
From Organotypic Mouse Brain Slices to Human Alzheimer Plasma Biomarkers: A Focus on Microglia
 , , ,
, , ,
Abstract
:1. Introduction
1.1. Biomarkers in Alzheimer’s Disease
1.2. Plasma Biomarkers in Alzheimer’s Disease
1.3. Microglia in Alzheimer’s Disease
1.4. Organotypic Brain Slices as a Tool to Search for Biomarkers
2. Materials and Methods
2.1. Human Plasma Samples
2.2. Microcontact Printing of Human Plasma
2.3. Organotypic Brain Slices
2.4. Immunofluorescence for Microglia
2.5. Mass Spectrometry
2.6. Data Analysis and Statistics
3. Results
3.1. Efficiency of Plasma Microcontact Printing
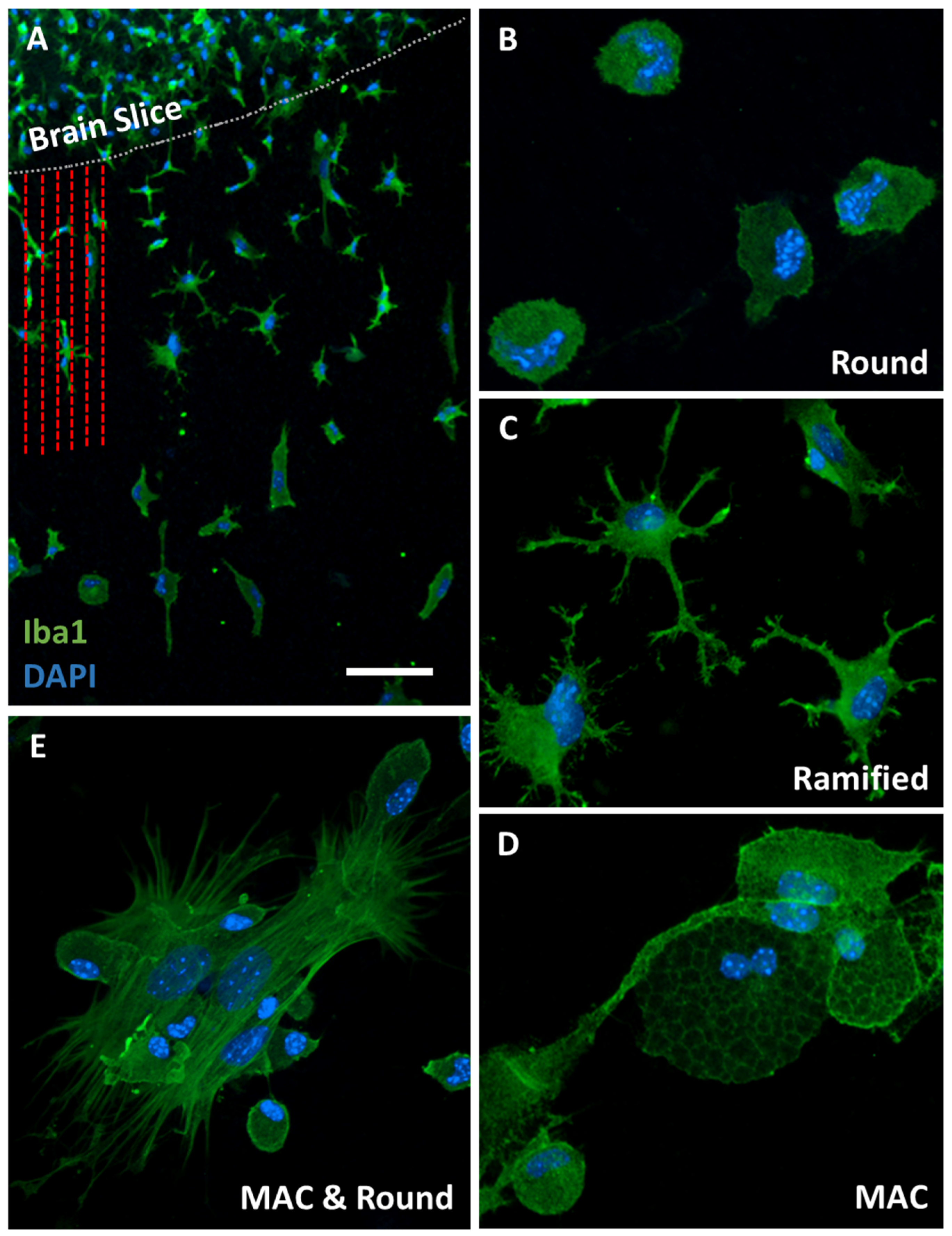
3.2. Iba1-Postive Microglial Immunoreactivity
3.3. Effects of MCP-1 on Microglial Migration
3.4. Epidemiology of the Patients in This Study
3.5. Effects of Plasma on Migration of Iba1-Positive Microglia
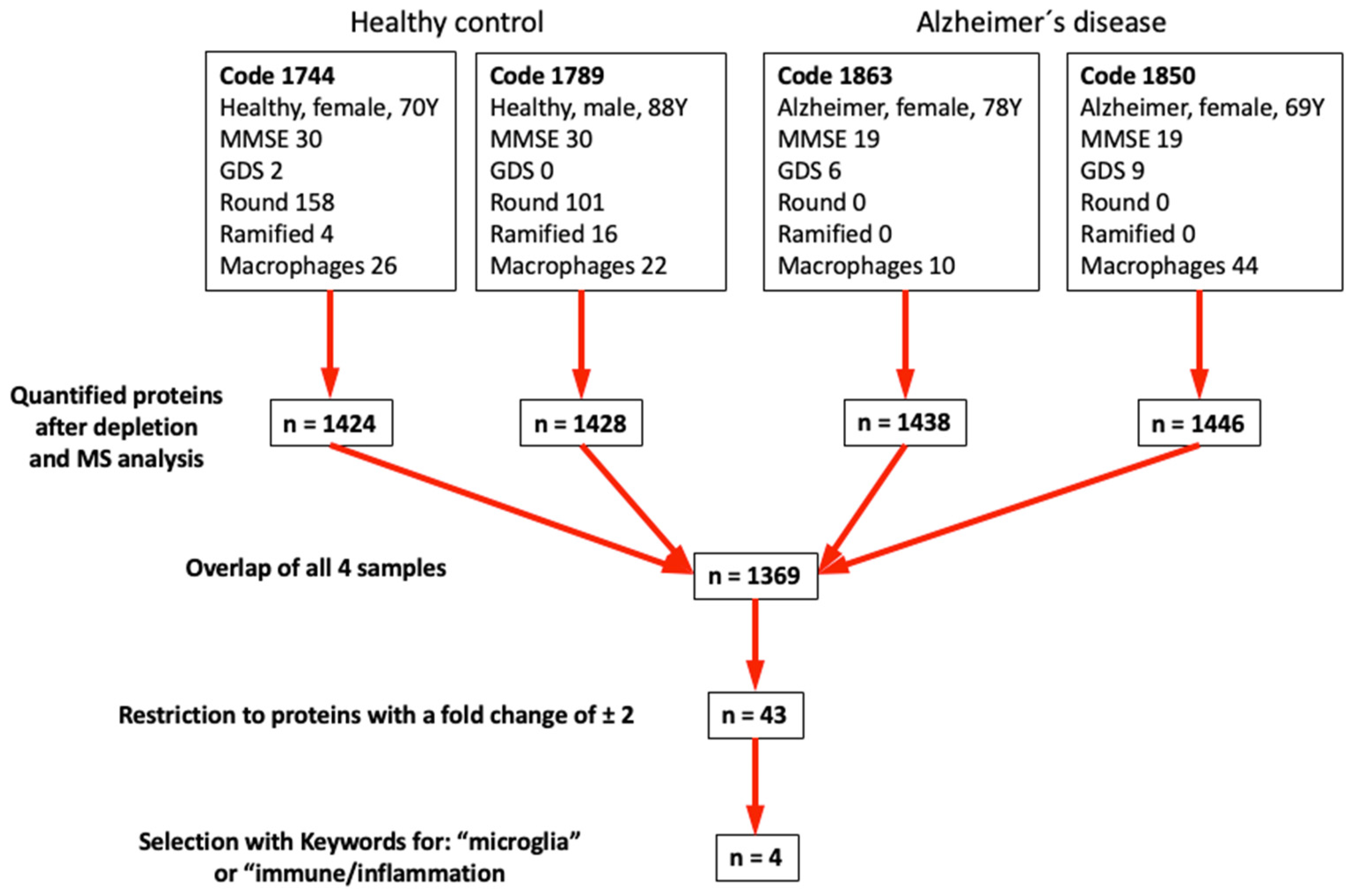
3.6. Mass Spectrometry
4. Discussion
4.1. Iba1-Positive Microglia in Organotypic Brain Slices
4.2. Microcontact Printing for Delivering Plasma to Brain Slices
4.3. Effects of Microcontact-Printed MCP-1 on Microglia
4.4. Effects of Microcontact-Printed Plasma on Microglia
4.5. Differential Mass Spectrometry
4.6. Limitations of the Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mulder, C.; Verwey, N.A.; van der Flier, W.M.; Bouwman, F.H.; Kok, A.; van Elk, E.J.; Scheltens, P.; Blankenstein, M.A. Amyloid-beta(1-42), total tau, and phosphorylated tau as cerebrospinal fluid biomarkers for the diagnosis of Alzheimer disease. Clin. Chem. 2010, 56, 248–253. [Google Scholar] [CrossRef]
- Ray, S.; Britschgi, M.; Herbert, C.; Takeda-Uchimura, Y.; Boxer, A.; Blennow, K.; Friedman, L.F.; Galasko, D.R.; Jutel, M.; Karydas, A.; et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat. Med. 2007, 13, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Bjorkqvist, M.; Ohlsson, M.; Minthon, L.; Hansson, O. Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer’s disease. PLoS ONE 2012, 7, e29868. [Google Scholar] [CrossRef]
- Kaneko, N.; Yamamoto, R.; Sato, T.A.; Tanaka, K. Identification and quantification of amyloid beta-related peptides in human plasma using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Pais, M.V.; Forlenza, O.V.; Diniz, B.S. Plasma Biomarkers of Alzheimer’s Disease: A Review of Available Assays, Recent Developments, and Implications for Clinical Practice. J. Alzheimer’s Dis. Rep. 2023, 7, 355–380. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef]
- Varesi, A.; Carrara, A.; Pires, V.G.; Floris, V.; Pierella, E.; Savioli, G.; Prasad, S.; Esposito, C.; Ricevuti, G.; Chirumbolo, S.; et al. Blood-Based Biomarkers for Alzheimer’s Disease Diagnosis and Progression: An Overview. Cells 2022, 11, 1367. [Google Scholar] [CrossRef]
- Gustaw-Rothenberg, K.A.; Siedlak, S.L.; Bonda, D.J.; Lerner, A.; Tabaton, M.; Perry, G.; Smith, M.A. Dissociated amyloid-beta antibody levels as a serum biomarker for the progression of Alzheimer’s disease: A population-based study. Exp. Gerontol. 2010, 45, 47–52. [Google Scholar] [CrossRef]
- Weldon Furr, J.; Morales-Scheihing, D.; Manwani, B.; Lee, J.; McCullough, L.D. Cerebral Amyloid Angiopathy, Alzheimer’s Disease and MicroRNA: miRNA as Diagnostic Biomarkers and Potential Therapeutic Targets. Neuromol. Med. 2019, 21, 369–390. [Google Scholar] [CrossRef]
- Kumar, P.; Dezso, Z.; MacKenzie, C.; Oestreicher, J.; Agoulnik, S.; Byrne, M.; Bernier, F.; Yanagimachi, M.; Aoshima, K.; Oda, Y. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS ONE 2013, 8, e69807. [Google Scholar] [CrossRef] [PubMed]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 253–259. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting neuroinflammation in Alzheimer’s disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Zurcher, C.; Humpel, C. Saliva: A challenging human fluid to diagnose brain disorders with a focus on Alzheimer’s disease. Neural Regen. Res. 2023, 18, 2606–2610. [Google Scholar] [CrossRef] [PubMed]
- Humpel, C. Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011, 29, 26–32. [Google Scholar] [CrossRef]
- Vidal-Itriago, A.; Radford, R.A.W.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Front. Immunol. 2022, 13, 997786. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- von Bernhardi, R.; Tichauer, J.E.; Eugenin, J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 2010, 112, 1099–1114. [Google Scholar] [CrossRef]
- Li, K.; Liu, S.; Yao, S.; Wang, B.; Dai, D.; Yao, L. Interaction between interleukin-8 and methylenetetrahydrofolate reductase genes modulates Alzheimer’s disease risk. Dement. Geriatr. Cogn. Disord. 2009, 27, 286–291. [Google Scholar] [CrossRef]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Suppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, Y.P.; Liu, Y.H.; Jiao, S.S.; Liu, L.; Wang, Y.J.; Fu, W.L. Diagnostic utility of VEGF and soluble CD40L levels in serum of Alzheimer’s patients. Clin. Chim. Acta 2016, 453, 154–159. [Google Scholar] [CrossRef]
- Alam, Q.; Alam, M.Z.; Mushtaq, G.; Damanhouri, G.A.; Rasool, M.; Kamal, M.A.; Haque, A. Inflammatory Process in Alzheimer’s and Parkinson’s Diseases: Central Role of Cytokines. Curr. Pharm. Des. 2016, 22, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Meri, S. Yin and Yang: Complement activation and regulation in Alzheimer’s disease. Prog. Neurobiol. 2003, 70, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Deitmer, J.W.; Rose, C.R. pH regulation and proton signalling by glial cells. Prog. Neurobiol. 1996, 48, 73–103. [Google Scholar] [CrossRef]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Humpel, C. Organotypic brain slice cultures: A review. Neuroscience 2015, 305, 86–98. [Google Scholar] [CrossRef]
- Steiner, K.; Humpel, C. Long-term organotypic brain slices cultured. on collagen-based microcontact prints: A perspective for a brain-on-a-chip. J. Neurosci. Methods 2023, 399, 109979. [Google Scholar] [CrossRef]
- Steiner, K.; Humpel, C. Effects of Ischemia on the Migratory Capacity of Microglia Along Collagen Microcontact Prints on Organotypic Mouse Cortex Brain Slices. Front. Cell Neurosci. 2022, 16, 858802. [Google Scholar] [CrossRef]
- Steiner, K.; Humpel, C. Beta-Amyloid Enhances Vessel Formation in Organotypic Brain Slices Connected to Microcontact Prints. Biomolecules 2023, 14, 3. [Google Scholar] [CrossRef]
- Steiner, K.; Humpel, C. Microcontact Printing of Cholinergic Neurons in Organotypic Brain Slices. Front. Neurol. 2021, 12, 775621. [Google Scholar] [CrossRef]
- Marksteiner, J.; Imarhiagbe, D.; Defrancesco, M.; Deisenhammer, E.A.; Kemmler, G.; Humpel, C. Analysis of 27 vascular-related proteins reveals that NT-proBNP is a potential biomarker for Alzheimer’s disease and mild cognitive impairment: A pilot-study. Exp. Gerontol. 2014, 50, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Weyer, Y.; Baumann, V.; Widerin, M.A.; Eising, S.; Angelova, M.; Schleiffer, A.; Kremser, L.; Lindner, H.; Peter, M.; et al. Endosome and Golgi-associated degradation (EGAD) of membrane proteins regulates sphingolipid metabolism. EMBO J. 2019, 38, e101433. [Google Scholar] [CrossRef] [PubMed]
- Indelicato, E.; Faserl, K.; Amprosi, M.; Nachbauer, W.; Schneider, R.; Wanschitz, J.; Sarg, B.; Boesch, S. Skeletal muscle proteome analysis underpins multifaceted mitochondrial dysfunction in Friedreich’s ataxia. Front. Neurosci. 2023, 17, 1289027. [Google Scholar] [CrossRef]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.R.N.; Lopez, R.; Butcher, S. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef] [PubMed]
- Croft, C.L.; Futch, H.S.; Moore, B.D.; Golde, T.E. Organotypic brain slice cultures to model neurodegenerative proteinopathies. Mol. Neurodegener. 2019, 14, 45. [Google Scholar] [CrossRef]
- Bahr, B.A. Long-term hippocampal slices: A model system for investigating synaptic mechanisms and pathologic processes. J. Neurosci. Res. 1995, 42, 294–305. [Google Scholar] [CrossRef]
- Stoppini, L.; Buchs, P.A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Huh, D.; Brickelmaier, M.; Burns, J.C.; Roberts, C.; Challa, R.; Raymond, N.; Cullen, P.; Carlile, T.M.; Ennis, K.A.; et al. Organotypic Brain Slice Culture Microglia Exhibit Molecular Similarity to Acutely-Isolated Adult Microglia and Provide a Platform to Study Neuroinflammation. Front. Cell Neurosci. 2020, 14, 592005. [Google Scholar] [CrossRef]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Moller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Abels, E.R.; Nieland, L.; Hickman, S.; Broekman, M.L.D.; El Khoury, J.; Maas, S.L.N. Comparative Analysis Identifies Similarities between the Human and Murine Microglial Sensomes. Int. J. Mol. Sci. 2021, 22, 1495. [Google Scholar] [CrossRef]
- Ogaki, A.; Ikegaya, Y.; Koyama, R. Replacement of Mouse Microglia With Human Induced Pluripotent Stem Cell (hiPSC)-Derived Microglia in Mouse Organotypic Slice Cultures. Front. Cell Neurosci. 2022, 16, 918442. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef]
- Lier, J.; Streit, W.J.; Bechmann, I. Beyond Activation: Characterizing Microglial Functional Phenotypes. Cells 2021, 10, 2236. [Google Scholar] [CrossRef]
- Xia, Y.; Whitesides, G.M. Soft Lithography. Angew. Chem. Int. Ed. Engl. 1998, 37, 550–575. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, Y.; Qu, Q.; Zhang, X.; Lu, T.; Xu, J.; Ma, W.; Zhu, M.; Huang, C.; Xiong, R. Biomaterials based on hyaluronic acid, collagen and peptides for three-dimensional cell culture and their application in stem cell differentiation. Int. J. Biol. Macromol. 2023, 226, 14–36. [Google Scholar] [CrossRef]
- Ucar, B. Natural biomaterials in brain repair: A focus on collagen. Neurochem. Int. 2021, 146, 105033. [Google Scholar] [CrossRef]
- Haw, R.T.; Tong, C.K.; Yew, A.; Lee, H.C.; Phillips, J.B.; Vidyadaran, S. A three-dimensional collagen construct to model lipopolysaccharide-induced activation of BV2 microglia. J. Neuroinflamm. 2014, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Balion, Z.; Svirskiene, N.; Svirskis, G.; Inokaitis, H.; Cepla, V.; Ulcinas, A.; Jelinskas, T.; Eimont, R.; Pauziene, N.; Valiokas, R.; et al. Comparison of Microglial Morphology and Function in Primary Cerebellar Cell Cultures on Collagen and Collagen-Mimetic Hydrogels. Biomedicines 2022, 10, 1023. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Banisadr, G.; Gosselin, R.D.; Mechighel, P.; Kitabgi, P.; Rostene, W.; Parsadaniantz, S.M. Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: Evidence for its colocalization with neurotransmitters and neuropeptides. J. Comp. Neurol. 2005, 489, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Boddeke, E.W.; Meigel, I.; Frentzel, S.; Gourmala, N.G.; Harrison, J.K.; Buttini, M.; Spleiss, O.; Gebicke-Harter, P. Cultured rat microglia express functional beta-chemokine receptors. J. Neuroimmunol. 1999, 98, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Meng, Y.; Li, W.; Yong, Y.; Fan, Z.; Ding, H.; Wei, Y.; Luo, J.; Ke, Z.J. Neuronal MCP-1 mediates microglia recruitment and neurodegeneration induced by the mild impairment of oxidative metabolism. Brain Pathol. 2011, 21, 279–297. [Google Scholar] [CrossRef]
- Hinojosa, A.E.; Garcia-Bueno, B.; Leza, J.C.; Madrigal, J.L. CCL2/MCP-1 modulation of microglial activation and proliferation. J. Neuroinflamm. 2011, 8, 77. [Google Scholar] [CrossRef]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Yan, Z.; Dixit, K.; Johnson, J.R.; Bouhaddou, M.; Meyer-Franke, A.; Shin, M.G.; Yong, Y.; Agrawal, A.; MacDonald, E.; et al. Defining blood-induced microglia functions in neurodegeneration through multiomic profiling. Nat. Immunol. 2023, 24, 1173–1187. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef]
- Keshavan, A.; Heslegrave, A.; Zetterberg, H.; Schott, J.M. Blood Biomarkers for Alzheimer’s Disease: Much Promise, Cautious Progress. Mol. Diagn. Ther. 2017, 21, 13–22. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Status Report on the Public Health Response to Dementia; World Health Organization (WHO): Geneva, Switzerland, 2021. [Google Scholar]
- Birhanu, A.G. Mass spectrometry-based proteomics as an emerging tool in clinical laboratories. Clin. Proteom. 2023, 20, 32. [Google Scholar] [CrossRef]
- Sjolander, A.; Minthon, L.; Nuytinck, L.; Vanmechelen, E.; Blennow, K.; Nilsson, S. Functional mannose-binding lectin haplotype variants are associated with Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 35, 121–127. [Google Scholar] [CrossRef]
- Vincent, M.B.; Bakken, I.J.; White, L.R. Endothelin-1 inhibits the vasodilation induced by substance P in isolated porcine ophthalmic artery. Funct. Neurol. 1992, 7, 475–480. [Google Scholar]
- Wang, L.; Yang, H.Y.; Zang, C.X.; Shang, J.M.; Liu, H.; Zhang, Z.H.; Yuan, F.Y.; Ju, C.; Li, F.Y.; Bao, X.Q.; et al. TLR2 Potentiates SR-Marco-Mediated Neuroinflammation by Interacting with the SRCR Domain. Mol. Neurobiol. 2021, 58, 5743–5755. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Wong, A.; Zhang, Z.; Na, H.; Tian, H.; Tao, Q.; Rajab, I.M.; Potempa, L.A.; Qiu, W.Q. Monomeric C-reactive protein induces the cellular pathology of Alzheimer’s disease. Alzheimer’s Dement. 2022, 8, e12319. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Alvin Ang, T.F.; Akhter-Khan, S.C.; Itchapurapu, I.S.; Killiany, R.; Zhang, X.; Budson, A.E.; Turk, K.W.; Goldstein, L.; Mez, J.; et al. Impact of C-Reactive Protein on Cognition and Alzheimer Disease Biomarkers in Homozygous APOE varepsilon4 Carriers. Neurology 2021, 97, e1243–e1252. [Google Scholar] [CrossRef] [PubMed]
- Larvie, M.; Shoup, T.; Chang, W.C.; Chigweshe, L.; Hartshorn, K.; White, M.R.; Stahl, G.L.; Elmaleh, D.R.; Takahashi, K. Mannose-binding lectin binds to amyloid beta protein and modulates inflammation. J. Biomed. Biotechnol. 2012, 2012, 929803. [Google Scholar] [CrossRef]
- Siew, J.J.; Chern, Y. Microglial Lectins in Health and Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 158. [Google Scholar] [CrossRef]
- Foidl, B.M.; Humpel, C. Can mouse models mimic sporadic Alzheimer’s disease? Neural Regen. Res. 2020, 15, 401–406. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | n | Round | Ramified | Macrophagic | Length |
|---|---|---|---|---|---|
| Minus medium | 5 | 0.2 ± 0.2 vs. | 0.2 ± 0.2 vs. | 0.8 ± 0.2 vs. | 255 ± 89 vs. |
| MCP-1 medium | 7 | 2.7 ± 1.7 ** | 6.7 ± 3.6 ** | 3.0 ± 0.7 ** | 364 ± 96 |
| µCP (-) | 5 | 5.8 ± 2.3 vs. | 8.4 ± 3.3 vs. | 7.0 ± 2.4 vs. | 591 ± 76 vs. |
| µCP (MCP-1) | 10 | 4.8 ± 1.7 | 28.2 ± 6.1 *** | 14.0 ± 3.2 * | 864 ± 26 |
| µCP (Plasma) | 6 | 13.1 ± 5.5 vs. | 16.0 ± 5.9 vs. | 16.0 ± 7.9 vs. | 721 ± 63 vs. |
| µCP (Plasma+MCP-1) | 10 | 11.3 ± 3.2 | 31.6 ± 6.2 ** | 9.4 ± 1.8 | 861 ± 63 |
| Patient | n | Male | Age | MMSE | GDS |
|---|---|---|---|---|---|
| Control | 23 | 13 | 73 ± 1.5 vs. | 29.6 ± 0.1 vs. | 3.8 ± 0.5 vs. |
| MCI | 25 | 9 | 75 ± 1.6 | 27.4 ± 0.2 * | 4.4 ± 0.6 |
| AD | 24 | 4 | 81 ± 1.5 ** | 18.9 ± 1.2 *** | 5.0 ± 0.6 |
| Group | n | Round | Ramified | Macrophagic | Length |
|---|---|---|---|---|---|
| Control | 23 | 49.6 ± 16 vs. | 12.5 ± 1.9 vs. | 12.5 ± 2.6 vs. | 400 ± 50 vs. |
| MCI | 25 | 27.1 ± 6 | 11.1 ± 2.6 | 10.5 ± 2.2 | 441 ± 51 |
| AD | 24 | 12.9 ± 3 p = 0.01 * | 10.0 ± 2.2 | 13.1 ± 3.0 | 424 ± 55 |
| Putative Biomarker Identified | Symbol | Acc. No. | MW (kDa) | # Unique Peptides | Change in AD | Identity (%) | Similarity (%) |
|---|---|---|---|---|---|---|---|
| Mannose-binding protein C | MBL2 | P11226 | 26.1 | 10 | 5.5 ↑ | 60.2 | 75.2 |
| Macrophage receptor MARCO | MARCO | Q9UEW3 | 52.6 | 4 | 0.5 ↓ | 66.9 | 77.1 |
| Complement factor H-related protein-3 | CFHR3 | Q02985 | 37.3 | 3 | 0.5 ↓ | 45.4 | 59.3 |
| C-reactive protein | CRP | P02741 | 25 | 6 | 0.3 ↓ | 70.1 | 83 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steiner, K.; Yilmaz, S.N.; Gern, A.; Marksteiner, J.; Faserl, K.; Villunger, M.; Sarg, B.; Humpel, C. From Organotypic Mouse Brain Slices to Human Alzheimer Plasma Biomarkers: A Focus on Microglia. Biomolecules 2024, 14, 1109. https://doi.org/10.3390/biom14091109
Steiner K, Yilmaz SN, Gern A, Marksteiner J, Faserl K, Villunger M, Sarg B, Humpel C. From Organotypic Mouse Brain Slices to Human Alzheimer Plasma Biomarkers: A Focus on Microglia. Biomolecules. 2024; 14(9):1109. https://doi.org/10.3390/biom14091109
Chicago/Turabian StyleSteiner, Katharina, Sakir Necat Yilmaz, Alessa Gern, Josef Marksteiner, Klaus Faserl, Mathias Villunger, Bettina Sarg, and Christian Humpel. 2024. "From Organotypic Mouse Brain Slices to Human Alzheimer Plasma Biomarkers: A Focus on Microglia" Biomolecules 14, no. 9: 1109. https://doi.org/10.3390/biom14091109