; Glamuzina, E.; Kornblum, C.; Tasca, G.; Hernandez-Lain, A.; Sandstedt, J.; Dellgren, G.; Roach, P. Glycogenin is dispensable for glycogen synthesis in human muscle, and glycogenin deficiency causes polyglucosan storage. J. Clin. Endocrinol. Metab. 2020, 105, 557–566. [Google Scholar] [CrossRef]
- Conte, F.; Morava, E.; Bakar, N.A.; Wortmann, S.B.; Poerink, A.J.; Grunewald, S.; Crushell, E.; Al-Gazali, L.; de Vries, M.C.; Mørkrid, L. Phosphoglucomutase-1 deficiency: Early presentation, metabolic management and detection in neonatal blood spots. Mol. Genet. Metab. 2020, 131, 135–146. [Google Scholar] [CrossRef]
- Altassan, R.; Radenkovic, S.; Edmondson, A.C.; Barone, R.; Brasil, S.; Cechova, A.; Coman, D.; Donoghue, S.; Falkenstein, K.; Ferreira, V. International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): Diagnosis, follow-up, and management. J. Inherit. Metab. Dis. 2021, 44, 148–163. [Google Scholar] [CrossRef]
- Grünert, S.C.; Schwab, K.O.; Pohl, M.; Sass, J.O.; Santer, R. Fanconi–Bickel syndrome: GLUT2 mutations associated with a mild phenotype. Mol. Genet. Metab. 2012, 105, 433–437. [Google Scholar] [CrossRef]
- Sharari, S.; Abou-Alloul, M.; Hussain, K.; Ahmad Khan, F. Fanconi–bickel syndrome: A review of the mechanisms that lead to dysglycaemia. Int. J. Mol. Sci. 2020, 21, 6286. [Google Scholar] [CrossRef] [PubMed]
- Matsumaru, S.; Oguni, H.; Ogura, H.; Shimojima, K.; Nagata, S.; Kanno, H.; Yamamoto, T. A novel PGK1 mutation associated with neurological dysfunction and the absence of episodes of hemolytic anemia or myoglobinuria. Intractable Rare Dis. Res. 2017, 6, 132–136. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Nadjar, Y.; Béhin, A.; Biancalana, V.; Piraud, M.; Malfatti, E.; Laforêt, P. Phosphoglycerate kinase deficiency: A nationwide multicenter retrospective study. J. Inherit. Metab. Dis. 2019, 42, 803–808. [Google Scholar] [CrossRef]
- Vega, R.B.; Horton, J.L.; Kelly, D.P. Maintaining ancient organelles: Mitochondrial biogenesis and maturation. Circ. Res. 2015, 116, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 310, G205–G214. [Google Scholar] [CrossRef]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Rodenburg, R.J. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Kurbatova, O.; Surkov, A.; Namazova-Baranova, L.; Polyakova, S.; Miroshkina, L.; Semenova, G.; Samokhina, I.; Kapustina, E.Y.; Dukhova, Z.; Potapov, A. Mitochondrial dysfunction in children with hepatic forms of glycogen storage disease. Ann. Russ. Acad. Med. Sci. 2014, 69, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Wary, C.; Nadaj-Pakleza, A.; Laforêt, P.; Claeys, K.G.; Carlier, R.; Monnet, A.; Fleury, S.; Baligand, C.; Eymard, B.; Labrune, P. Investigating glycogenosis type III patients with multi-parametric functional NMR imaging and spectroscopy. Neuromuscular Disord. 2010, 20, 548–558. [Google Scholar] [CrossRef]
- Preisler, N.; Pradel, A.; Husu, E.; Madsen, K.L.; Becquemin, M.-H.; Mollet, A.; Labrune, P.; Petit, F.; Hogrel, J.-Y.; Jardel, C. Exercise intolerance in glycogen storage disease type III: Weakness or energy deficiency? Mol. Genet. Metab. 2013, 109, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Hennis, P.J.; Murphy, E.; Meijer, R.I.; Lachmann, R.H.; Ramachandran, R.; Bordoli, C.; Rayat, G.; Tomlinson, D.J. Aerobic capacity and skeletal muscle characteristics in glycogen storage disease IIIa: An observational study. Orphanet J. Rare Dis. 2022, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, L.; Theimer, J.; Wingert, V.; Klotz, K.; Bierschenk, I.; Nitschke, R.; Spiekerkoetter, U.; Gruenert, S.C. Metabolic profiling in human fibroblasts enables subtype clustering in glycogen storage disease. Front. Endocrinol. 2020, 11, 579981. [Google Scholar] [CrossRef]
- Rossi, A.; Ruoppolo, M.; Formisano, P.; Villani, G.; Albano, L.; Gallo, G.; Crisci, D.; Moccia, A.; Parenti, G.; Strisciuglio, P. Insulin-resistance in glycogen storage disease type Ia: Linking carbohydrates and mitochondria? J. Inherit. Metab. Dis. 2018, 41, 985–995. [Google Scholar] [CrossRef]
- Saavedra, H.; Yu, A.; Rodriguez-Buritica, D. The use of alanine, free carnitine and IGFBP-1 as potential biomarkers for glycogen storage disease type I. Mol. Genet. Metab. 2022, 136, S18. [Google Scholar] [CrossRef]
- Rossi, A.; Assunto, A.; Rosano, C.; Tucci, S.; Ruoppolo, M.; Caterino, M.; Pirozzi, F.; Strisciuglio, P.; Parenti, G.; Melis, D. Mitochondrial reprogramming in peripheral blood mononuclear cells of patients with glycogen storage disease type Ia. Genes Nutr. 2023, 18, 10. [Google Scholar] [CrossRef]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial dysfunction plays central role in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, B.; Summers, S.A. Ceramides–lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Trivedi, M.; Singh, A.; Talekar, M.; Amiji, M. Mitochondrial biology, targets, and drug delivery. J. Control. Release 2015, 207, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Hasani, S.; Young, L.E.A.; Van Nort, W.; Banerjee, M.; Rivas, D.R.; Kim, J.; Xiong, X.; Sun, R.C.; Gentry, M.S.; Sesaki, H.; et al. Inhibition of mitochondrial fission activates glycogen synthesis to support cell survival in colon cancer. Cell Death Dis. 2023, 14, 664. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M. Pompe disease diagnosis and management guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Vander Roest, A.S.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac energy metabolism in heart failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-P.; Chen, P.-H.; Hwu, W.-L.; Chuang, C.-Y.; Chien, Y.-H.; Stone, L.; Chien, C.-L.; Li, L.-T.; Chiang, S.-C.; Chen, H.-F. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum. Mol. Genet. 2011, 20, 4851–4864. [Google Scholar] [CrossRef]
- Lim, J.-A.; Li, L.; Kakhlon, O.; Myerowitz, R.; Raben, N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015, 11, 385–402. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Venditti, P. Mitochondrial management of reactive oxygen species. Antioxidants 2021, 10, 1824. [Google Scholar] [CrossRef]
- Bou-Teen, D.; Kaludercic, N.; Weissman, D.; Turan, B.; Maack, C.; Di Lisa, F.; Ruiz-Meana, M. Mitochondrial ROS and mitochondria-targeted antioxidants in the aged heart. Free. Radic. Biol. Med. 2021, 167, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Vujic, A.; Koo, A.N.; Prag, H.A.; Krieg, T. Mitochondrial redox and TCA cycle metabolite signaling in the heart. Free. Radic. Biol. Med. 2021, 166, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat Commun. 2019, 10, 1704. [Google Scholar] [CrossRef]
- Kim, J.; Bai, H. Peroxisomal stress response and inter-organelle communication in cellular homeostasis and aging. Antioxidants 2022, 11, 192. [Google Scholar] [CrossRef]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Lee, B.-C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 446–462. [Google Scholar] [CrossRef]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Gonçalves, V.F. Mitochondrial Genetics. In Mitochondria in Health and in Sickness; Urbani, A., Babu, M., Eds.; Springer: Singapore, 2019; pp. 247–255. [Google Scholar] [CrossRef]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α is a master regulator of mitochondrial lifecycle and ROS stress response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Islam, H.; Edgett, B.A.; Gurd, B.J. Coordination of mitochondrial biogenesis by PGC-1α in human skeletal muscle: A re-evaluation. Metabolism 2018, 79, 42–51. [Google Scholar] [CrossRef]
- Jannig, P.R.; Dumesic, P.A.; Spiegelman, B.M.; Ruas, J.L. SnapShot: Regulation and biology of PGC-1α. Cell 2022, 185, 1444.e1. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Julia Xu, X.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Carling, D.; Viollet, B. Beyond energy homeostasis: The expanding role of AMP-activated protein kinase in regulating metabolism. Cell Metab. 2015, 21, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Devin, A. Reactive oxygen species-mediated control of mitochondrial biogenesis. Int J Cell Biol. 2012, 2012, 403870. [Google Scholar] [CrossRef]
- Cho, J.-H.; Kim, G.-Y.; Mansfield, B.C.; Chou, J.Y. Sirtuin signaling controls mitochondrial function in glycogen storage disease type Ia. J. Inherit. Metab. Dis. 2018, 41, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Majeed, S.T.; Majeed, R.; Andrabi, K.I. Expanding the view of the molecular mechanisms of autophagy pathway. J. Cell. Physiol. 2022, 237, 3257–3277. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria–lysosome crosstalk: From physiology to neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef]
- Audano, M.; Schneider, A.; Mitro, N. Mitochondria, lysosomes, and dysfunction: Their meaning in neurodegeneration. J. Neurochem. 2018, 147, 291–309. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Zhuang, N.; Li, L.; Chen, S.; Wang, T. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 2016, 7, e2501. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhu, H.; Liu, X.; Huang, X.; Huang, A.; Huang, Y. Mitophagy in diabetic cardiomyopathy: Roles and mechanisms. Front. Cell Dev. Biol. 2021, 9, 750382. [Google Scholar] [CrossRef]
- Nakamura, K.; Miyoshi, T.; Yoshida, M.; Akagi, S.; Saito, Y.; Ejiri, K.; Matsuo, N.; Ichikawa, K.; Iwasaki, K.; Naito, T. Pathophysiology and treatment of diabetic cardiomyopathy and heart failure in patients with diabetes mellitus. Int. J. Mol. Sci. 2022, 23, 3587. [Google Scholar] [CrossRef]
- Farah, B.L.; Landau, D.J.; Sinha, R.A.; Brooks, E.D.; Wu, Y.; Fung, S.Y.S.; Tanaka, T.; Hirayama, M.; Bay, B.-H.; Koeberl, D.D. Induction of autophagy improves hepatic lipid metabolism in glucose-6-phosphatase deficiency. J. Hepatol. 2016, 64, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-H.; Kim, G.-Y.; Pan, C.-J.; Anduaga, J.; Choi, E.-J.; Mansfield, B.C.; Chou, J.Y. Downregulation of SIRT1 signaling underlies hepatic autophagy impairment in glycogen storage disease type Ia. PLoS Genet. 2017, 13, e1006819. [Google Scholar] [CrossRef] [PubMed]
- Farah, B.L.; Yen, P.M.; Koeberl, D.D. Links between autophagy and disorders of glycogen metabolism–perspectives on pathogenesis and possible treatments. Mol. Genet. Metab. 2020, 129, 3–12. [Google Scholar] [CrossRef]
- Mitra, S.; Gumusgoz, E.; Minassian, B.A. Lafora disease: Current biology and therapeutic approaches. Rev. Neurol. 2022, 178, 315–325. [Google Scholar] [CrossRef]
- Lahuerta, M.; Aguado, C.; Sánchez-Martín, P.; Sanz, P.; Knecht, E. Degradation of altered mitochondria by autophagy is impaired in Lafora disease. FEBS J. 2018, 285, 2071–2090. [Google Scholar] [CrossRef]
- Klemmensen, M.M.; Borrowman, S.H.; Pearce, C.; Pyles, B.; Chandra, B. Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics 2024, 21, e00292. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Vallette, F.; Vayssiere, J.-L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012, 942, 157–183. [Google Scholar] [PubMed]
- Sun, B.; Li, S.; Yang, L.; Damodaran, T.; Desai, D.; Diehl, A.M.; Alzate, O.; Koeberl, D.D. Activation of glycolysis and apoptosis in glycogen storage disease type Ia. Mol. Genet. Metab. 2009, 97, 267–271. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A. Myopathies related to glycogen metabolism disorders. Neurotherapeutics 2018, 15, 915–927. [Google Scholar] [CrossRef]
- Tsai, L.-K.; Hwu, W.-L.; Lee, N.-C.; Huang, P.-H.; Chien, Y.-H. Clinical features of Pompe disease with motor neuronopathy. Neuromuscular Disord. 2019, 29, 903–906. [Google Scholar] [CrossRef]
- De Feo, P.; Di Loreto, C.; Lucidi, P.; Murdolo, G.; Parlanti, N.; De Cicco, A.; Piccioni, F.; Santeusanio, F. Metabolic response to exercise. J. Endocrinol. Investig. 2003, 26, 851–854. [Google Scholar] [CrossRef]
- Santalla, A.; Valenzuela, P.L.; Rodriguez-Lopez, C.; Rodríguez-Gómez, I.; Nogales-Gadea, G.; Pinós, T.; Arenas, J.; Martín, M.A.; Santos-Lozano, A.; Morán, M. Long-Term Exercise Intervention in Patients with McArdle Disease: Clinical and Aerobic Fitness Benefits. Med. Sci. Sports Exerc. 2022, 54, 1231–1241. [Google Scholar] [CrossRef]
- Munguía-Izquierdo, D.; Santalla, A.; Lucia, A. Cardiorespiratory fitness, physical activity, and quality of life in patients with McArdle disease. Med. Sci. Sports Exerc. 2015, 47, 799–808. [Google Scholar] [CrossRef]
- Scalco, R.S.; Lucia, A.; Santalla, A.; Martinuzzi, A.; Vavla, M.; Reni, G.; Toscano, A.; Musumeci, O.; Voermans, N.C.; Kouwenberg, C.V. Data from the European registry for patients with McArdle disease and other muscle glycogenoses (EUROMAC). Orphanet J. Rare Dis. 2020, 15, 330. [Google Scholar] [CrossRef]
- Santalla, A.; Nogales-Gadea, G.; Encinar, A.B.; Vieitez, I.; González-Quintana, A.; Serrano-Lorenzo, P.; Consuegra, I.G.; Asensio, S.; Ballester-Lopez, A.; Pintos-Morell, G. Genotypic and phenotypic features of all Spanish patients with McArdle disease: A 2016 update. BMC Genom. 2017, 18, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Pizzamiglio, C.; Mahroo, O.A.; Khan, K.N.; Patasin, M.; Quinlivan, R. Phenotype and genotype of 197 British patients with McArdle disease: An observational single-centre study. J. Inherit. Metab. Dis. 2021, 44, 1409–1418. [Google Scholar] [CrossRef]
- De Stefano, N.; Argov, Z.; Matthews, P.M.; Karpati, G.; Arnold, D.L. Impairment of muscle mitochondrial oxidative metabolism in McArdle’s disease. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 1996, 19, 764–769. [Google Scholar] [CrossRef]
- Moslemi, A.-R.; Lindberg, C.; Nilsson, J.; Tajsharghi, H.; Andersson, B.; Oldfors, A. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N. Engl. J. Med. 2010, 362, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1. [Google Scholar] [CrossRef] [PubMed]
- Hannah, W.B.; Derks, T.G.; Drumm, M.L.; Grünert, S.C.; Kishnani, P.S.; Vissing, J. Glycogen storage diseases. Nat. Rev. Dis. Primers 2023, 9, 46. [Google Scholar] [CrossRef]
- Keutzer, J.M. Establishing Pompe disease newborn screening: The role of industry. Int. J. Neonatal Screen. 2020, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Coppede, F.; Migliore, L.; Siciliano, G.; Murri, L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J. Alzheimer’s Dis. 2006, 10, 59–73. [Google Scholar] [CrossRef]
- Rodenburg, R.J.T. Biochemical diagnosis of mitochondrial disorders. J. Inherit. Metab. Dis. 2011, 34, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Ermani, M.; Salviati, L.; Angelini, C. Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion 2011, 11, 893–904. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef]
- Medja, F.; Allouche, S.; Frachon, P.; Jardel, C.; Malgat, M.; De Camaret, B.M.; Slama, A.; Lunardi, J.; Mazat, J.-P.; Lombès, A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Winkel, L.P.; Kamphoven, J.H.; Van Den Hout, H.J.; Severijnen, L.A.; Van Doorn, P.A.; Reuser, A.J.; Van Der Ploeg, A.T. Morphological changes in muscle tissue of patients with infantile Pompe’s disease receiving enzyme replacement therapy. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2003, 27, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Koeberl, D.D.; Koch, R.L.; Lim, J.A.; Brooks, E.D.; Arnson, B.D.; Sun, B.; Kishnani, P.S. Gene therapy for glycogen storage diseases. J. Inherit. Metab. Dis. 2024, 47, 93–118. [Google Scholar] [CrossRef]
- Jauze, L.; Monteillet, L.; Mithieux, G.; Rajas, F.; Ronzitti, G. Challenges of gene therapy for the treatment of glycogen storage diseases type I and type III. Hum. Gene Ther. 2019, 30, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Sun, B.; Koeberl, D.D. Gene therapy for glycogen storage diseases. Hum. Mol. Genet. 2019, 28, R31–R41. [Google Scholar] [CrossRef] [PubMed]
- Polyak, E.; Ostrovsky, J.; Peng, M.; Dingley, S.D.; Tsukikawa, M.; Kwon, Y.J.; McCormack, S.E.; Bennett, M.; Xiao, R.; Seiler, C. N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol. Genet. Metab. 2018, 123, 449–462. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Crozier, M.; Di Carlo, A.; Xhuti, D.; Manta, K.; Roik, L.J.; Bujak, A.L.; Nederveen, J.P.; Tarnopolsky, M.G.; Hettinga, B. Nutritional co-therapy with 1, 3-butanediol and multi-ingredient antioxidants enhances autophagic clearance in Pompe disease. Mol. Genet. Metab. 2022, 137, 228–240. [Google Scholar] [CrossRef]
- Andreadi, A.; Bellia, A.; Di Daniele, N.; Meloni, M.; Lauro, R.; Della-Morte, D.; Lauro, D. The molecular link between oxidative stress, insulin resistance, and type 2 diabetes: A target for new therapies against cardiovascular diseases. Curr. Opin. Pharmacol. 2022, 62, 85–96. [Google Scholar] [CrossRef]
- Grünert, S.C.; Elling, R.; Maag, B.; Wortmann, S.B.; Derks, T.G.; Hannibal, L.; Schumann, A.; Rosenbaum-Fabian, S.; Spiekerkoetter, U. Improved inflammatory bowel disease, wound healing and normal oxidative burst under treatment with empagliflozin in glycogen storage disease type Ib. Orphanet J. Rare Dis. 2020, 15, 218. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Van Hove, J.L.; Derks, T.G.; Chevalier, N.; Knight, V.; Koller, A.; Oussoren, E.; Mayr, J.A.; van Spronsen, F.J.; Lagler, F.B. Treating neutropenia and neutrophil dysfunction in glycogen storage disease type Ib with an SGLT2 inhibitor. Blood J. Am. Soc. Hematol. 2020, 136, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Maiorana, A.; Tagliaferri, F.; Dionisi-Vici, C. Current understanding on pathogenesis and effective treatment of glycogen storage disease type Ib with empagliflozin: New insights coming from diabetes for its potential implications in other metabolic disorders. Front. Endocrinol. 2023, 14, 1145111. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorders (Common Name) | Enzyme | Gene/Chromosome/-Inheritance | Clinical Manifestation |
|---|---|---|---|
| GSD 0a | Glycogen synthase in Liver | GYS2/12p12.1/AR | Fasting ketotic hypoglycemia, reactive hyperglycemia and lactate elevation [15,16] |
| GSD 0b | Glycogen synthase in muscle | GYS1/19q13.33/AR | Hypertrophic cardiomyopathy exercise intolerance, and adult-onset myopathy without cardiomyopathy [17,18] |
| GSD Ia (von Gierke disease) | Glucose-6-phosphatase | G6PC/17q21.31/AR | Hypoglycemia, lactic acidosis, Hypertriglyceridemia, hepatomegaly, renal dysfunction [19] |
| GSD Ib | Glucose-6-phosphate translocase | SLC37A4/11q23.3/AR | Neutropenia, neutrophil dysfunction and inflammatory bowel disease [20] |
| GSD II (PD) | alpha-1,4-glucosidase | GAA/17q25.3/AR | Hypertrophic cardiomyopathy, hypotonia, and motor delay [21] |
| GSD III (Cori or Forbes disease) | Glycogen debranching (amylo-1,6 glucosidase) | AGL/1p21.2/AR | Hypoglycemia, elevated ketosis in hyperlipidemia, hepatomegaly, elevated liver enzyme myopathy, variable muscle and cardiac phenotype [11] |
| GSD IV (Andersen or Adult polyglucosan body disease) | Glycogen branching enzyme- amylo (1,4-1,6) transglucosidase | GBE1/3p12.2/AR | Hepatosplenomegaly, liver dysfunction, progressive cirrhosis, cardiomyopathy, hypotonia, gait difficulty, progressive neurogenic bladder, autonomic dysfunction, sensory loss, and variable cognitive difficulty [22,23,24] |
| GSD 5 (McArdle disease) | Myophosphorylase | PYGM/11q13.1/AR | Exercise induces fatigue, cramps, tachypnoea and tachycardia, rhabdo-myolysis, myoglobinuria [25] |
| GSD 6 (Hers disease) | Liver glycogen phosphorylase | PYGL/14q22.1/AR | Hepatomegaly, hypoglycemia, with ketosis, elevated liver transaminases, hyperlipidemia, osteoporosis, and liver fibrosis [26,27] |
| GSD 7 (Tarui disease) | Muscle phosphofructokinase | PFKM/12q13.11/AR | Hemolytic anemia, muscle weakness, exercise-induced muscle cramping, exertional Myopathy, and gout/hyperuricemia [28,29] |
| GSD 9A1 (formerly GSD 8) | Alpha-2 subunit of liver phosphorylase kinase | PHKA2/Xp22.13 XL | Hepatomegaly, growth retardation, motor developmental delay. Hypercholesterolmia, hypertriglyceridemia, elevated liver enzymes; fasting hyperketosis [30] |
| GSD 9B (GSD IXb) | Beta subunit of liver and muscle phosphorylase | PHKB/16q12.1/AR | Short stature, hepatomegaly, diarrhea, muscle weakness, hypotonia [3,31] |
| GSD 9C (GSD IXc) | Hepatic and testis isoform gamma subunit of phosphorylase kinase | PHKG2/16p11.2/AR | Growth retardation, hepatomegaly, hypotonia; cognitive delay [32,33] |
| GSD 9D (GSD IXd) | Alpha subunit of muscle phosphorylase kinase | PHKA1/Xq13.1/XL | Muscle weakness, exercise-induced muscle pain and stiffness, muscle atrophy, elevated CK [3,30,32] |
| GSD 10 (GSD X) | Muscle phosphoglycerate mutase | PGAM2/7p13/AR | Exercise intolerance with cramp or pain, rhabdo-myolysis, myoglobinuria, hyperuricemia, coronary arteriosclerosis [2,34] |
| GSD 11 (GSD XI) | Lactate dehydrogenase A | LDHA/11p15.1/AR | Exercise-induced muscle cramps and pain, uterine muscle stiffness in pregnancy, psoriatic skin lesions, Elevated serum CK during myoglobinuria, with low serum lactate dehydrogenase [35,36,37] |
| GSD 12 (GSD XII) | Fructose-1,6- bisphosphate aldolase | ALDOA/16p11.2/AR | Short stature dysmorphic face, myopathy; mental retardation; delayed puberty; hemolytic anemia, hepatosplenomegaly; rhabdomyolysis with febrile illness [38,39] |
| GSD 13 (GSD XIII) | Beta-enolase | ENO3/17p13.2/AR | Exercise intolerance, myalgia, rhabdomyo-lysis with fatty infiltration [40,41] |
| GSD 15 (GSD XV) | Glycogenin-1 | GYG1/3q24/AR | Weakness, arrhythmias, skeletal myopathy, cardiomyopathy [42,43] |
| GSD 14 (Previously GSDXIV) | Phosphoglucomutase-1 | PGM1/1p31.3/AR | Hematological anomalies, hypoglycemia, growth retardation, and dilated cardio-myopathy [44,45] |
| Fanconi–Bickel syndrome (GSD XI) | SLC2A2 | GLUT2/3q26.2/AR | Postprandial elevations of glucose and galactose, fasting hypoglycemia, hepatomegaly, proximal tubular nephropathy, glucosuria, short stature [46,47] |
| PGK deficiency | PGK1 | PGK/Xq21.1/XL | Nonspherocytic hemolytic anemia, myopathy with rhabdomyolysis and neurological features, myopathy, rhabdomyolysis [48,49] |
| Type of GSDs | General Lab Test and Imaging | Diagnostic Test | Genetic Test | Treatment and Management |
|---|---|---|---|---|
| GSD 0a | NA | NA | Biallelic PLP GYS2 variants | Protein-rich meals and bedtime consumption of uncooked cornstarch |
| GSD 0b | NA | NA | Biallelic PLP GYS1 variants | |
| GSD I | Kidney and liver function tests, lipids and uric acid levels, a complete blood count, and iron studies. Liver and kidney imaging, echocardiography | GSD Ia, G6Pase enzyme activity in liver tissue | Biallelic PLP G6PC1 variants (GSD Ia), biallelic PLP SLC37A4 (GSD Ib) | Angiotensin receptor blockers, low-purine diet, and Allopurinol for gout, G-CSF, and empagliflozin Liver transplantation |
| GSD II | CK, AST, ALT, urine Glc4, BNP, and, when receiving ERT, IgG against recombinant protein levels. ECG, Echo, Chest X-Ray | GAA activity testing in blood, fibroblast, or muscle | Biallelic PLP variants in GAA | ERT, Respiratory muscle training, maximize clearance of airway secretions |
| GSD III | Liver function tests, coagulation, Lipid, CK levels, echocardiography | GDE activity assay for liver and muscle, can be measured in leukocytes, erythrocytes, or cultured fibroblasts | Biallelic PLP AGL variants | Dietary management, hypoglycemia monitoring and muscle health support |
| GSD IV | liver function tests, PT/INR, albumin, renal function panel, complete blood count, ammonia levels, AFP levels, USG, Bone density scan, physical therapy/occupational therapy | GBE activity assay in on fibroblast, liver or muscle and PB accumulation by PAS staining | Biallelic PLP GBE1 variants | Hepatitis A and B vaccinations. Prophylactic antibiotics for small bowel infections and urinary tract infections. Physical therapy and occupational therapy. Routinely screen for dysphagia (difficulty swallowing). Liver transplant if required |
| GSD 5 | Serum CK and uric acid levels, Hb A1c, lipid profile. weakness and muscle evaluation. Assess ADLs and QoL | Myophosphorylase activity | Biallelic PLP PYGM | Carbohydrate rich diet, Sucrose to improve exercise tolerance. Exercise is helpful for chronic pain |
| GSD 6 | AST, ALT, serum albumin and γ-glutamyl transferase levels, coagulation, blood glucose, and serum β-hydroxybutyrate monitoring | Phosphorylase activity in liver specimens | Biallelic PLP variants in PYGL | Dietary and symptomatic treatment |
| GSD 7 | Phosphofructokinase activity in muscle | Biallelic PLP PFKM variants | Low-carbohydrate ketogenic diet. Exercise to avoid chronic pain | |
| GSD 9 | AST, ALT, serum albumin and γ-glutamyl transferase levels, coagulation DEXA, and heart imaging. Blood glucose and serum β-hydroxybutyrate monitoring | Phosphorylase b kinase activity can be measured in liver, blood, and muscle (based on subtype). Enzyme activity in blood can be normal | Hemizygous PLP variant in PHKA2 (GSD IX A) biallelic PLP variants in PHKB (GSD IX B) | Dietary and symptomatic treatment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, K.; Kakhlon, O. Mitochondrial Dysfunction in Glycogen Storage Disorders (GSDs). Biomolecules 2024, 14, 1096. https://doi.org/10.3390/biom14091096
Mishra K, Kakhlon O. Mitochondrial Dysfunction in Glycogen Storage Disorders (GSDs). Biomolecules. 2024; 14(9):1096. https://doi.org/10.3390/biom14091096
Chicago/Turabian StyleMishra, Kumudesh, and Or Kakhlon. 2024. "Mitochondrial Dysfunction in Glycogen Storage Disorders (GSDs)" Biomolecules 14, no. 9: 1096. https://doi.org/10.3390/biom14091096