

New BDNF and NT-3 Cyclic Mimetics Concur with Copper to Activate Trophic Signaling Pathways as Potential Molecular Entities to Protect Old Brains from Neurodegeneration

 , , , , , , , and
, , , , , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Design and Synthesis
2.2. NMR Measurements
2.3. Potentiometric Titrations
2.4. Ultraviolet-Visible (UV-Vis) Measurements
2.5. Circular Dichroism (CD) Measurements
2.6. Inductively Coupled Plasma—Optical Emission Spectroscopy
2.7. Cell Culture Reagents and Antibodies
2.8. Cell Culture and Treatment Conditions
2.9. Neuronal Differentiation of SH-SY5Y by Neurite Outgrowth Analysis
2.10. Determination of Ctr1 Translocation by Immunofluorescence Assay
2.11. Protein Lysate Preparation and Immunoblotting
2.12. Sandwich ELISA Assay
2.13. Statistical Analysis
3. Results
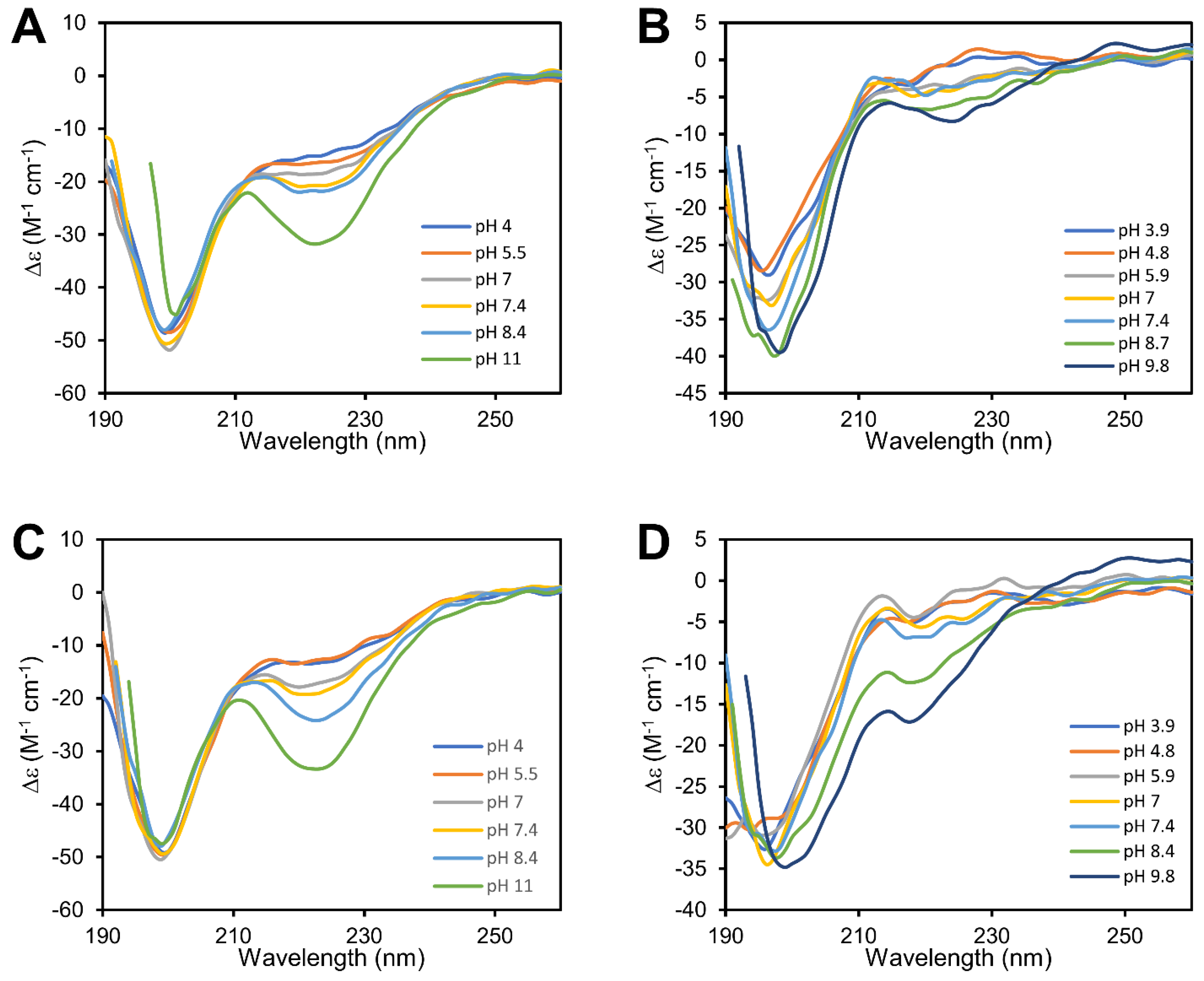
3.1. cBDNF(1-12) and cNT3(1−13) Are Cyclic Peptides Which Show Different Secondary Structures
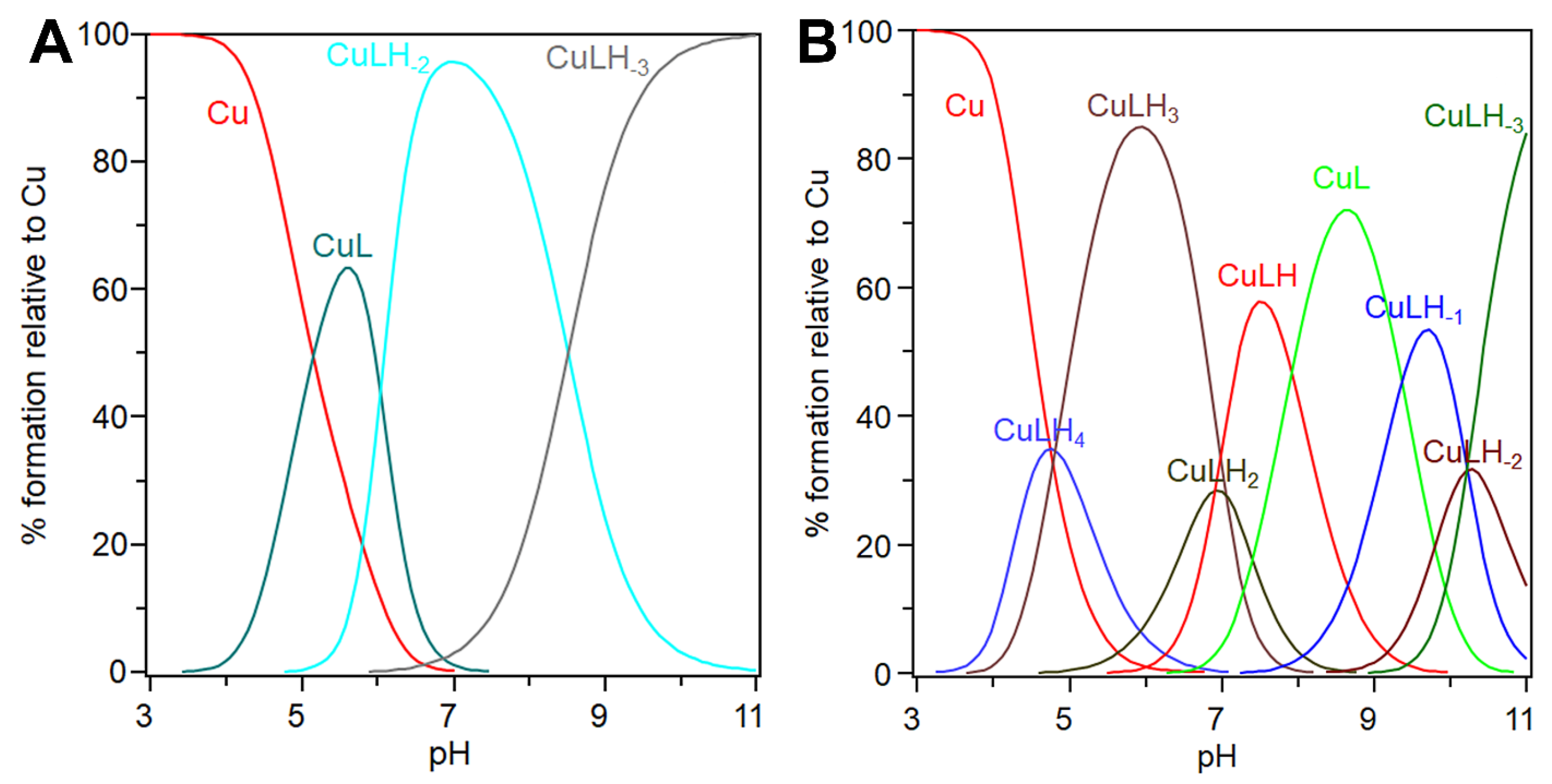
3.2. cBDNF(1-12) and cNT3(1-13) Show Different Speciation and Binding Affinity for the Copper(II) Ion
3.3. Conditional Dissociation Constant Values (cKd) Allow for a Comparison of Different Affinity of the Two Peptides
3.4. Copper(II) Ion Is Present at Sub-Micromolar Concentration in Culture Media Supplemented with Serum
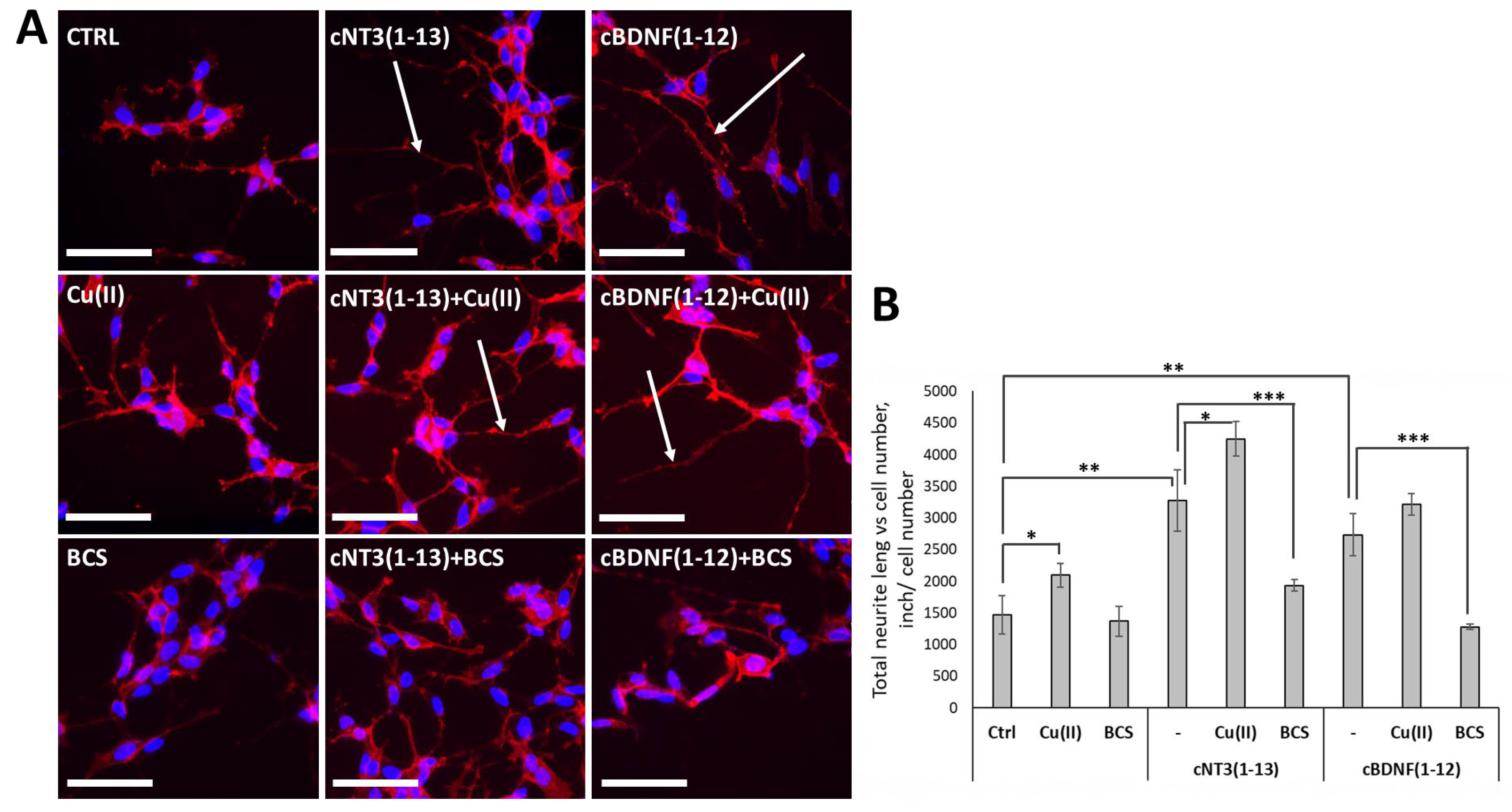
3.5. cNT3(1-13) and cBDNF(1-12) Mimic the Neuronal Differentiation Capability of NTs in SH-SY5Y Neuroblastoma Cells
3.6. cNT-3(1-13) and cBDNF(1-12) Are Ionophore Peptides and Induce a Decrease of Ctr1 Localization on Cell Membrane
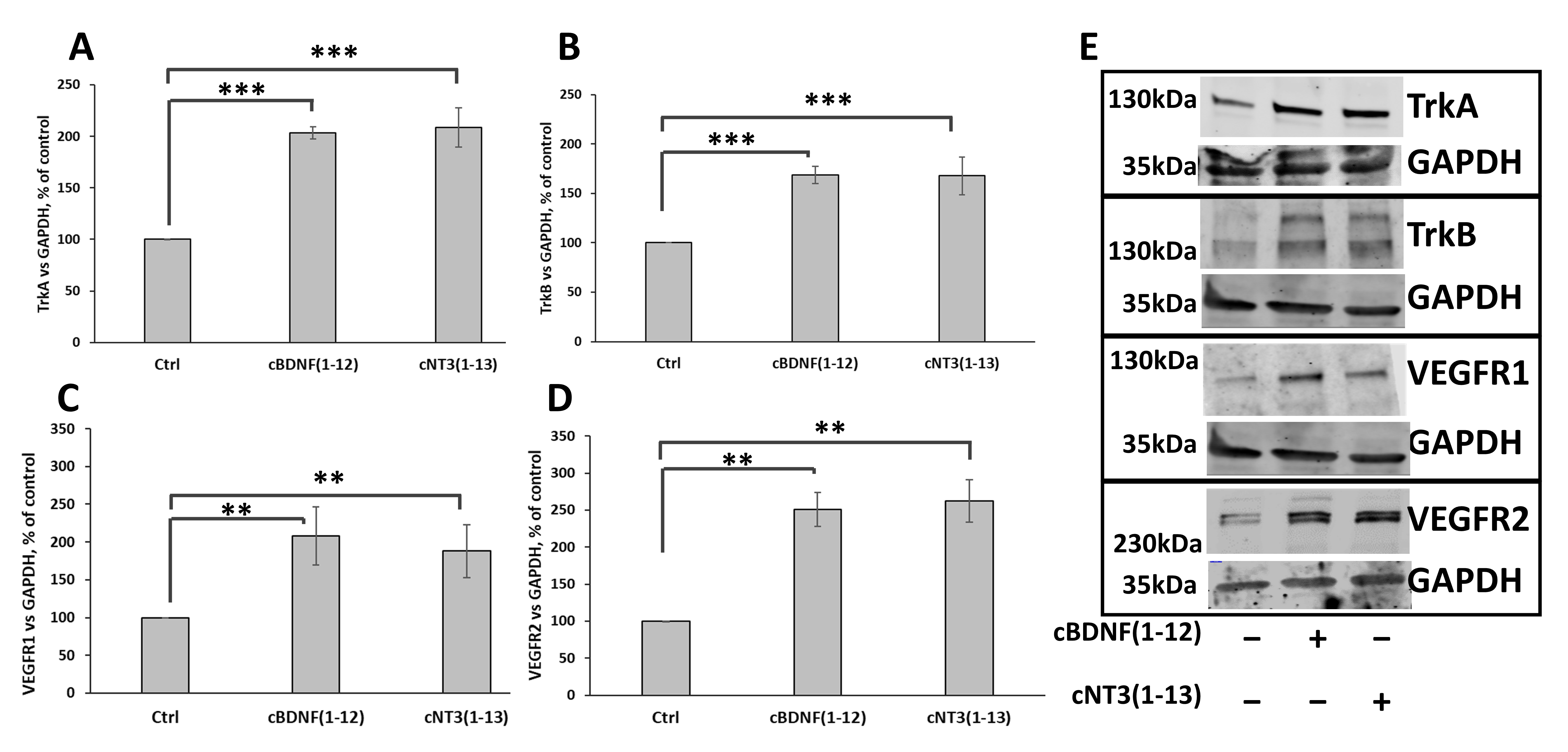
3.7. cNT3(1-13) and cBDNF(1-12) Increase TrkA and TrkB as Well as VEGFR1 and VEGFR2 Receptors Expression
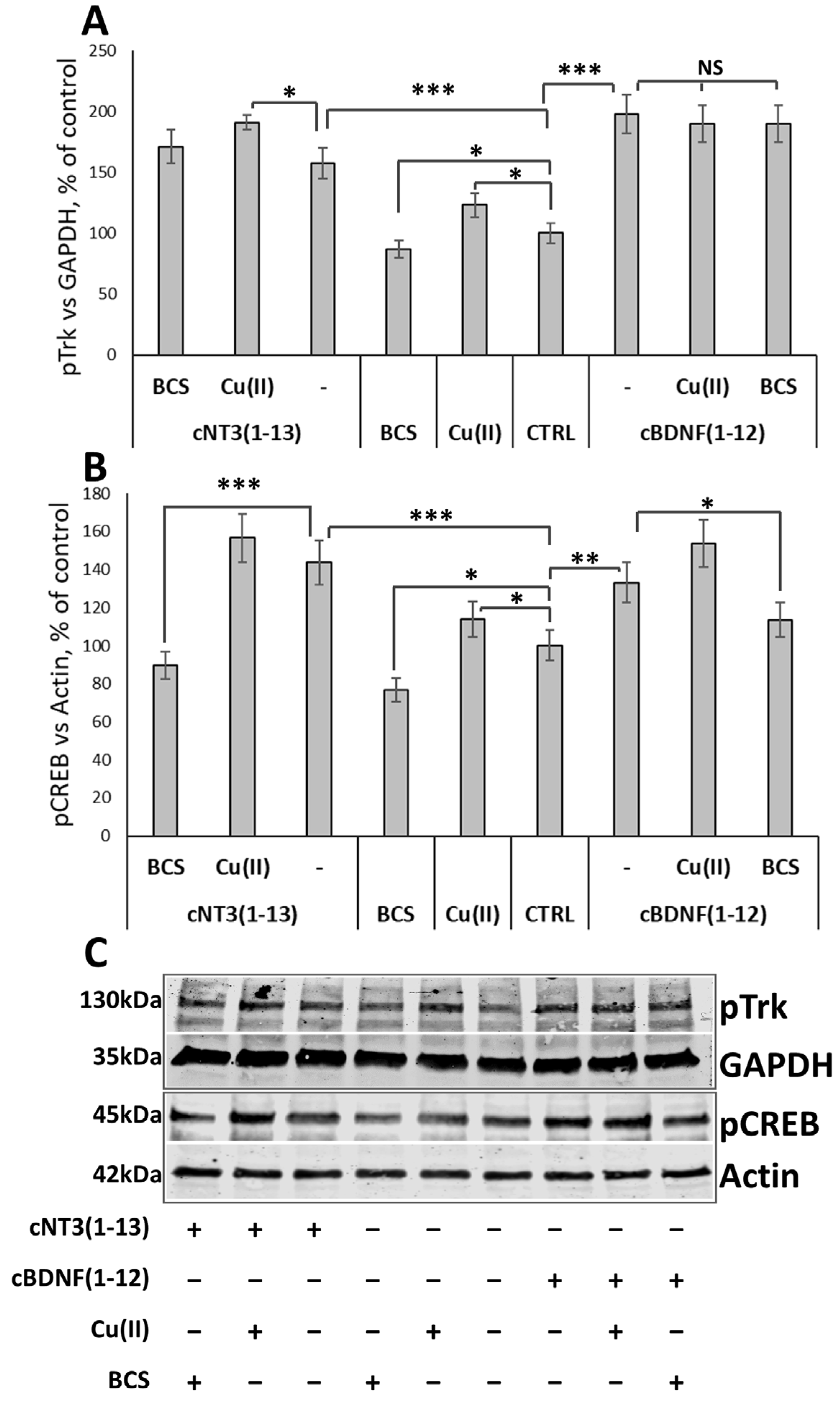
3.8. cNT3(1-13) and cBDNF(1-12) Promote Trk Signaling Cascade by CREB Phosphorylation in a Copper Dependent Manner
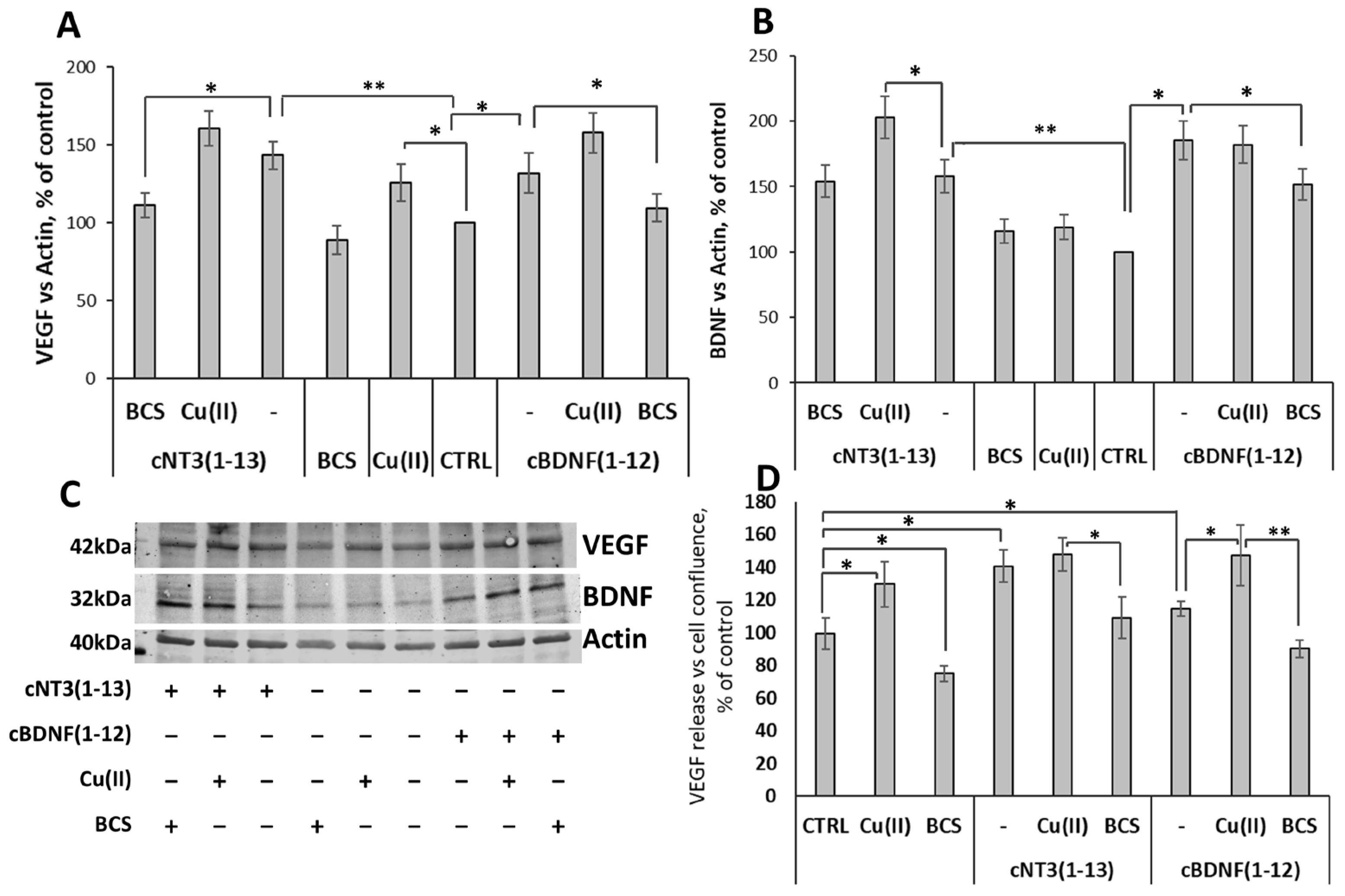
3.9. cNT3(1-13)/ and cBDNF(1-12)/Trk Pathway Stimulate Expression of Trophic Factors and the Release of VEGF
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Tokuda, T.; Kasai, T.; Ishigami, N.; Hidaka, H.; Kondo, M.; Allsop, D.; Nakagawa, M. High-molecular-weight Β-amyloid Oligomers Are Elevated in Cerebrospinal Fluid of Alzheimer Patients. FASEB J. 2010, 24, 2716–2726. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Kuhn, P.; Haass, C.; Kennedy, M.E.; Rajendran, L.; Wong, P.C.; Lichtenthaler, S.F. Function, Therapeutic Potential and Cell Biology of BACE Proteases: Current Status and Future Prospects. J. Neurochem. 2014, 130, 4–28. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of Tau Protein in Health and Disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hardy, J. Alzheimer’s Disease: The Amyloid Cascade Hypothesis: An Update and Reappraisal. J. Alzheimer’s Dis. 2006, 9, 151–153. [Google Scholar] [CrossRef]
- Appel, S.H. A Unifying Hypothesis for the Cause of Amyotrophic Lateral Sclerosis, Parkinsonism, and Alzheimer Disease. Ann. Neurol. 1981, 10, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Hefti, F.; Weiner, W.J. Nerve Growth Factor and Alzheimer’s Disease. Ann. Neurol. 1986, 20, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Cuello, A.C. Nerve Growth Factor Metabolic Dysfunction in Alzheimer’s Disease and Down Syndrome. Trends Pharmacol. Sci. 2014, 35, 338–348. [Google Scholar] [CrossRef]
- Cattaneo, A.; Calissano, P. Nerve Growth Factor and Alzheimer’s Disease: New Facts for an Old Hypothesis. Mol. Neurobiol. 2012, 46, 588–604. [Google Scholar] [CrossRef]
- Capsoni, S.; Ugolini, G.; Comparini, A.; Ruberti, F.; Berardi, N.; Cattaneo, A. Alzheimer-like Neurodegeneration in Aged Antinerve Growth Factor Transgenic Mice. Proc. Natl. Acad. Sci. USA 2000, 97, 6826–6831. [Google Scholar] [CrossRef]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. The Human Brain NGF Metabolic Pathway Is Impaired in the Pre-Clinical and Clinical Continuum of Alzheimers Disease. Mol. Psychiatry 2021, 26, 6023–6037. [Google Scholar] [CrossRef]
- Jiao, F.; Jiang, D.; Li, Y.; Mei, J.; Wang, Q.; Li, X. Amyloidogenesis and Neurotrophic Dysfunction in Alzheimer’s Disease: Do They Have a Common Regulating Pathway? Cells 2022, 11, 3201. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Garcez, M.L.; Zugno, A.I. The Involvement of BDNF, NGF and GDNF in Aging and Alzheimer’s Disease. Aging Dis. 2015, 6, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Ginsberg, S.D.; Mahady, L.; Perez, S.E.; Massa, S.M.; Longo, F.M.; Ikonomovic, M.D. Nerve Growth Factor Pathobiology During the Progression of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 533. [Google Scholar] [CrossRef]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF Signaling Control Amyloidogenic Route and Aβ Production in Hippocampal Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef]
- Zou, L.; Wang, Z.; Shen, L.; Bin Bao, G.; Wang, T.; Kang, J.H.; Pei, G. Receptor Tyrosine Kinases Positively Regulate BACE Activity and Amyloid-Β Production through Enhancing BACE Internalization. Cell Res. 2007, 17, 389–401. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Liu, Y.; Zhao, Y.; Liao, F.-F.; Xu, H. APP Regulates NGF Receptor Trafficking and NGF-Mediated Neuronal Differentiation and Survival. PLoS ONE 2013, 8, e80571. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. Neurotrophic Factors: An Overview. Methods Mol. Biol. 2018, 1727, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Keefe, K.M.; Sheikh, I.S.; Smith, G.M. Targeting Neurotrophins to Specific Populations of Neurons: NGF, BDNF, and NT-3 and Their Relevance for Treatment of Spinal Cord Injury. Int. J. Mol. Sci. 2017, 18, 548. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Schindowski, K.; Belarbi, K.; Buée, L. Neurotrophic Factors in Alzheimer’s Disease: Role of Axonal Transport. Genes Brain Behav. 2008, 7, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Chao, M. V Neurotrophins and Their Receptors: A Convergence Point for Many Signalling Pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Girotra, P.; Behl, T.; Sehgal, A.; Singh, S.; Bungau, S. Investigation of the Molecular Role of Brain-Derived Neurotrophic Factor in Alzheimer’s Disease. J. Mol. Neurosci. 2022, 72, 173–186. [Google Scholar] [CrossRef]
- Narisawa-Saito, M.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Nawa, H. Regional Specificity of Alterations in NGF, BDNF and NT-3 Levels in Alzheimer’s Disease. NeuroReport 1996, 7, 2925–2928. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Herbrüggen, O.; Eckart, S.; Deicke, U.; Kühl, A.; Otten, U.; Danker-Hopfe, H.; Abramowski, D.; Staufenbiel, M.; Hellweg, R. Age-Dependent Time Course of Cerebral Brain-Derived Neurotrophic Factor, Nerve Growth Factor, and Neurotrophin-3 in APP23 Transgenic Mice. J. Neurosci. Res. 2008, 86, 2774–2783. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective Effects of Brain-Derived Neurotrophic Factor in Rodent and Primate Models of Alzheimer’s Disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef]
- Sampaio, T.B.; Savall, A.S.; Gutierrez, M.E.Z.; Pinton, S. Neurotrophic Factors in Alzheimer’s and Parkinson’s Diseases: Implications for Pathogenesis and Therapy. Neural Regen. Res. 2017, 12, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.J.; Wang, J.L.; Jin, W.L. The Emerging Therapeutic Role of NGF in Alzheimer’s Disease. Neurochem. Res. 2016, 41, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Fragkouli, A.; Tzinia, A.K.; Charalampopoulos, I.; Gravanis, A.; Tsilibary, E.C. Matrix Metalloproteinase-9 Participates in NGF-Induced α-Secretase Cleavage of Amyloid-β Protein Precursor in PC12 Cells. J. Alzheimer’s Dis. 2011, 24, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Liu, Y.; Ni, X.; Li, N.; Zhang, B.; Fang, X. Enhancement of the Nonamyloidogenic Pathway by Exogenous NGF in an Alzheimer Transgenic Mouse Model. Neuropeptides 2014, 48, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Xiao, Z.; Huang, J. C6 Glioma-Secreted NGF and FGF2 Regulate Neuronal APP Processing Through Up-Regulation of ADAM10 and Down-Regulation of BACE1, Respectively. J. Mol. Neurosci. 2016, 59, 334–342. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Costantini, C.; Scrable, H.; Puglielli, L. An Aging Pathway Controls the TrkA to P75NTR Receptor Switch and Amyloid β-Peptide Generation. EMBO J. 2006, 25, 1997–2006. [Google Scholar] [CrossRef]
- Roßner, S.; Ueberham, U.; Schliebs, R.; Regino Perez-Polo, J.; Bigl, V. The Regulation of Amyloid Precursor Protein Metabolism by Cholinergic Mechanisms and Neurotrophin Receptor Signaling. Prog. Neurobiol. 1998, 56, 541–569. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Weindruch, R.; Della Valle, G.; Puglielli, L. A TrkA-to-P75NTR Molecular Switch Activates Amyloid β-Peptide Generation during Aging. Biochem. J. 2005, 391, 59–67. [Google Scholar] [CrossRef]
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF Steers Microglia toward a Neuroprotective Phenotype. GLIA 2018, 66, 1395–1416. [Google Scholar] [CrossRef]
- Baydyuk, M.; Xu, B. BDNF Signaling and Survival of Striatal Neurons. Front. Cell. Neurosci. 2014, 8, 254. [Google Scholar] [CrossRef]
- Mitra, S.; Behbahani, H.; Eriksdotter, M. Innovative Therapy for Alzheimer’s Disease-with Focus on Biodelivery of NGF. Front. Neurosci. 2019, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Tuszynski, M.H. Potential Therapeutic Uses of BDNF in Neurological and Psychiatric Disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef]
- Gupta, V.; You, Y.; Gupta, V.; Klistorner, A.; Graham, S. TrkB Receptor Signalling: Implications in Neurodegenerative, Psychiatric and Proliferative Disorders. Int. J. Mol. Sci. 2013, 14, 10122–10142. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor Form of Brain-Derived Neurotrophic Factor and Mature Brain-Derived Neurotrophic Factor Are Decreased in the Pre-Clinical Stages of Alzheimer’s Disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Malek-Ahmadi, M.H.; Alldred, M.J.; Chen, Y.; Chen, K.; Chao, M.V.; Counts, S.E.; Mufson, E.J. Brain-Derived Neurotrophic Factor (BDNF) and TrkB Hippocampal Gene Expression Are Putative Predictors of Neuritic Plaque and Neurofibrillary Tangle Pathology. Neurobiol. Dis. 2019, 132, 104540. [Google Scholar] [CrossRef]
- De Pins, B.; Cifuentes-Díaz, C.; Thamila Farah, A.; López-Molina, L.; Montalban, E.; Sancho-Balsells, A.; López, A.; Ginés, S.; Delgado-García, J.M.; Alberch, J.; et al. Conditional BDNF Delivery from Astrocytes Rescues Memory Deficits, Spine Density, and Synaptic Properties in the 5xFAD Mouse Model of Alzheimer Disease. J. Neurosci. 2019, 39, 2441–2458. [Google Scholar] [CrossRef]
- Mitroshina, E.V.; Yarkov, R.S.; Mishchenko, T.A.; Krut’, V.G.; Gavrish, M.S.; Epifanova, E.A.; Babaev, A.A.; Vedunova, M.V. Brain-Derived Neurotrophic Factor (BDNF) Preserves the Functional Integrity of Neural Networks in the β-Amyloidopathy Model in Vitro. Front. Cell Dev. Biol. 2020, 8, 582. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult Hippocampal Neurogenesis Is Abundant in Neurologically Healthy Subjects and Drops Sharply in Patients with Alzheimer’s Disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Waterhouse, E.G.; An, J.J.; Orefice, L.L.; Baydyuk, M.; Liao, G.Y.; Zheng, K.; Lu, B.; Xu, B. BDNF Promotes Differentiation and Maturation of Adult-Born Neurons through GABArgic Transmission. J. Neurosci. 2012, 32, 14318–14330. [Google Scholar] [CrossRef] [PubMed]
- Harward, S.C.; Hedrick, N.G.; Hall, C.E.; Parra-Bueno, P.; Milner, T.A.; Pan, E.; Laviv, T.; Hempstead, B.L.; Yasuda, R.; McNamara, J.O. Autocrine BDNF-TrkB Signalling within a Single Dendritic Spine. Nature 2016, 538, 99–103. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, Y.; Dai, C.L.; Baazaoui, N.; Tung, Y.C.; Liu, F.; Iqbal, K. Neurotrophic Treatment Initiated during Early Postnatal Development Prevents the Alzheimer-Like Behavior and Synaptic Dysfunction. J. Alzheimer’s Dis. 2021, 82, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Ma, Y. Molecular Mechanisms of Altered Adult Hippocampal Neurogenesis in Alzheimer’s Disease. Mech. Ageing Dev. 2021, 195, 111452. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF Signaling in Context: From Synaptic Regulation to Psychiatric Disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Shi, X.; Wang, H.; Si, C.; Liu, Q.; Du, Y. Neurotrophin-3 Promotes the Neuronal Differentiation of BMSCs and Improves Cognitive Function in a Rat Model of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 629356. [Google Scholar] [CrossRef]
- Ateaque, S.; Merkouris, S.; Wyatt, S.; Allen, N.D.; Xie, J.; DiStefano, P.S.; Lindsay, R.M.; Barde, Y.A. Selective Activation and Down-Regulation of Trk Receptors by Neurotrophins in Human Neurons Co-Expressing TrkB and TrkC. J. Neurochem. 2022, 161, 463–477. [Google Scholar] [CrossRef]
- Lesné, S.; Gabrieli, C.; Nelson, D.A.; White, E.; MacKenzie, E.T.; Vivien, D.; Buisson, A. Akt-Dependent Expression of NAIP-1 Protects Neurons against Amyloid-β Toxicity. J. Biol. Chem. 2005, 280, 24941–24947. [Google Scholar] [CrossRef]
- Santos, C.Y.; Snyder, P.J.; Wu, W.C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic Relationship between Alzheimer’s Disease, Cerebrovascular Disease, and Cardiovascular Risk: A Review and Synthesis. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 7, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Bracko, O.; Cruz Hernández, J.C.; Park, L.; Nishimura, N.; Schaffer, C.B. Causes and Consequences of Baseline Cerebral Blood Flow Reductions in Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2021, 41, 1501–1516. [Google Scholar] [CrossRef]
- Nation, D.A.; Wierenga, C.E.; Clark, L.R.; Dev, S.I.; Stricker, N.H.; Jak, A.J.; Salmon, D.P.; Delano-Wood, L.; Bangen, K.J.; Rissman, R.A.; et al. Cortical and Subcortical Cerebrovascular Resistance Index in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 36, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Blais, M.; Lévesque, P.; Bellenfant, S.; Berthod, F. Nerve Growth Factor, Brain-Derived Neurotrophic Factor, Neurotrophin-3 and Glial-Derived Neurotrophic Factor Enhance Angiogenesis in a Tissue-Engineered in Vitro Model. Tissue Eng. Part A 2013, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Mangieri, D.; Benagiano, V.; Crivellato, E.; Ribatti, D. Nerve Growth Factor as an Angiogenic Factor. Microvasc. Res. 2008, 75, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Moser, K.V.; Reindl, M.; Blasig, I.; Humpel, C. Brain Capillary Endothelial Cells Proliferate in Response to NGF, Express NGF Receptors and Secrete NGF after Inflammation. Brain Res. 2004, 1017, 53–60. [Google Scholar] [CrossRef]
- Julio-Pieper, M.; Lozada, P.; Tapia, V.; Vega, M.; Miranda, C.; Vantman, D.; Ojeda, S.R.; Romero, C. Nerve Growth Factor Induces Vascular Endothelial Growth Factor Expression in Granulosa Cells via a TrkA Receptor/Mitogen-Activated Protein Kinase-Extracellularly Regulated Kinase 2-Dependent Pathway. J. Clin. Endocrinol. Metab. 2009, 94, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Martin, K.C.; Jackson, J.K.; Beppu, K.; Woo, C.W.; Thiele, C.J. Brain-Derived Neurotrophic Factor Activation of TrkB Induces Vascular Endothelial Growth Factor Expression via Hypoxia-Inducible Factor-1α in Neuroblastoma Cells. Cancer Res. 2006, 66, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Y.; Zhou, Z.; Shi, H.; Qiu, X.; Xiong, J.; Chen, Y. BDNF Regulates the Expression and Secretion of VEGF from Osteoblasts via the TrkB/ERK1/2 Signaling Pathway during Fracture Healing. Mol. Med. Rep. 2017, 15, 1362–1367. [Google Scholar] [CrossRef]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. VEGFs and Receptors Involved in Angiogenesis versus Lymphangiogenesis. Curr. Opin. Cell Biol. 2009, 21, 154–165. [Google Scholar] [CrossRef]
- Lange, C.; Storkebaum, E.; De Almodóvar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular Endothelial Growth Factor: A Neurovascular Target in Neurological Diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The Biology of VEGF and Its Receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Dudley, A.C.; Griffioen, A.W. Pathological Angiogenesis: Mechanisms and Therapeutic Strategies. Angiogenesis 2023, 26, 313–347. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Martin, L.; Bouvet, P.; Chounlamountri, N.; Watrin, C.; Besançon, R.; Pinatel, D.; Meyronet, D.; Honnorat, J.; Buisson, A.; Salin, P.A.; et al. VEGF Counteracts Amyloid-β-Induced Synaptic Dysfunction. Cell Rep. 2021, 35, 109121. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-J.; Park, M.H.; Han, C.; Yoon, K.; Koh, Y.H. VEGFR2 Alteration in Alzheimer’s Disease. Sci. Rep. 2017, 7, 17713. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Greenberg, S.M. β-Amyloid, Blood Vessels, and Brain Function. Stroke 2009, 40, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s Disease: A Matter of Blood-Brain Barrier Dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Paris, D.; Townsend, K.; Quadros, A.; Humphrey, J.; Sun, J.; Brem, S.; Wotoczek-Obadia, M.; DelleDonne, A.; Patel, N.; Obregon, D.F.; et al. Inhibition of Angiogenesis by Aβ Peptides. Angiogenesis 2004, 7, 75–85. [Google Scholar] [CrossRef]
- Religa, P.; Cao, R.; Religa, D.; Xue, Y.; Bogdanovic, N.; Westaway, D.; Marti, H.H.; Winblad, B.; Cao, Y. VEGF Significantly Restores Impaired Memory Behavior in Alzheimer’s Mice by Improvement of Vascular Survival. Sci. Rep. 2013, 3, 2053. [Google Scholar] [CrossRef]
- Patel, N.S.; Mathura, V.S.; Bachmeier, C.; Beaulieu-Abdelahad, D.; Laporte, V.; Weeks, O.; Mullan, M.; Paris, D. Alzheimer’s β-Amyloid Peptide Blocks Vascular Endothelial Growth Factor Mediated Signaling via Direct Interaction with VEGFR-2. J. Neurochem. 2010, 112, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.P.; Bae, D.G.; Kang, H.J.; Gwag, B.J.; Gho, Y.S.; Chae, C.B. Co-Accumulation of Vascular Endothelial Growth Factor with β-Amyloid in the Brain of Patients with Alzheimer’s Disease. Neurobiol. Aging 2004, 25, 283–290. [Google Scholar] [CrossRef]
- Patel, N.S.; Quadros, A.; Brem, S.; Wotoczek-Obadia, M.; Mathura, V.S.; Laporte, V.; Mullan, M.; Paris, D. Potent Anti-Angiogenic Motifs within the Alzheimer β-Amyloid Peptide. Amyloid 2008, 15, 5–19. [Google Scholar] [CrossRef]
- Wei, M.; Wu, T.; Chen, N. Bridging Neurotrophic Factors and Bioactive Peptides to Alzheimer’s Disease. Ageing Res. Rev. 2024, 94, 102177. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; U, H.S.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; et al. A Phase 1 Clinical Trial of Nerve Growth Factor Gene Therapy for Alzheimer Disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M.H.; Yang, J.H.; Barba, D.; Hoi-Sang, U.; Bakay, R.A.E.; Pay, M.M.; Masliah, E.; Conner, J.M.; Kobalka, P.; Roy, S.; et al. Nerve Growth Factor Gene Therapy Activation of Neuronal Responses in Alzheimer Disease. JAMA Neurol. 2015, 72, 1139–1147. [Google Scholar] [CrossRef]
- Mitra, S.; Gera, R.; Linderoth, B.; Lind, G.; Wahlberg, L.; Almqvist, P.; Behbahani, H.; Eriksdotter, M. A Review of Techniques for Biodelivery of Nerve Growth Factor (NGF) to the Brain in Relation to Alzheimer’s Disease. Adv. Exp. Med. Biol. 2021, 1331, 167–191. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, L. Enhancement of Neural Regeneration as a Therapeutic Strategy for Alzheimer’s Disease (Review). Exp. Ther. Med. 2023, 26, 444. [Google Scholar] [CrossRef] [PubMed]
- Bahlakeh, G.; Rahbarghazi, R.; Mohammadnejad, D.; Abedelahi, A.; Karimipour, M. Current Knowledge and Challenges Associated with Targeted Delivery of Neurotrophic Factors into the Central Nervous System: Focus on Available Approaches. Cell Biosci. 2021, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Houlton, J.; Abumaria, N.; Hinkley, S.F.R.; Clarkson, A.N. Therapeutic Potential of Neurotrophins for Repair after Brain Injury: A Helping Hand from Biomaterials. Front. Genet. 2019, 10, 790. [Google Scholar] [CrossRef]
- Ceci, C.; Lacal, P.M.; Barbaccia, M.L.; Mercuri, N.B.; Graziani, G.; Ledonne, A. The VEGFs/VEGFRs System in Alzheimer’s and Parkinson’s Diseases: Pathophysiological Roles and Therapeutic Implications. Pharmacol. Res. 2024, 201, 107101. [Google Scholar] [CrossRef]
- Teleanu, R.I.; Preda, M.D.; Niculescu, A.G.; Vladâcenco, O.; Radu, C.I.; Grumezescu, A.M.; Teleanu, D.M. Current Strategies to Enhance Delivery of Drugs across the Blood–Brain Barrier. Pharmaceutics 2022, 14, 987. [Google Scholar] [CrossRef]
- Longo, F.M.; Massa, S.M. Small-Molecule Modulation of Neurotrophin Receptors: A Strategy for the Treatment of Neurological Disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef]
- Gascon, S.; Jann, J.; Langlois-Blais, C.; Plourde, M.; Lavoie, C.; Faucheux, N. Peptides Derived from Growth Factors to Treat Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6071. [Google Scholar] [CrossRef] [PubMed]
- Gudasheva, T.A.; Povarnina, P.Y.; Tarasiuk, A.V.; Seredenin, S.B. Low-Molecular Mimetics of Nerve Growth Factor and Brain-Derived Neurotrophic Factor: Design and Pharmacological Properties. Med. Res. Rev. 2021, 41, 2746–2774. [Google Scholar] [CrossRef]
- Shih, A.; Laramee, G.R.; Schmelzer, C.H.; Burton, L.E.; Winslow, J.W. Mutagenesis Identifies Amino-Terminal Residues of Nerve Growth Factor Necessary for Trk Receptor Binding and Biological Activity. J. Biol. Chem. 1994, 269, 27679–27686. [Google Scholar] [CrossRef] [PubMed]
- Berrera, M.; Cattaneo, A.; Carloni, P. Molecular Simulation of the Binding of Nerve Growth Factor Peptide Mimics to the Receptor Tyrosine Kinase A. Biophys. J. 2006, 91, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Travaglia, A.; Pietropaolo, A.; Di Martino, R.; Nicoletti, V.G.; La Mendola, D.; Calissano, P.; Rizzarelli, E. A Small Linear Peptide Encompassing the NGF N-Terminus Partly Mimics the Biological Activities of the Entire Neurotrophin in PC12 Cells. ACS Chem. Neurosci. 2015, 6, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Satriano, C.; Pietropaolo, A.; Gianì, F.; Travaglia, A.; La Mendola, D.; Nicoletti, V.G.; Rizzarelli, E. The Inorganic Side of NGF: Copper(II) and Zinc(II) Affect the NGF Mimicking Signaling of the N-Terminus Peptides Encompassing the Recognition Domain of TrkA Receptor. Front. Neurosci. 2016, 10, 569. [Google Scholar] [CrossRef]
- Travaglia, A.; La Mendola, D.; Magrì, A.; Nicoletti, V.G.; Pietropaolo, A.; Rizzarelli, E. Copper, BDNF and Its N-Terminal Domain: Inorganic Features and Biological Perspectives. Chem. A Eur. J. 2012, 18, 15618–15631. [Google Scholar] [CrossRef]
- Naletova, I.; Grasso, G.I.; Satriano, C.; Travaglia, A.; La Mendola, D.; Arena, G.; Rizzarelli, E. Copper Complexes of Synthetic Peptides Mimicking Neurotrophin-3 Enhance Neurite Outgrowth and CREB Phosphorylation. Metallomics 2019, 11, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- Naletova, I.; Satriano, C.; Pietropaolo, A.; Gianì, F.; Pandini, G.; Triaca, V.; Amadoro, G.; Latina, V.; Calissano, P.; Travaglia, A.; et al. The Copper(II)-Assisted Connection between NGF and BDNF by Means of Nerve Growth Factor-Mimicking Short Peptides. Cells 2019, 8, 301. [Google Scholar] [CrossRef]
- Russo, L.; Giacomelli, C.; Fortino, M.; Marzo, T.; Ferri, G.; Calvello, M.; Viegi, A.; Magrì, A.; Pratesi, A.; Pietropaolo, A.; et al. Neurotrophic Activity and Its Modulation by Zinc Ion of a Dimeric Peptide Mimicking the Brain-Derived Neurotrophic Factor N-Terminal Region. ACS Chem. Neurosci. 2022, 13, 3453–3463. [Google Scholar] [CrossRef]
- La Mendola, D.; Arena, G.; Pietropaolo, A.; Satriano, C.; Rizzarelli, E. Metal Ion Coordination in Peptide Fragments of Neurotrophins: A Crucial Step for Understanding the Role and Signaling of These Proteins in the Brain. Coord. Chem. Rev. 2021, 435, 213790. [Google Scholar] [CrossRef]
- Tomasello, B.; Bellia, F.; Naletova, I.; Magrì, A.; Tabbì, G.; Attanasio, F.; Tomasello, M.F.; Cairns, W.R.L.; Fortino, M.; Pietropaolo, A.; et al. BDNF- and VEGF-Responsive Stimulus to an NGF Mimic Cyclic Peptide with Copper Ionophore Capability and Ctr1/CCS-Driven Signaling. ACS Chem. Neurosci. 2024, 15, 1755–1769. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Pietropaolo, A.; Tabbì, G.; La Mendola, D.; Rizzarelli, E. From Peptide Fragments to Whole Protein: Copper(II) Load and Coordination Features of IAPP. Chem. A Eur. J. 2017, 23, 17898–17902. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad Simulation and Speciation (HySS): A Utility Program for the Investigation of Equilibria Involving Soluble and Partially Soluble Species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Ranjbar, B.; Gill, P. Circular Dichroism Techniques: Biomolecular and Nanostructural Analyses—A Review. Chem. Biol. Drug Des. 2009, 74, 101–120. [Google Scholar] [CrossRef]
- Lopes, J.L.S.; Miles, A.J.; Whitmore, L.; Wallace, B.A. Distinct Circular Dichroism Spectroscopic Signatures of Polyproline II and Unordered Secondary Structures: Applications in Secondary Structure Analyses. Protein Sci. 2014, 23, 1765–1772. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Tóth, G.; Lovas, S.; Hirst, J.D. Influence of Tyrosine on the Electronic Circular Dichroism of Helical Peptides. J. Phys. Chem. B 2003, 107, 8682–8688. [Google Scholar] [CrossRef]
- Várnagy, K.; Szabó, J.; Sóvágó, I.; Malandrinos, G.; Hadjiliadis, N.; Sanna, D.; Micera, G. Equilibrium and Structural Studies on Copper(II) Complexes of Tetra-, Penta- And Hexa-Peptides Containing Histidyl Residues at the C-Termini. J. Chem. Soc. Dalton Trans. 2000, 4, 467–472. [Google Scholar] [CrossRef]
- Kowalik-Jankowska, T.; Ruta-Dolejsz, M.; Wiśniewska, K.; Lankiewicz, L. Cu(II) Interaction with N-Terminal Fragments of Human and Mouse β-Amyloid Peptide. J. Inorg. Biochem. 2001, 86, 535–545. [Google Scholar] [CrossRef]
- Kowalik-Jankowska, T.; Ruta-Dolejsz, M.; Wiśniewska, K.; Łankiewicz, L. Coordination of Copper(II) Ions by the 11-20 and 11-28 Fragments of Human and Mouse β-Amyloid Peptide. J. Inorg. Biochem. 2002, 92, 1–10. [Google Scholar] [CrossRef] [PubMed]
- La Mendola, D.; Magrì, A.; Vagliasindi, L.I.; Hansson, Ö.; Bonomo, R.P.; Rizzarelli, E. Copper(II) Complex Formation with a Linear Peptide Encompassing the Putative Cell Binding Site of Angiogenin. Dalton Trans. 2010, 39, 10678. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Tabbì, G.; Giuffrida, A.; Pappalardo, G.; Satriano, C.; Naletova, I.; Nicoletti, V.G.; Attanasio, F. Influence of the N-Terminus Acetylation of Semax, a Synthetic Analog of ACTH(4-10), on Copper(II) and Zinc(II) Coordination and Biological Properties. J. Inorg. Biochem. 2016, 164, 59–69. [Google Scholar] [CrossRef]
- Kowalik-Jankowska, T.; Ruta, M.; Wiśniewska, K.; Łankiewicz, L. Coordination Abilities of the 1-16 and 1-28 Fragments of β-Amyloid Peptide towards Copper(II) Ions: A Combined Potentiometric and Spectroscopic Study. J. Inorg. Biochem. 2003, 95, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; La Mendola, D.; Rizzarelli, E. Nerve Growth Factor Peptides Bind Copper(Ii) with High Affinity: A Thermodynamic Approach to Unveil Overlooked Neurotrophin Roles. Int. J. Mol. Sci. 2021, 22, 5085. [Google Scholar] [CrossRef] [PubMed]
- Vanýsek, P.; Hovancová, J.; Oriňaková, R. Electrical Impedance Spectroscopy and Its Application in the Study of Electrochemical Sensors. ECS Meet. Abstr. 2019, MA2019-01, 1933. [Google Scholar] [CrossRef]
- Brasili, D.; Watly, J.; Simonovsky, E.; Guerrini, R.; Barbosa, N.A.; Wieczorek, R.; Remelli, M.; Kozlowski, H.; Miller, Y. The Unusual Metal Ion Binding Ability of Histidyl Tags and Their Mutated Derivatives. Dalton Trans. 2016, 45, 5629–5639. [Google Scholar] [CrossRef]
- La Mendola, D.; Magrì, A.; Santoro, A.M.; Nicoletti, V.G.; Rizzarelli, E. Copper(II) Interaction with Peptide Fragments of Histidine-Proline-Rich Glycoprotein: Speciation, Stability and Binding Details. J. Inorg. Biochem. 2012, 111, 59–69. [Google Scholar] [CrossRef]
- Atrián-Blasco, E.; Del Barrio, M.; Faller, P.; Hureau, C. Ascorbate Oxidation by Cu(Amyloid-β) Complexes: Determination of the Intrinsic Rate as a Function of Alterations in the Peptide Sequence Revealing Key Residues for Reactive Oxygen Species Production. Anal. Chem. 2018, 90, 5909–5915. [Google Scholar] [CrossRef]
- Arena, G.; Pappalardo, G.; Sovago, I.; Rizzarelli, E. Copper(II) Interaction with Amyloid-β: Affinity and Speciation. Coord. Chem. Rev. 2012, 256, 3–12. [Google Scholar] [CrossRef]
- Tõugu, V.; Karafin, A.; Palumaa, P. Binding of Zinc(II) and Copper(II) to the Full-Length Alzheimer’s Amyloid-β Peptide. J. Neurochem. 2008, 104, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, H.; Łuczkowski, M.; Remelli, M. Prion Proteins and Copper Ions. Biological and Chemical Controversies. Dalton Trans. 2010, 39, 6371–6385. [Google Scholar] [CrossRef] [PubMed]
- Falcone, E.; Okafor, M.; Vitale, N.; Raibaut, L.; Sour, A.; Faller, P. Extracellular Cu2+ Pools and Their Detection: From Current Knowledge to next-Generation Probes. Coord. Chem. Rev. 2021, 433, 213727. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Trace Metal Contamination Initiates the Apparent Auto-Aggregation, Amyloidosis, and Oligomerization of Alzheimer’s Aβ Peptides. J. Biol. Inorg. Chem. 2004, 9, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Milardi, D.; Rizzarelli, E. Neurodegeneration; Milardi, D., Rizzarelli, E., Eds.; Drug Discovery; Royal Society of Chemistry: Cambridge, UK, 2011; ISBN 978-1-84973-050-1. [Google Scholar]
- Grasso, G.; Giuffrida, M.L.; Rizzarelli, E. Metallostasis and Amyloid β-Degrading Enzymes. Metallomics 2012, 4, 937. [Google Scholar] [CrossRef] [PubMed]
- Keenan, J.; Horgan, K.; Clynes, M.; Sinkunaite, I.; Ward, P.; Murphy, R.; O’Sullivan, F. Unexpected Fluctuations of Trace Element Levels in Cell Culture Medium in Vitro: Caveat Emptor. Vitr. Cell. Dev. Biol. Anim. 2018, 54, 555–558. [Google Scholar] [CrossRef]
- Kermani, P.; Rafii, D.; Jin, D.K.; Whitlock, P.; Schaffer, W.; Chiang, A.; Vincent, L.; Friedrich, M.; Shido, K.; Hackett, N.R.; et al. Neurotrophins Promote Revascularization by Local Recruitment of TrkB+ Endothelial Cells and Systemic Mobilization of Hematopoietic Progenitors. J. Clin. Investig. 2005, 115, 653–663. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-Regulated Signalling Pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef]
- Atkinson, E.; Dickman, R. Growth Factors and Their Peptide Mimetics for Treatment of Traumatic Brain Injury. Bioorganic Med. Chem. 2023, 90, 117368. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Li, B.; Yamamori, H.; Tatebayashi, Y.; Shafit-Zagardo, B.; Tanimukai, H.; Chen, S.; Iqbal, K.; Grundke-Iqbal, I. Failure of Neuronal Maturation in Alzheimer Disease Dentate Gyrus. J. Neuropathol. Exp. Neurol. 2008, 67, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Baazaoui, N.; Flory, M.; Iqbal, K. Synaptic Compensation as a Probable Cause of Prolonged Mild Cognitive Impairment in Alzheimer’s Disease: Implications from a Transgenic Mouse Model of the Disease. J. Alzheimer’s Dis. 2017, 56, 1385–1401. [Google Scholar] [CrossRef] [PubMed]
- Kelleher-Andersson, J. Neurogenesis as a Potential Therapeutic Strategy for Neurodegenerative Disorders. J. Alzheimer’s Dis. 2004, 6, S19–S25. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Kazim, S.; Bolognin, S.; Blanchard, J. Shifting Balance from Neurodegeneration to Regeneration of the Brain: A Novel Therapeutic Approach to Alzheimer′s Disease and Related Neurodegenerative Conditions. Neural Regen. Res. 2014, 9, 1518. [Google Scholar] [CrossRef] [PubMed]
- Baazaoui, N.; Iqbal, K. Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules 2022, 12, 1409. [Google Scholar] [CrossRef]
- Saragovi, H.U.; Gehring, K. Development of Pharmacological Agents for Targeting Neurotrophins and Their Receptors. Trends Pharmacol. Sci. 2000, 21, 93–98. [Google Scholar] [CrossRef]
- Soppet, D.; Escandon, E.; Maragos, J.; Middlemas, D.S.; Raid, S.W.; Blair, J.; Burton, L.E.; Stanton, B.R.; Kaplan, D.R.; Hunter, T.; et al. The Neurotrophic Factors Brain-Derived Neurotrophic Factor and Neurotrophin-3 Are Ligands for the TrkB Tyrosine Kinase Receptor. Cell 1991, 65, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Squinto, S.P.; Stitt, T.N.; Aldrich, T.H.; Davis, S.; Blanco, S.M.; RadzieJewski, C.; Glass, D.J.; Masiakowski, P.; Furth, M.E.; Valenzuela, D.M.; et al. TrkB Encodes a Functional Receptor for Brain-Derived Neurotrophic Factor and Neurotrophin-3 but Not Nerve Growth Factor. Cell 1991, 65, 885–893. [Google Scholar] [CrossRef]
- Ip, N.Y.; Stitt, T.N.; Tapley, P.; Klein, R.; Glass, D.J.; Fandl, J.; Greene, L.A.; Barbacid, M.; Yancopoulos, G.D. Similarities and Differences in the Way Neurotrophins Interact with the Trk Receptors in Neuronal and Nonneuronal Cells. Neuron 1993, 10, 137–149. [Google Scholar] [CrossRef]
- Lindsay, R.M.; Wiegand, S.J.; Anthony Altar, C.; DiStefano, P.S. Neurotrophic Factors: From Molecule to Man. Trends Neurosci. 1994, 17, 182–190. [Google Scholar] [CrossRef]
- Schnell, L.; Schneider, R.; Kolbeck, R.; Barde, Y.A.; Schwab, M.E. Neurotrophin-3 Enhances Sprouting of Corticospinal Tract during Development and after Adult Spinal Cord Lesion. Nature 1994, 367, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Carnahan, J.; Greenberg, M.E. Requirement for BDNF in Activity-Dependent Survival of Cortical Neurons. Science 1994, 263, 1618–1623. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Hwang, P.K.; Mobley, W.C.; Fletterick, R.J. Crystal Structure of Neurotrophin-3 Homodimer Shows Distinct Regions Are Used to Bind Its Receptors. Biochemistry 1998, 37, 16846–16852. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Ghosh, A.P.; Ho, M.N.; Li, C.; Huang, X.; Pentelute, B.L.; Baleja, J.D.; Lin, Y.-S. Assessing the Performance of Peptide Force Fields for Modeling the Solution Structural Ensembles of Cyclic Peptides. J. Phys. Chem. B 2024, 128, 5281–5292. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Min, J.; Wang, F. Copper Homeostasis and Cuproptosis in Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Q.; Lu, L.; Su, Y.; Shi, W.; Zhang, H.; Liu, R.; Pu, Y.; Yin, L. Copper Induces Cognitive Impairment in Mice via Modulation of Cuproptosis and CREB Signaling. Nutrients 2023, 15, 972. [Google Scholar] [CrossRef]
- Scholefield, M.; Church, S.J.; Xu, J.; Cooper, G.J.S. Metallomic Analysis of Brain Tissues Distinguishes between Cases of Dementia with Lewy Bodies, Alzheimer’s Disease, and Parkinson’s Disease Dementia. Front. Neurosci. 2024, 18, 1412356. [Google Scholar] [CrossRef]
- Roy, S.; Lutsenko, S. Mechanism of Cu Entry into the Brain: Many Unanswered Questions. Neural Regen. Res. 2024, 19, 2421–2429. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, H.; Zhu, K.; Chang, C.; Lv, W.; Li, R.; Li, X.; Ye, T.; Cao, D. SLC31A1 Identifying a Novel Biomarker with Potential Prognostic and Immunotherapeutic Potential in Pan-Cancer. Biomedicines 2023, 11, 2884. [Google Scholar] [CrossRef]
- Rosenzweig, A. Structure and Chemistry of the Copper Chaperone Proteins. Curr. Opin. Chem. Biol. 2000, 4, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S. Dynamic and Cell-Specific Transport Networks for Intracellular Copper Ions. J. Cell Sci. 2021, 134, jcs240523. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C. Ceruloplasmin and Other Copper Binding Components of Blood Plasma and Their Functions: An Update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Da Costa Ferreira, A.M.; Ciriolo, M.R.; Marcocci, L.; Rotilio, G. Copper(I) Transfer into Metallothionein Mediated by Glutathione. Biochem. J. 1993, 292, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Tavera-Montañez, C.; Hainer, S.J.; Cangussu, D.; Gordon, S.J.V.; Xiao, Y.; Reyes-Gutierrez, P.; Imbalzano, A.N.; Navea, J.G.; Fazzio, T.G.; Padilla-Benavides, T. The Classic Metal-Sensing Transcription Factor MTF1 Promotes Myogenesis in Response to Copper. FASEB J. 2019, 33, 14556–14574. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Klomp, L.W.J. ATOX1: A Novel Copper-Responsive Transcription Factor in Mammals? Int. J. Biochem. Cell Biol. 2009, 41, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J.; Barnham, K.J. Therapeutic Redistribution of Metal Ions to Treat Alzheimer’s Disease. Acc. Chem. Res. 2012, 45, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Du, T.; Laughton, K.M.; Volitakis, I.; Sharples, R.A.; Xilinas, M.E.; Hoke, D.E.; Holsinger, R.M.D.; Evin, G.; Cherny, R.A.; et al. Degradation of the Alzheimer Disease Amyloid β-Peptide by Metal-Dependent up-Regulation of Metalloprotease Activity. J. Biol. Chem. 2006, 281, 17670–17680. [Google Scholar] [CrossRef]
- Caragounis, A.; Du, T.; Filiz, G.; Laughton, K.M.; Volitakis, I.; Sharples, R.A.; Cherny, R.A.; Masters, C.L.; Drew, S.C.; Hill, A.F.; et al. Differential Modulation of Alzheimer’s Disease Amyloid β-Peptide Accumulation by Diverse Classes of Metal Ligands. Biochem. J. 2007, 407, 435–450. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Caragounis, A.; Du, T.; Laughton, K.M.; Volitakis, I.; Cherny, R.A.; Sharples, R.A.; Hill, A.F.; Li, Q.X.; Masters, C.L.; et al. Selective Intracellular Release of Copper and Zinc Ions from Bis(Thiosemicarbazonato) Complexes Reduces Levels of Alzheimer Disease Amyloid-β Peptide. J. Biol. Chem. 2008, 283, 4568–4577. [Google Scholar] [CrossRef]
- Bica, L.; Liddell, J.R.; Donnelly, P.S.; Duncan, C.; Caragounis, A.; Volitakis, I.; Paterson, B.M.; Cappai, R.; Grubman, A.; Camakaris, J.; et al. Neuroprotective Copper Bis(Thiosemicarbazonato) Complexes Promote Neurite Elongation. PLoS ONE 2014, 9, e90070. [Google Scholar] [CrossRef]
- Niciu, M.J.; Ma, X.M.; El Meskini, R.; Ronnett, G.V.; Mains, R.E.; Eipper, B.A. Developmental Changes in the Expression of ATP7A during a Critical Period in Postnatal Neurodevelopment. Neuroscience 2006, 139, 947–964. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, C.; Méndez, G.M.; Bhattacharjee, S.; Vrailas-Mortimer, A.D.; Zlatic, S.A.; Freeman, A.A.H.; Gokhale, A.; Concilli, M.; Werner, E.; Savas, C.S.; et al. Golgi-Dependent Copper Homeostasis Sustains Synaptic Development and Mitochondrial Content. J. Neurosci. 2021, 41, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Dame, C.; Horn, D.; Schomburg, L.; Grünhagen, J.; Chillon, T.S.; Tietze, A.; Vogt, A.; Bührer, C. Fatal Congenital Copper Transport Defect Caused by a Homozygous Likely Pathogenic Variant of SLC31A1. Clin. Genet. 2023, 103, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.F.; Xapelli, S. Intervention of Brain-Derived Neurotrophic Factor and Other Neurotrophins in Adult Neurogenesis. Adv. Exp. Med. Biol. 2021, 1331, 95–115. [Google Scholar] [CrossRef]
- Hollands, C.; Tobin, M.K.; Hsu, M.; Musaraca, K.; Yu, T.S.; Mishra, R.; Kernie, S.G.; Lazarov, O. Depletion of Adult Neurogenesis Exacerbates Cognitive Deficits in Alzheimer’s Disease by Compromising Hippocampal Inhibition. Mol. Neurodegener. 2017, 12, 64. [Google Scholar] [CrossRef]
- Richetin, K.; Leclerc, C.; Toni, N.; Gallopin, T.; Pech, S.; Roybon, L.; Rampon, C. Genetic Manipulation of Adult-Born Hippocampal Neurons Rescues Memory in a Mouse Model of Alzheimer’s Disease. Brain 2015, 138, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Caillé, I.; Allinquant, B.; Dupont, E.; Bouillot, C.; Langer, A.; Müller, U.; Prochiantz, A. Soluble Form of Amyloid Precursor Protein Regulates Proliferation of Progenitors in the Adult Subventricular Zone. Development 2004, 131, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Young, J.K. Neurogenesis Makes a Crucial Contribution to the Neuropathology of Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2020, 4, 365–371. [Google Scholar] [CrossRef]
- Choi, S.H.; Tanzi, R.E. Is Alzheimer’s Disease a Neurogenesis Disorder? Cell Stem Cell 2019, 25, 7–8. [Google Scholar] [CrossRef]
- Mu, Y.; Gage, F.H. Adult Hippocampal Neurogenesis and Its Role in Alzheimer’s Disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef]
- Hatori, Y.; Yan, Y.; Schmidt, K.; Furukawa, E.; Hasan, N.M.; Yang, N.; Liu, C.N.; Sockanathan, S.; Lutsenko, S. Neuronal Differentiation Is Associated with a Redox-Regulated Increase of Copper Flow to the Secretory Pathway. Nat. Commun. 2016, 7, 10640. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; O’Neal, S.; Hong, L.; Jiang, W.; Zheng, W. Elevated Adult Neurogenesis in Brain Subventricular Zone Following in Vivo Manganese Exposure: Roles of Copper and DMT1. Toxicol. Sci. 2015, 143, 482–498. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; van Rijn, R.M.; Zheng, W. Copper Modulates Adult Neurogenesis in Brain Subventricular Zone. Int. J. Mol. Sci. 2022, 23, 9888. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R.; Till, J.H. Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, T. Crystal Structures of Neurotrophin Receptors Kinase Domain. Vitam. Horm. 2017, 104, 1–18. [Google Scholar] [CrossRef] [PubMed]
- McTigue, M.A.; Wickersham, J.A.; Pinko, C.; Showalter, R.E.; Parast V, C.; Tempczyk-Russell, A.; Gehring, M.R.; Mroczkowski, B.; Chen-Chen, K.; Villafranca, J.E.; et al. Crystal Structure of the Kinase Domain of Human Vascular Endothelial Growth Factor Receptor 2: A Key Enzyme in Angiogenesis. Structure 1999, 7, 319–330. [Google Scholar] [CrossRef]
- Martin, C.R.; Preedy, V.R.; Rajendram, R. Front Matter. In Factors Affecting Neurodevelopment; Elsevier: Amsterdam, The Netherlands, 2022; p. iii. ISBN 9780128179864. [Google Scholar]
- Numakawa, T.; Odaka, H. Brain-Derived Neurotrophic Factor Signaling in the Pathophysiology of Alzheimer’s Disease: Beneficial Effects of Flavonoids for Neuroprotection. Int. J. Mol. Sci. 2021, 22, 5719. [Google Scholar] [CrossRef]
- He, F.; Chang, C.; Liu, B.; Li, Z.; Li, H.; Cai, N.; Wang, H.-H. Copper (II) Ions Activate Ligand-Independent Receptor Tyrosine Kinase (RTK) Signaling Pathway. BioMed Res. Int. 2019, 2019, 4158415. [Google Scholar] [CrossRef]
- Woo, S.B.; Neet, K.E. Characterization of Histidine Residues Essential for Receptor Binding and Activity of Nerve Growth Factor. J. Biol. Chem. 1996, 271, 24433–24441. [Google Scholar] [CrossRef]
- Bartolotti, N.; Lazarov, O. CREB Signals as PBMC-Based Biomarkers of Cognitive Dysfunction: A Novel Perspective of the Brain-Immune Axis. Brain Behav. Immun. 2019, 78, 9–20. [Google Scholar] [CrossRef]
- Lisman, J.; Cooper, K.; Sehgal, M.; Silva, A.J. Memory Formation Depends on Both Synapse-Specific Modifications of Synaptic Strength and Cell-Specific Increases in Excitability. Nat. Neurosci. 2018, 21, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Simmons, M.S.; Perry, R.T.; Wiener, H.W.; Harrell, L.E.; Go, R.C.P. Genetic Association of Neurotrophic Tyrosine Kinase Receptor Type 2 (NTRK2) with Alzheimer’s Disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.; Miners, J.S.; Allen, S.; Love, S. VEGFR1 and VEGFR2 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 61, 741–752. [Google Scholar] [CrossRef] [PubMed]
- El-Hashim, A.Z.; Jaffal, S.M.; Al-Rashidi, F.T.; Luqmani, Y.A.; Akhtar, S. Nerve Growth Factor Enhances Cough via a Central Mechanism of Action. Pharmacol. Res. 2013, 74, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.-T.; Groves, M.J.; Scaravilli, F.; Sugden, D.; An, S.F. Neurotrophin-3 Administration Alters Neurotrophin, Neurotrophin Receptor and Nestin MRNA Expression in Rat Dorsal Root Ganglia Following Axotomy. Neuroscience 2007, 147, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.; Akhavan, M.M.; Kamyab, K.; Ebrahimi, S.A. Maternal Voluntary Exercise Ameliorates Learning Deficit in Rat Pups Exposed, in Utero, to Valproic Acid; Role of BDNF and VEGF and Their Receptors. Neuropeptides 2018, 71, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Solvsten, C.A.E.; Daugaard, T.F.; Luo, Y.; de Paoli, F.; Christensen, J.H.; Nielsen, A.L. The Effects of Voluntary Physical Exercise-Activated Neurotrophic Signaling in Rat Hippocampus on MRNA Levels of Downstream Signaling Molecules. J. Mol. Neurosci. 2017, 62, 142–153. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, C.; Jiang, H.; Li, Y.; Zhang, L.; Robin, A.; Katakowski, M.; Lu, M.; Chopp, M. Atorvastatin Induction of VEGF and BDNF Promotes Brain Plasticity after Stroke in Mice. J. Cereb. Blood Flow Metab. 2005, 25, 281–290. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Company and Code | Dilution and Application |
|---|---|---|
| phospho-Trk | SantaCruz (Santa Cruz, CA, USA), sc-8058 | 1:500 for western blotting assay |
| phospho-CREB | SantaCruz (Santa Cruz, CA, USA), sc-8058 | 1:500 for western blotting assay |
| VEGFR1 | SantaCruz (Santa Cruz, CA, USA), sc-271789 | 1:500 for western blotting assay |
| VEGFR2 | SantaCruz (Santa Cruz, CA, USA), sc-6251 | 1:500 for western blotting assay |
| TrkA | Cell Signaling (Danvers, MA, USA), #2505 | 1:800 for western blotting assay |
| TrkB | Abcam Waltham, MA, USA), ab33655 | 1:1000 for western blotting assay |
| anti-Ctr1 | Abcam Waltham, MA, USA), ab129067 | 1:3000 for western blotting assay 2.5 µg/mL for immunofluorescence assay |
| anti-VEGF | SantaCruz, sc-72693 | 1:500 for western blotting assay 1:50 for ELISA |
| anti-BDNF | SantaCruz, sc-65513 | 1:500 for western blotting assay |
| anti-GAPDH | Abcam (Waltham, MA, USA), ab8245 | 1:3000 for western blotting assay |
| anti-Actin | Sigma Aldrich (St. Louis, MO, USA), A3853 | 1:2000 for western blotting assay |
| Species [LqHr] c | cBDNF(1-12) | cNT3(1-13) | ||
|---|---|---|---|---|
| log β d | pK | log β d | pK | |
| 11 | 6.60 (3) | 6.60 | - | - |
| 12 | 11.31 (4) | 4.71 | 20.01 (1) | 10.00 × 2 |
| 13 | 15.07 (4) | 3.76 | 29.11 (2) | 9.10 |
| 14 | - | - | 35.78 (3) | 6.67 |
| 15 | - | - | 41.81 (3) | 6.03 |
| 16 | - | - | 46.36 (4) | 4.55 |
| 17 | - | - | 50.31 (3) | 3.95 |
| Species [CupLqHr] c | cBDNF(1-12) | cNT3(1-13) | ||
|---|---|---|---|---|
| log β d | pK | log β d | pK | |
| 114 | - | - | 40.58 (1) | - |
| 113 | - | - | 35.77 (1) | 4.81 |
| 112 | - | - | 28.73 (3) | 7.04 |
| 111 | - | - | 21.82 (1) | 6.91 |
| 110 | 4.77 (5) | - | 13.90 (2) | 7.92 |
| 11-1 | - | 4.57 (3) | 9.34 | |
| 11-2 | −7.30 (4) | 6.03 × 2 | −5.68 (2) | 10.24 |
| 11-3 | −15.81 (5) | 8.51 | −15.88 (2) | 10.20 |
| L | Species [CupLqHr] | UV-Vis λ (nm) (ε, M−1 cm−1) | CD λ (nm) (Δε, M−1 cm−1) |
|---|---|---|---|
| CuL | 686 (75) | 254 (+1.11); 337 (+0.13) | |
| cBDNF(1-12) | CuLH−2 | 590 (99) | 254 (+3.17); 340 (+1.33); 578 (−0.78) |
| CuLH−3 | 546 (117) | 270 (+3.54), 308 (+1.51), 479 (+0.38), 555 (−1.31) | |
| CuLH3 | 688 (55) | 260 (+1.31) | |
| CuLH | 596 (65) | 260 (+2.01), 316 (+0.15), 356 (−0.20), 475 (−0.19), 553 (+0.10) | |
| cNT3(1-13) | CuL | 512 (113) | 260 (+4.22), 319 (+0.90), 358 (−0.15), 488 (−1.07), 612 (+0.81) |
| CuLH−1 | 512 (120) | 260 (+4.50), 319 (+0.95), 358 (−0.12), 490 (−1.13), 614 (+0.86) | |
| CuLH−3 | 515 (95) | 260 (+4.60), 319 (+0.92), 358 (−0.12), 496 (−0.94), 617 (+0.54) |
| Peptide | cKd (M−1) |
|---|---|
| cNT3(1-13) | 1.99 × 10−8 |
| cBDNF(1-12) | 3.42 × 10−8 |
| cNGF(1-14) | 1.49 × 10−7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magrì, A.; Tomasello, B.; Naletova, I.; Tabbì, G.; Cairns, W.R.L.; Greco, V.; Sciuto, S.; La Mendola, D.; Rizzarelli, E. New BDNF and NT-3 Cyclic Mimetics Concur with Copper to Activate Trophic Signaling Pathways as Potential Molecular Entities to Protect Old Brains from Neurodegeneration. Biomolecules 2024, 14, 1104. https://doi.org/10.3390/biom14091104
Magrì A, Tomasello B, Naletova I, Tabbì G, Cairns WRL, Greco V, Sciuto S, La Mendola D, Rizzarelli E. New BDNF and NT-3 Cyclic Mimetics Concur with Copper to Activate Trophic Signaling Pathways as Potential Molecular Entities to Protect Old Brains from Neurodegeneration. Biomolecules. 2024; 14(9):1104. https://doi.org/10.3390/biom14091104
Chicago/Turabian StyleMagrì, Antonio, Barbara Tomasello, Irina Naletova, Giovanni Tabbì, Warren R. L. Cairns, Valentina Greco, Sebastiano Sciuto, Diego La Mendola, and Enrico Rizzarelli. 2024. "New BDNF and NT-3 Cyclic Mimetics Concur with Copper to Activate Trophic Signaling Pathways as Potential Molecular Entities to Protect Old Brains from Neurodegeneration" Biomolecules 14, no. 9: 1104. https://doi.org/10.3390/biom14091104
APA StyleMagrì, A., Tomasello, B., Naletova, I., Tabbì, G., Cairns, W. R. L., Greco, V., Sciuto, S., La Mendola, D., & Rizzarelli, E. (2024). New BDNF and NT-3 Cyclic Mimetics Concur with Copper to Activate Trophic Signaling Pathways as Potential Molecular Entities to Protect Old Brains from Neurodegeneration. Biomolecules, 14(9), 1104. https://doi.org/10.3390/biom14091104