
Enhancing Missense Variant Pathogenicity Prediction with MissenseNet: Integrating Structural Insights and ShuffleNet-Based Deep Learning Techniques

Abstract
:1. Introduction
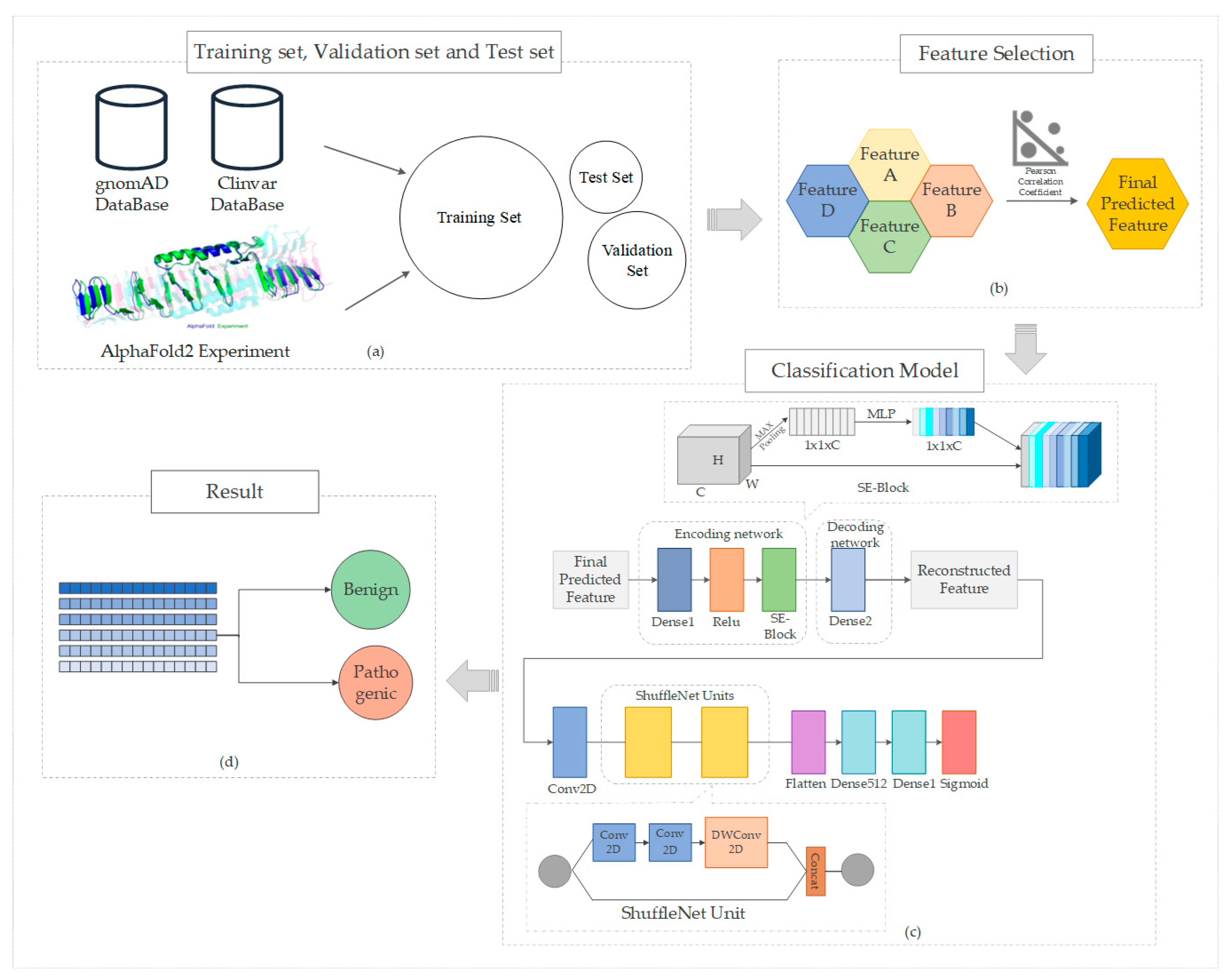
2. Feature Extractions
2.1. Commonly Used Predictive Features
2.2. AlphFold2 Structural Features
3. Data Processing
3.1. Data Set Composition
3.2. Data Acquisition
3.3. Data Imbalance Treatment
4. Construction of the Model
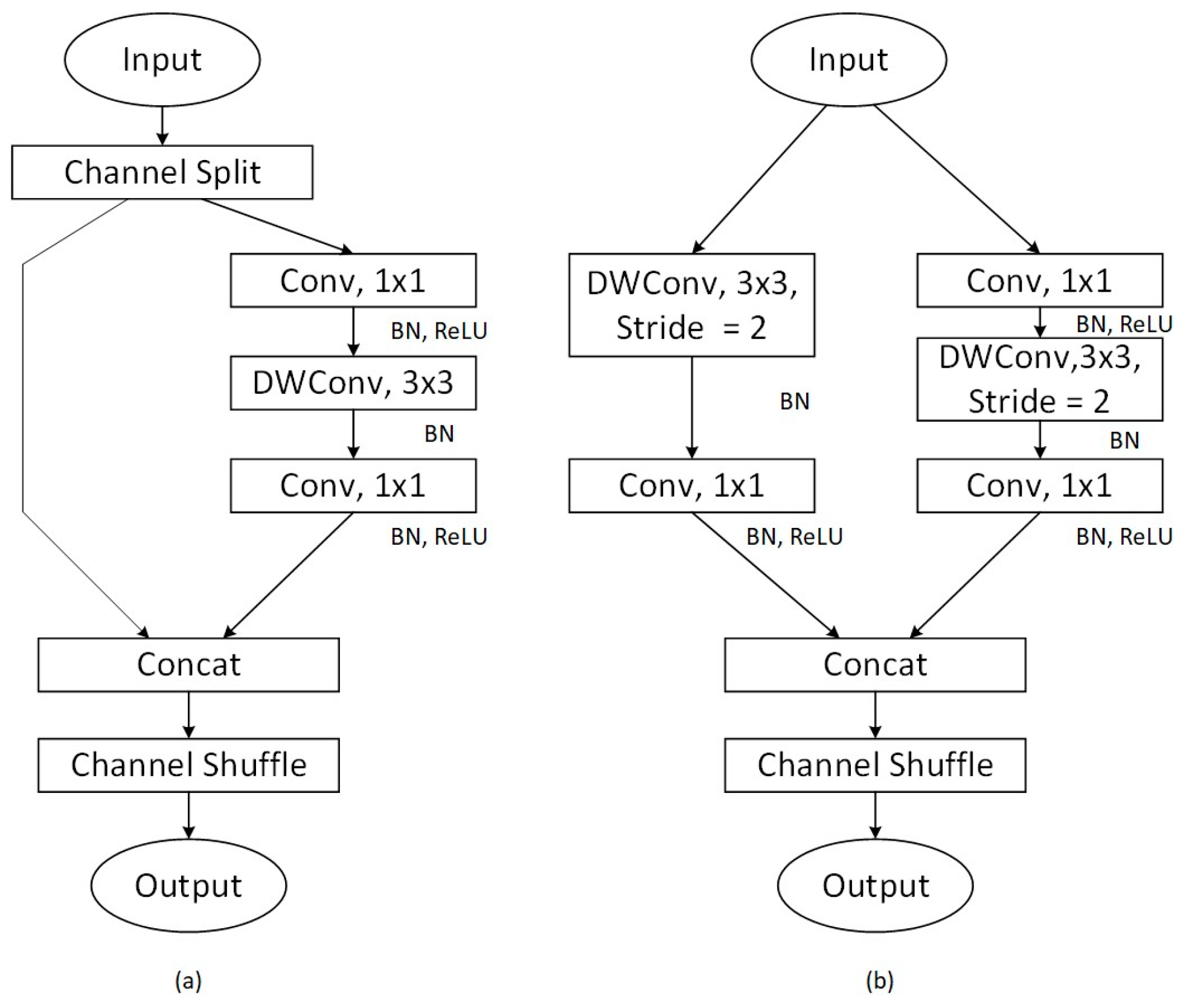
4.1. ShuffleNet
4.2. Squeeze-and-Excitation Module
4.3. Propose the Model MissenseNet
5. Model Evaluation Index
6. Results
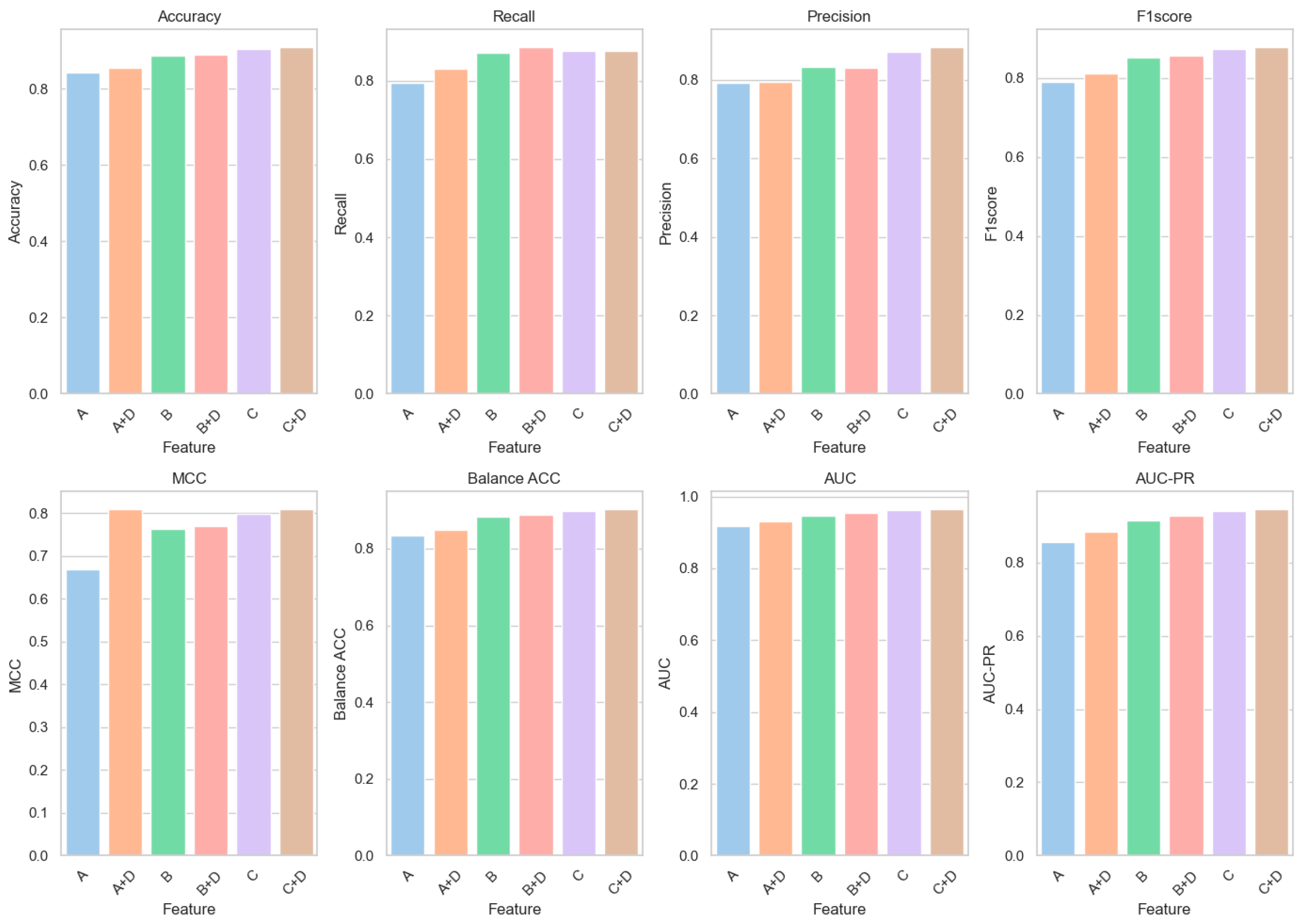
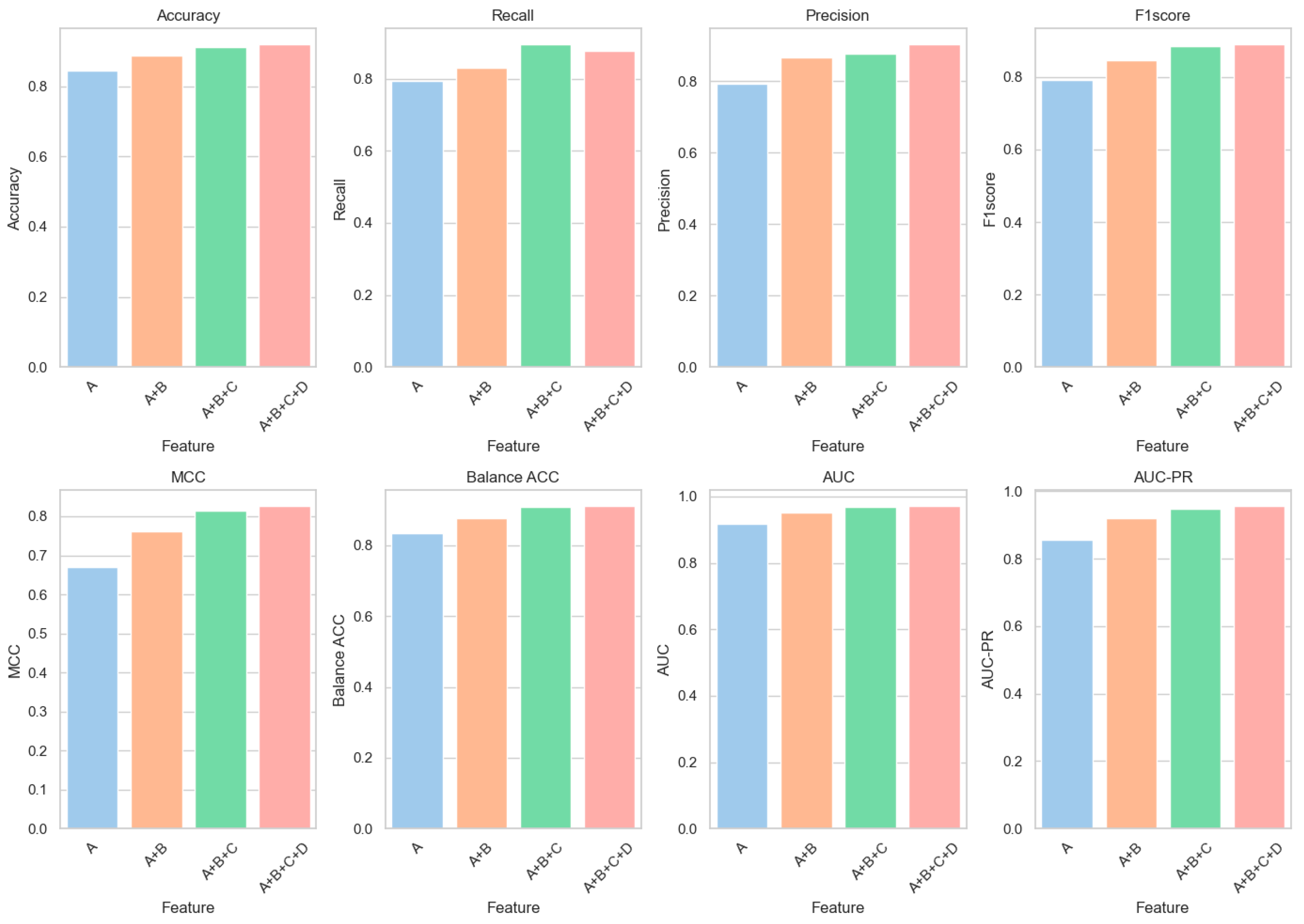
6.1. Feature Ablation Experiments
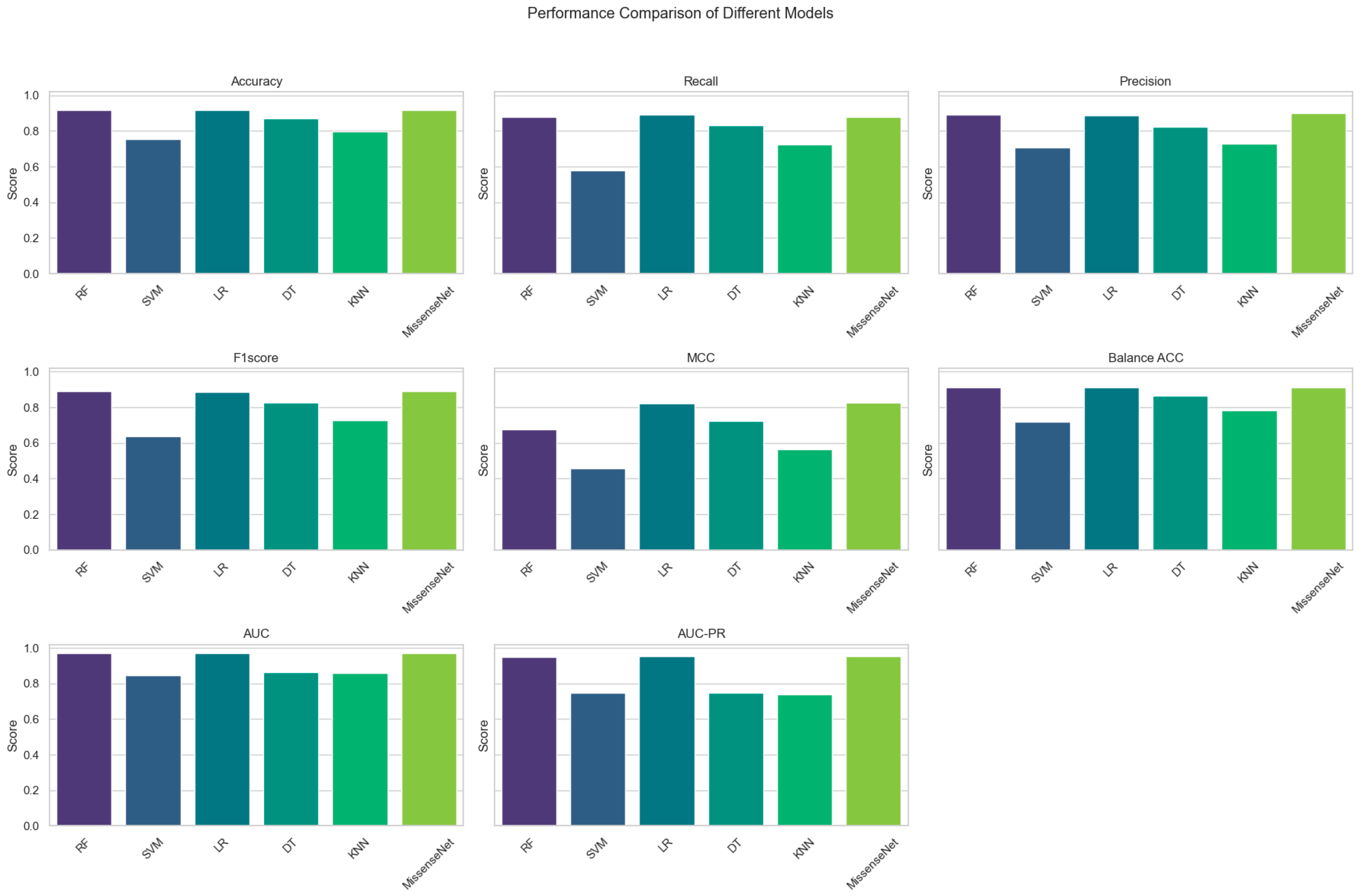
6.2. Comparison with Commonly Used Machine Learning Models
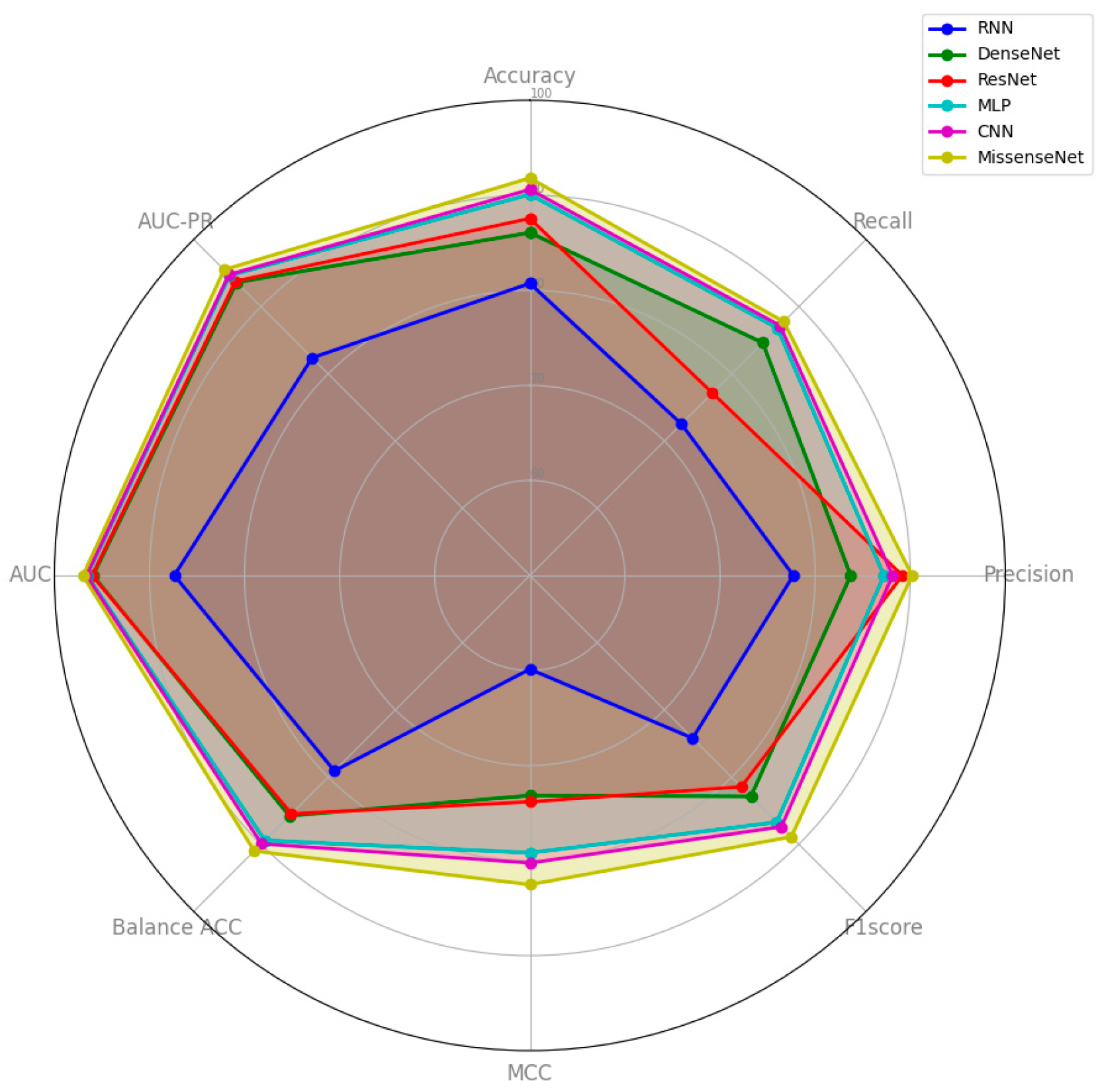
6.3. Comparison with Typical Deep Learning Models
6.4. Results of Ablation Experiments with SE and Encoder-Decoder Modules
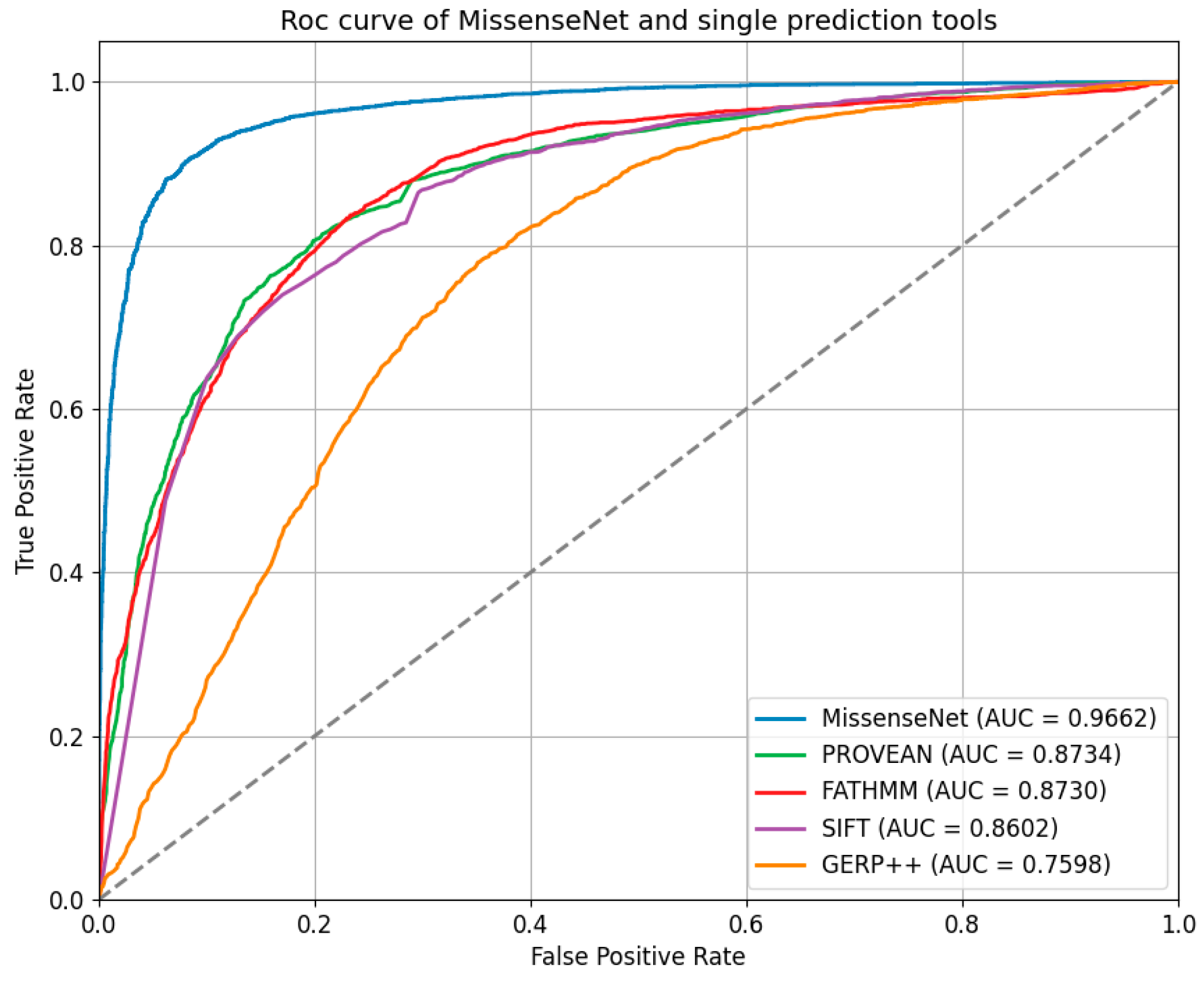
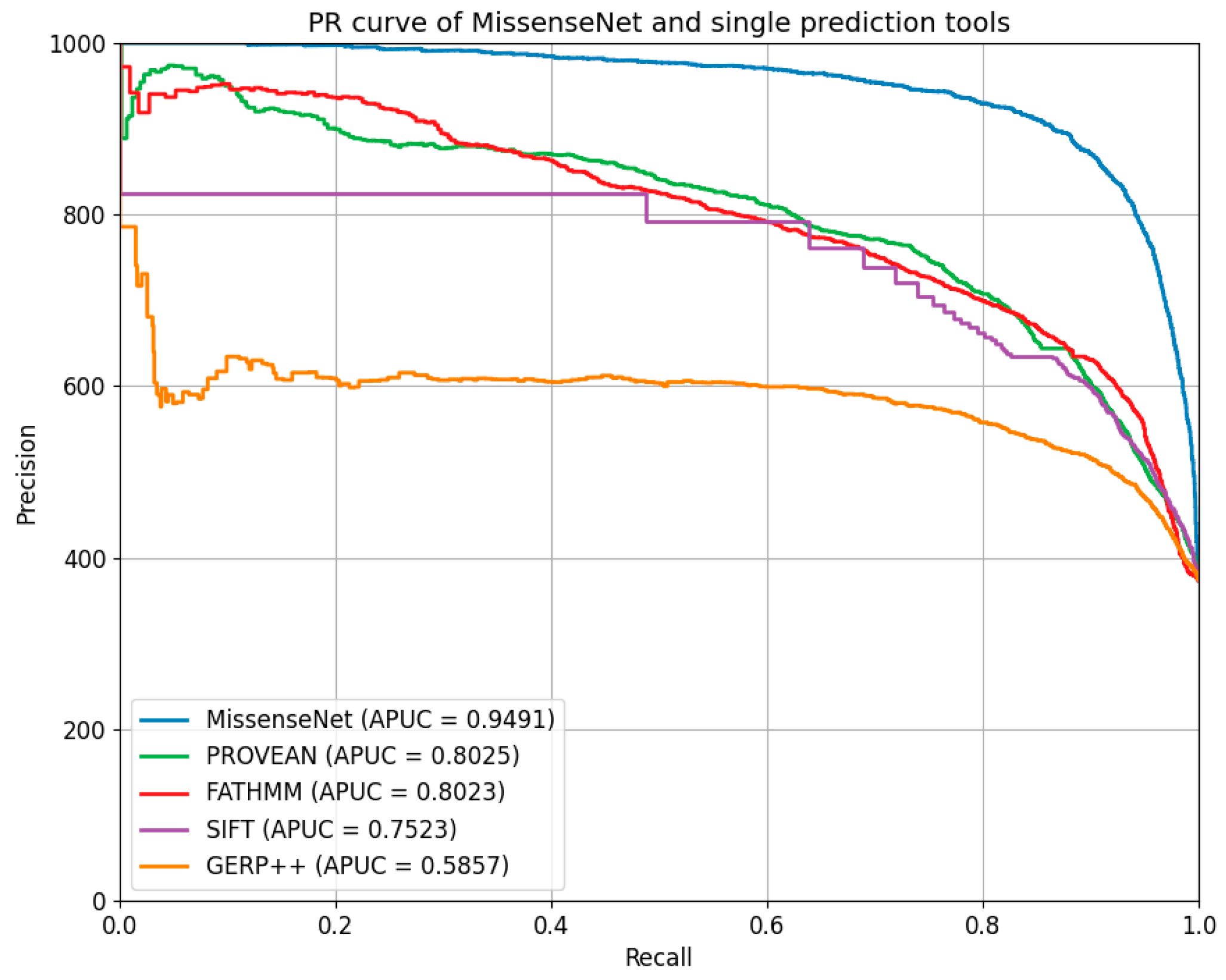
6.5. Comparison with Single-Type Forecasting Tools
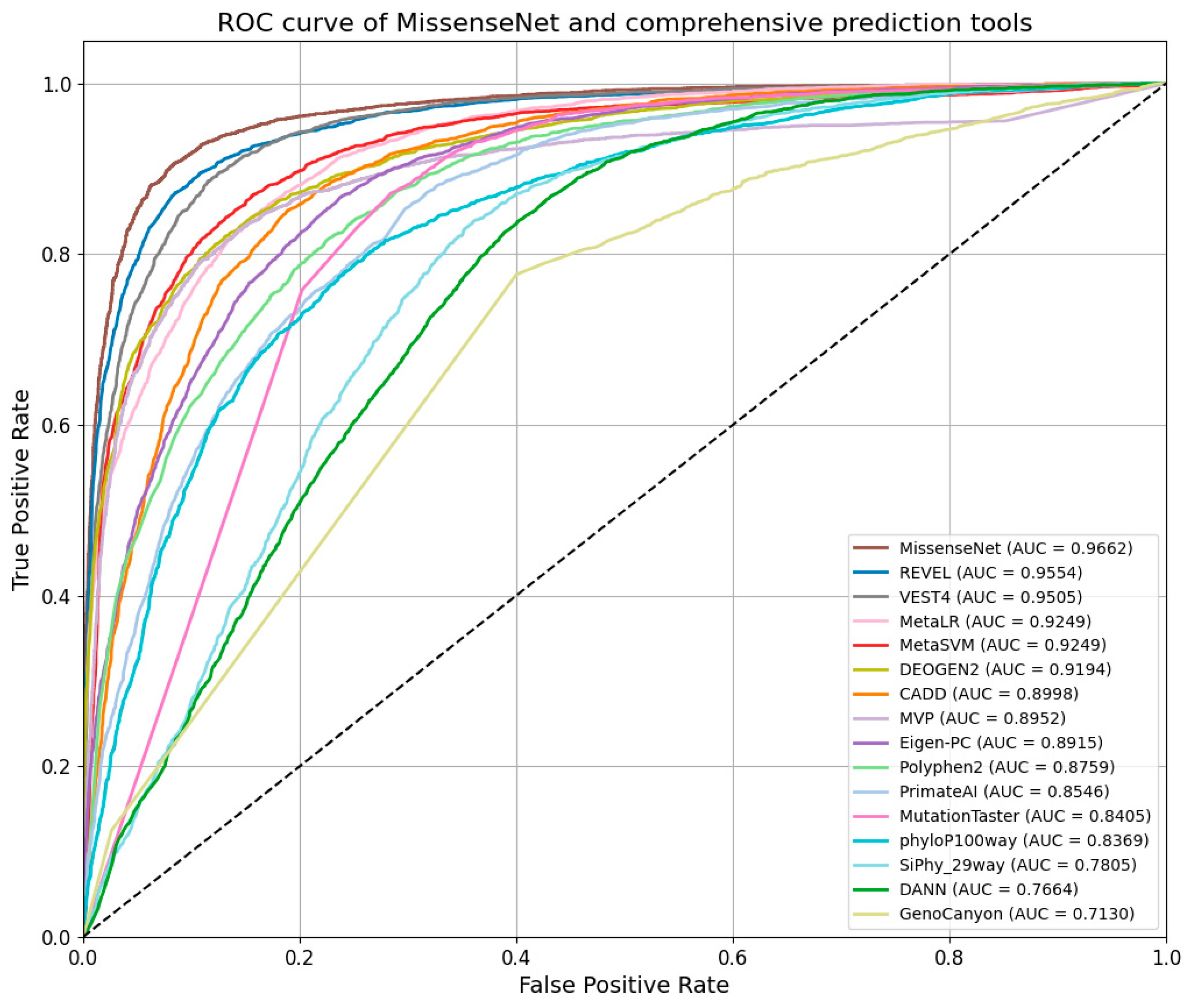
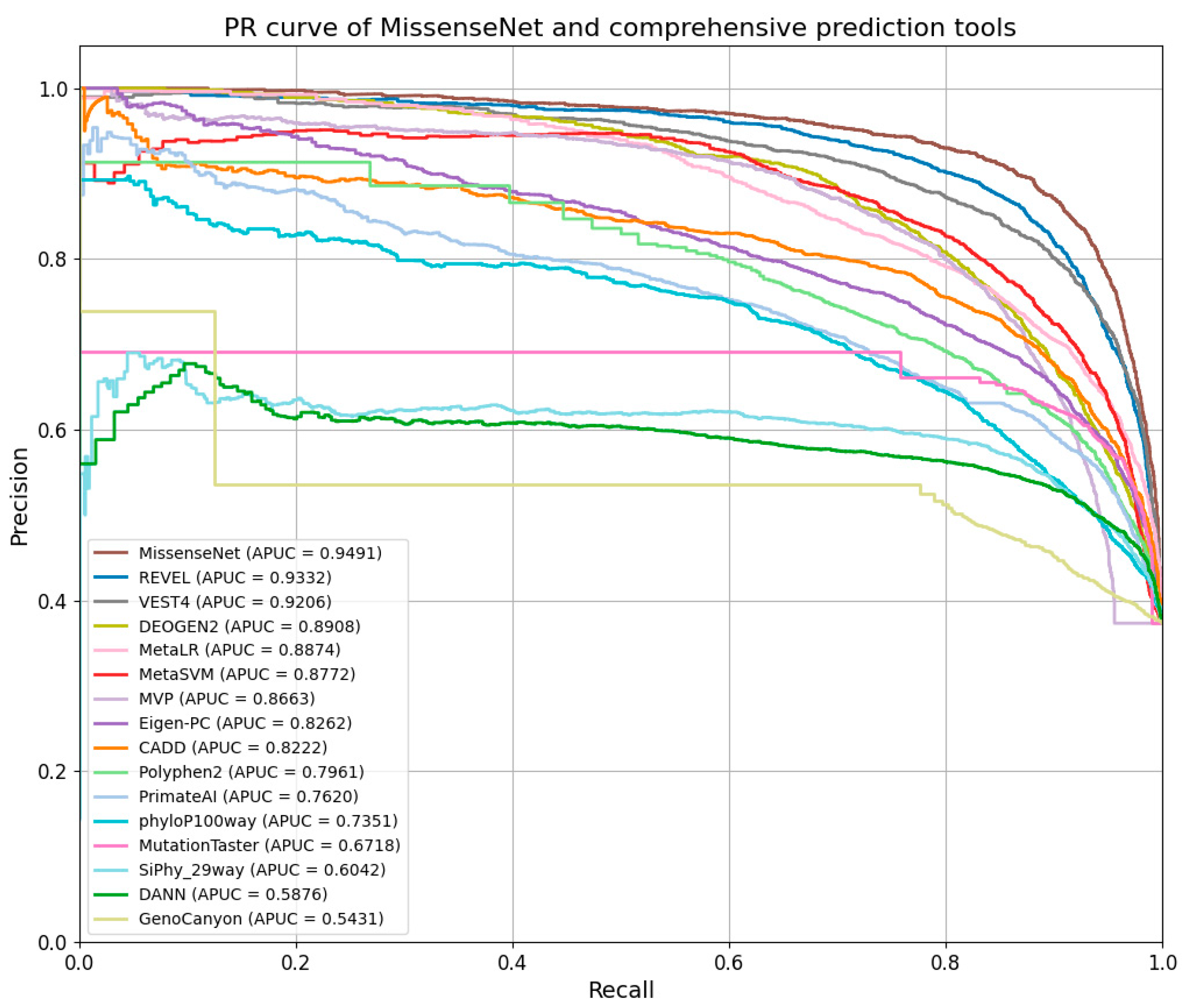
6.6. Comparison with Integrated Forecasting Tools
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.; Zendulka, J.; Brezovsky, J.; Damborský, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Su, Z.; Ma, Z.-Q.; Slebos, R.J.C.; Halvey, P.; Tabb, D.L.; Liebler, D.C.; Pao, W.; Zhang, B. A Bioinformatics Workflow for Variant Peptide Detection in Shotgun Proteomics. Mol. Cell. Proteom. 2011, 10, M110.006536. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L. Nutrigenomics. Mol. Diagn. Ther. 2006, 10, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kaput, J.; Evelo, C.; Perozzi, G.; Ommen, B.; Cotton, R. Connecting the Human Variome Project to nutrigenomics. Genes Nutr. 2010, 5, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kaminker, J.; Zhang, Y.; Waugh, A.; Haverty, P.M.; Peters, B.; Sebisanovic, D.; Stinson, J.; Forrest, W.; Bazan, J.; Seshagiri, S.; et al. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007, 67, 465–473. [Google Scholar] [CrossRef]
- Petrosino, M.; Novak, L.; Pasquo, A.; Chiaraluce, R.; Turina, P.; Capriotti, E.; Consalvi, V. Analysis and Interpretation of the Impact of Missense Variants in Cancer. Int. J. Mol. Sci. 2021, 22, 5416. [Google Scholar] [CrossRef] [PubMed]
- Cimmaruta, C.; Citro, V.; Andreotti, G.; Liguori, L.; Cubellis, M.; Mele, B.H. Challenging popular tools for the annotation of genetic variations with a real case, pathogenic mutations of lysosomal alpha-galactosidase. BMC Bioinform. 2018, 19, 433. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.N.; Douville, C.; Kim, D.; Stenson, P.D.; Cooper, D.N.; Chakravarti, A.; Karchin, R. Proteins linked to autosomal dominant and autosomal recessive disorders harbor characteristic rare missense mutation distribution patterns. Hum. Mol. Genet. 2015, 24, 5995–6002. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef]
- Fang, G.; Jun, H.; Yiheng, Z.; Dongjun, Y. Review on pathogenicity prediction studies of non-synonymous single nucleotide variations. J. Nanjing Univ. Sci. Technol. 2021, 45, 1–17. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.; Lin, G.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Bao, L.; Zhou, M.; Cui, Y. nsSNPAnalyzer: Identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005, 33, W480–W482. [Google Scholar] [CrossRef]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Bromberg, Y.; Rost, B. SNAP: Predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef]
- Ng, P.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef]
- Diwan, G.D.; Gonzalez-Sanchez, J.C.; Apic, G.; Russell, R. Next generation protein structure predictions and genetic variant interpretation. J. Mol. Biol. 2021, 433, 167180. [Google Scholar] [CrossRef]
- Somody, J.; MacKinnon, S.; Windemuth, A. Structural coverage of the proteome for pharmaceutical applications. Drug Discov. Today 2017, 22, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Röner, S.; Mai, K.; Klinkhammer, H.; Kircher, M.; Ludwig, K.U. Predicting the Pathogenicity of Missense Variants Using Features Derived from AlphaFold2. Bioinformatics 2023, 39, btad280. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Frazer, J.; Notin, P.; Dias, M.; Gomez, A.N.; Min, J.K.; Brock, K.P.; Gal, Y.; Marks, D. Disease variant prediction with deep generative models of evolutionary data. Nature 2021, 599, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Šikić, M.; Ng, P. SIFT missense predictions for genomes. Nat. Protoc. 2015, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Takeda, J.; Nanatsue, K.; Yamagishi, R.; Ito, M.; Haga, N.; Hirata, H.; Ogi, T.; Ohno, K. InMeRF: Prediction of pathogenicity of missense variants by individual modeling for each amino acid substitution. NAR Genom. Bioinform. 2020, 2, lqaa038. [Google Scholar] [CrossRef]
- Davydov, E.; Goode, D.; Sirota, M.; Cooper, G.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Pollard, K.; Hubisz, M.; Rosenbloom, K.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2009, 20, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Guttman, M.; Clamp, M.E.; Zody, M.; Friedman, N.; Xie, X. Identifying novel constrained elements by ex-ploiting biased substitution patterns. Bioinformatics 2009, 25, i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the Vari-ant Effect Scoring Tool. BMC Genom. 2013, 14, S3. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Hu, Y.; Sun, J.; Cheng, Y.; Cheung, K.; Zhao, H. A Statistical Framework to Predict Functional Non-Coding Regions in the Human Genome Through Integrated Analysis of Annotation Data. Sci. Rep. 2015, 5, 10576. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.; Pejaver, V.; Middha, S.; McDonnell, S.; Baheti, S.; Musolf, A.M.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.; Jain, P.; O’Roak, B.; Cooper, G.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef]
- Raimondi, D.; Tanyalcin, I.; Ferté, J.; Gazzo, A.M.; Orlando, G.; Lenaerts, T.; Rooman, M.; Vranken, W. DEOGEN2: Prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. 2017, 45, W201–W206. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, L.; Gao, H.; Padigepati, S.; McRae, J.; Li, Y.; Kosmicki, J.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018, 50, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; McCallum, K.; Xu, B.; Buxbaum, J.D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016, 48, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Ittisoponpisan, S.; Islam, S.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Halperin, I.; Glazer, D.S.; Wu, S.; Altman, R. The FEATURE framework for protein function annotation: Modeling new functions, improving performance, and extending to novel applications. BMC Genom. 2008, 9, S2. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.; Francioli, L.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.S.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2013, 42, D980–D985. [Google Scholar] [CrossRef]
- Qi, H.; Zhang, H.; Zhao, Y.; Chen, C.; Long, J.J.; Chung, W.K.; Guan, Y.; Shen, Y. MVP predicts the pathogenicity of missense variants by deep learning. Nat. Commun. 2021, 12, 510. [Google Scholar] [CrossRef]
- Zhu, T.; Liu, X.; Zhu, E. Oversampling with Reliably Expanding Minority Class Regions for Imbalanced Data Learning. IEEE Trans. Knowl. Data Eng. 2022, 35, 6167–6181. [Google Scholar] [CrossRef]
- Chollet, F. Xception: Deep Learning with Depthwise Separable Convolutions. In Proceedings of the the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Honolulu, HI, USA, 21–26 July 2017. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Zhang, X.; Zhou, X.; Lin, M.; Sun, J. ShuffleNet: An Extremely Efficient Convolutional Neural Network for Mobile Devices. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Salt Lake City, UT, USA, 18–23 June 2018. [Google Scholar]
- Chen, Z.; Yang, J.; Chen, L.; Jiao, H. Garbage classification system based on improved ShuffleNet v2. Resour. Conserv. Recycl. 2022, 178, 106090. [Google Scholar] [CrossRef]
- Hu, J.; Shen, L.; Sun, G. Squeeze-and-Excitation Networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Salt Lake City, UT, USA, 18–23 June 2018. [Google Scholar]
- Tang, Y.; Sun, J.; Wang, C.; Zhong, Y.; Jiang, A.; Liu, G.; Liu, X. ADHD classification using auto-encoding neural network and binary hypothesis testing. Artif. Intell. Med. 2022, 123, 102209. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- LaValley, M.P. Logistic Regression. Circulation 2008, 117, 2395–2399. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Minsky, M.; Papert, S.A. Perceptrons, Reissue of the 1988 Expanded Edition with a New Foreword by Léon Bottou: An Introduction to Computational Geometry; MIT Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Buel, G.; Walters, K. Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 2022, 29, 1–2. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Single-type forecasting tool | Sequence-based prediction methods (A) | SIFT [18] |
| SIFT4GSIF [28] | ||
| PROVEAN [27] | ||
| GERP++ [30] | ||
| phyloP100way [31] | ||
| phyloP30way [32] | ||
| SiPhy [33] | ||
| Methods integrating structural and functional features (B) | Polyphen2_HDIV [14] | |
| Polyphen2_HVAR [14] | ||
| MutationTaster2 [34] | ||
| VEST4 [35] | ||
| Fathmm [36] | ||
| GenoCanyon [37] | ||
| 29Ensembletools (C) | REVEL [38] | |
| CADD [39] | ||
| DANN [40] | ||
| MetaSVM [41] | ||
| MetaLR [41] | ||
| DEOGEN2 [42] | ||
| PrimateAI [43] | ||
| Eigen [44] | ||
| Feature | Accuracy | Recall | Precision | F1 Score | MCC | Balance ACC | AUC | AUC-PR |
|---|---|---|---|---|---|---|---|---|
| A | 0.8436 | 0.7938 | 0.7930 | 0.7905 | 0.6690 | 0.8335 | 0.9167 | 0.8554 |
| B | 0.8868 | 0.8717 | 0.8343 | 0.8516 | 0.7618 | 0.8837 | 0.9474 | 0.9148 |
| C | 0.9048 | 0.8762 | 0.8715 | 0.8727 | 0.7980 | 0.8990 | 0.9625 | 0.9411 |
| D | 0.7053 | 0.4943 | 0.6736 | 0.5302 | 0.3523 | 0.6626 | 0.7834 | 0.6806 |
| Feature | Accuracy | Recall | Precision | F1 Score | MCC | Balance ACC | AUC | AUC-PR |
|---|---|---|---|---|---|---|---|---|
| A + D | 0.8543 | 0.8306 | 0.7940 | 0.8121 | 0.8095 | 0.8495 | 0.9306 | 0.8831 |
| B + D | 0.8898 | 0.8867 | 0.8305 | 0.8571 | 0.7694 | 0.8892 | 0.9552 | 0.9265 |
| C + D | 0.9101 | 0.8773 | 0.8841 | 0.8792 | 0.8095 | 0.9035 | 0.9655 | 0.9461 |
| Feature | Accuracy | Recall | Precision | F1 Score | MCC | Balance ACC | AUC | AUC-PR |
|---|---|---|---|---|---|---|---|---|
| A | 0.8436 | 0.7938 | 0.7930 | 0.7905 | 0.6690 | 0.8335 | 0.9167 | 0.8554 |
| A + B | 0.8886 | 0.8306 | 0.8668 | 0.8471 | 0.7613 | 0.8768 | 0.9528 | 0.9191 |
| A + B + C | 0.9126 | 0.8945 | 0.8762 | 0.8843 | 0.8154 | 0.9089 | 0.9676 | 0.9492 |
| A + B + C + D | 0.9182 | 0.8776 | 0.9020 | 0.8889 | 0.8253 | 0.9100 | 0.9701 | 0.9549 |
| Model | Accuracy | Recall | Precision | F1 Score | MCC | Balance ACC | AUC | AUC-PR |
|---|---|---|---|---|---|---|---|---|
| RF | 0.9178 | 0.8804 | 0.8916 | 0.8896 | 0.6741 | 0.9118 | 0.9686 | 0.9506 |
| SVM | 0.7528 | 0.5807 | 0.7055 | 0.6368 | 0.4573 | 0.7181 | 08465 | 0.7480 |
| LR | 0.9160 | 0.8899 | 0.8861 | 0.8877 | 0.8209 | 0.9107 | 0.9692 | 0.9523 |
| DT | 0.8705 | 0.8301 | 0.8245 | 0.8271 | 0.7238 | 0.8623 | 0.8623 | 0.7479 |
| KNN | 0.7954 | 0.7245 | 0.7269 | 0.7255 | 0.5628 | 0.7811 | 0.8586 | 0.7401 |
| MissenseNet | 0.9182 | 0.8776 | 0.9020 | 0.8889 | 0.8253 | 0.9100 | 0.9701 | 0.9549 |
| Model | Accuracy | Recall | Precision | F1 Score | MCC | Balance ACC | AUC | AUC-PR |
|---|---|---|---|---|---|---|---|---|
| MLP | 0.9009 | 0.8682 | 0.8724 | 0.8673 | 0.7919 | 0.8943 | 0.9658 | 0.9466 |
| RNN | 0.8073 | 0.7254 | 0.7771 | 0.7422 | 0.5986 | 0.7908 | 0.8739 | 0.8239 |
| CNN | 0.9061 | 0.8719 | 0.8815 | 0.8740 | 0.8025 | 0.8992 | 0.9666 | 0.9477 |
| DenseNet | 0.8607 | 0.8463 | 0.8374 | 0.8288 | 0.7316 | 0.8577 | 0.9594 | 0.9361 |
| ResNet | 0.8756 | 0.7715 | 0.8918 | 0.8145 | 0.7382 | 0.8546 | 0.9605 | 0.9379 |
| MissenseNet | 0.9182 | 0.8776 | 0.9020 | 0.8889 | 0.8253 | 0.9100 | 0.9701 | 0.9549 |
| Model | Factor | Acc | Recall | Precision | F1 Score | AUC | AUC-PR | |
|---|---|---|---|---|---|---|---|---|
| SE Module | Encoder-Decoder Module | |||||||
| MissenseNet (Baseline) | / | / | 0.8981 | 0.8459 | 0.8892 | 0.8606 | 0.9683 | 0.9512 |
| MissenseNet-(EN) | / | √ | 0.9167 | 0.8714 | 0.9034 | 0.8864 | 0.9700 | 0.9540 |
| MissenseNet (SE + EN) | √ | √ | 0.9182 | 0.8776 | 0.9020 | 0.8889 | 0.9701 | 0.9549 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Chen, Y.; Huang, K.; Guan, X. Enhancing Missense Variant Pathogenicity Prediction with MissenseNet: Integrating Structural Insights and ShuffleNet-Based Deep Learning Techniques. Biomolecules 2024, 14, 1105. https://doi.org/10.3390/biom14091105
Liu J, Chen Y, Huang K, Guan X. Enhancing Missense Variant Pathogenicity Prediction with MissenseNet: Integrating Structural Insights and ShuffleNet-Based Deep Learning Techniques. Biomolecules. 2024; 14(9):1105. https://doi.org/10.3390/biom14091105
Chicago/Turabian StyleLiu, Jing, Yingying Chen, Kai Huang, and Xiao Guan. 2024. "Enhancing Missense Variant Pathogenicity Prediction with MissenseNet: Integrating Structural Insights and ShuffleNet-Based Deep Learning Techniques" Biomolecules 14, no. 9: 1105. https://doi.org/10.3390/biom14091105
APA StyleLiu, J., Chen, Y., Huang, K., & Guan, X. (2024). Enhancing Missense Variant Pathogenicity Prediction with MissenseNet: Integrating Structural Insights and ShuffleNet-Based Deep Learning Techniques. Biomolecules, 14(9), 1105. https://doi.org/10.3390/biom14091105