

Screening of Anti-Prion Compounds Using the Protein Misfolding Cyclic Amplification Technology
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
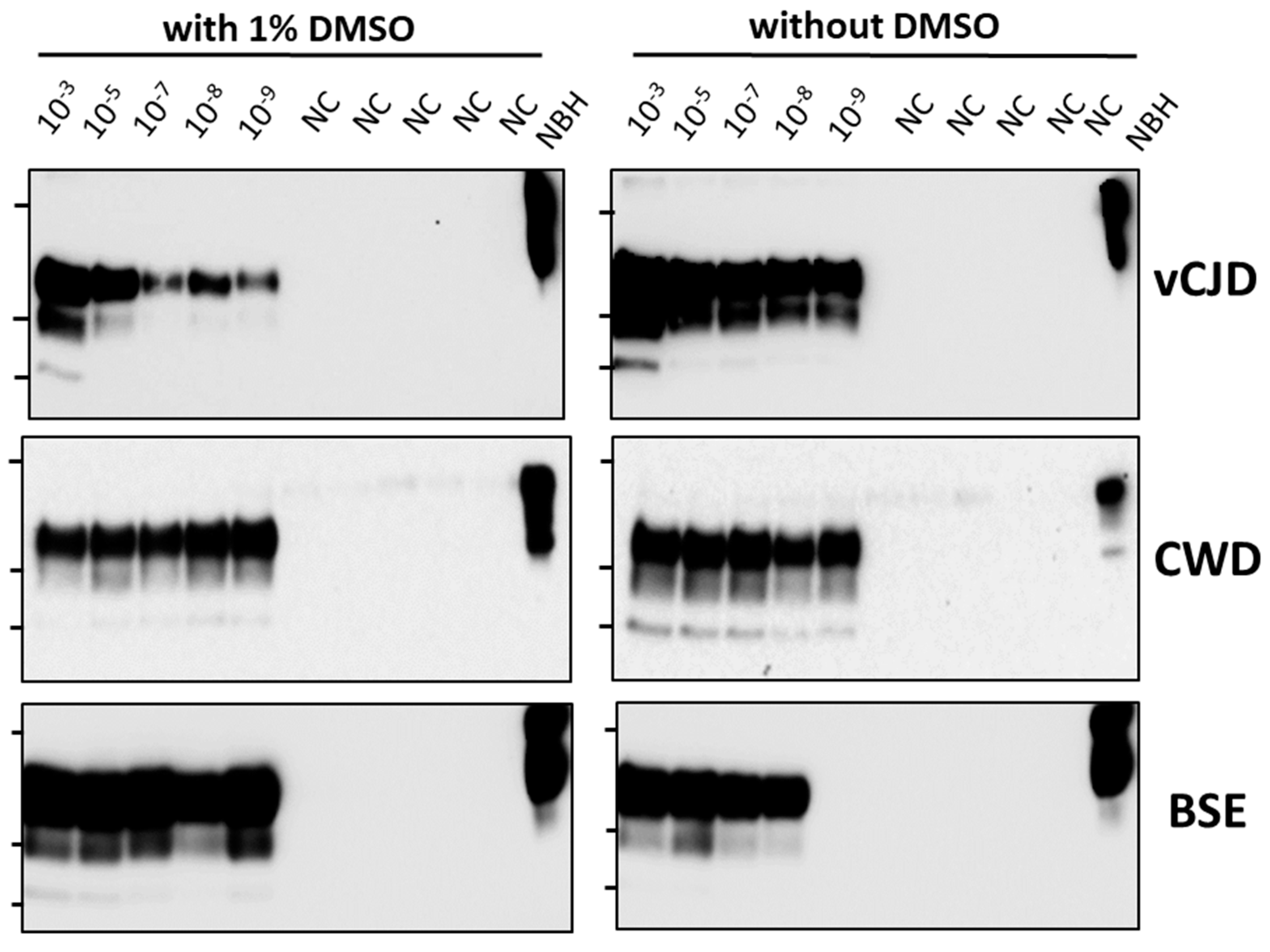
3.1. Screening of a Small Selection of Anti-Prion Compounds
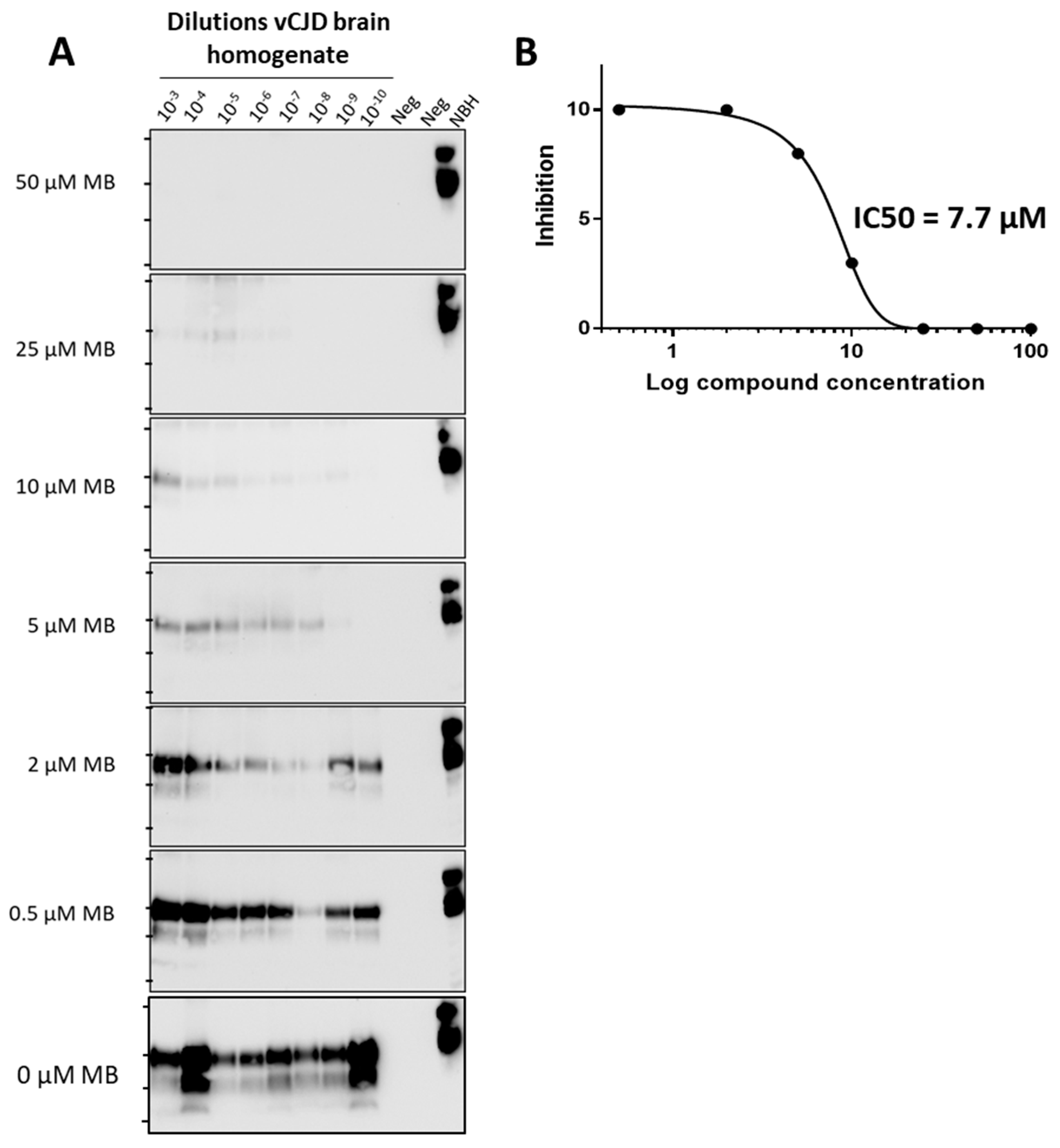
3.2. Estimation of IC50 for Compounds’ Activity
3.3. Initial Structure-Activity Relationship Studies
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pritzkow, S.; Gorski, D.; Ramirez, F.; Soto, C. Prion Dissemination through the Environment and Medical Practices: Facts and Risks for Human Health. Clin. Microbiol. Rev. 2021, 34, e0005919. [Google Scholar] [CrossRef]
- Prusiner, S.B. The prion diseases. Brain Pathol. 1998, 8, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Transmissible Proteins: Expanding the Prion Heresy. Cell 2012, 149, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 2021, 81, 4540–4551. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Telling, G.C. Insights into Mechanisms of Transmission and Pathogenesis from Transgenic Mouse Models of Prion Diseases. Methods Mol. Biol. 2017, 1658, 219–252. [Google Scholar] [CrossRef]
- Zafar, S.; Noor, A.; Zerr, I. Therapies for prion diseases. Handb. Clin. Neurol. 2019, 165, 47–58. [Google Scholar] [CrossRef]
- Zattoni, M.; Legname, G. Tackling prion diseases:A review of the patent landscape. Expert. Opin. Ther. Pat. 2021, 31, 1097–1115. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Morales, R.; Duran-Aniotz, C.; Diaz-Espinoza, R.; Camacho, M.V.; Soto, C. Protein misfolding cyclic amplification of infectious prions. Nat. Protoc. 2012, 7, 1397–1409. [Google Scholar] [CrossRef]
- Wang, F.; Pritzkow, S.; Soto, C. PMCA for ultrasensitive detection of prions and to study disease biology. Cell Tissue Res. 2023, 392, 307–321. [Google Scholar] [CrossRef]
- Telling, G. Protein-based PCR for prion diseases? Nat. Med. 2001, 7, 778–779. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Saá, P.; Hetz, C.; Soto, C. In vitro generation of infectious scrapie prions. Cell 2005, 121, 195–206. [Google Scholar] [CrossRef]
- Concha-Marambio, L.; Pritzkow, S.; Moda, F.; Tagliavini, F.; Ironside, J.; Schulz, P.; Soto, C. Detection of prions in blood from patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 2016, 8, 370ra183. [Google Scholar] [CrossRef]
- Castilla, J.; Saa, P.; Soto, C. Detection of prions in blood. Nat. Med. 2005, 11, 982–985. [Google Scholar] [CrossRef]
- Castilla, J.; Morales, R.; Saa, P.; Barria, M.; Gambetti, P.; Soto, C. Cell-free propagation of prion strains. EMBO J. 2008, 27, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Gonzalez-Romero, D.; Saa, P.; Morales, R.; De, C.J.; Soto, C. Crossing the species barrier by PrP (Sc) replication in vitro generates unique infectious prions. Cell 2008, 134, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Barria, M.A.; Mukherjee, A.; Gonzalez-Romero, D.; Morales, R.; Soto, C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog. 2009, 5, e1000421. [Google Scholar] [CrossRef]
- do Carmo Ferreira, N.; Caughey, B. Cell-free prion protein conversion assays in screening for anti-prion drug candidates. Curr. Opin. Pharmacol. 2019, 44, 1–7. [Google Scholar] [CrossRef]
- Manka, S.W.; Wenborn, A.; Collinge, J.; Wadsworth, J.D.F. Prion strains viewed through the lens of cryo-EM. Cell Tissue Res. 2023, 392, 167–178. [Google Scholar] [CrossRef]
- Terry, C.; Harniman, R.L.; Sells, J.; Wenborn, A.; Joiner, S.; Saibil, H.R.; Miles, M.J.; Collinge, J.; Wadsworth, J.D.F. Structural features distinguishing infectious ex vivo mammalian prions from non-infectious fibrillar assemblies generated in vitro. Sci. Rep. 2019, 9, 376. [Google Scholar] [CrossRef]
- Schmidt, C.; Fizet, J.; Properzi, F.; Batchelor, M.; Sandberg, M.K.; Edgeworth, J.A.; Afran, L.; Ho, S.; Badhan, A.; Klier, S.; et al. A systematic investigation of production of synthetic prions from recombinant prion protein. Open Biol. 2015, 5, 150165. [Google Scholar] [CrossRef] [PubMed]
- Barret, A.; Tagliavini, F.; Forloni, G.; Bate, C.; Salmona, M.; Colombo, L.; De Luigi, A.; Limido, L.; Suardi, S.; Rossi, G.; et al. Evaluation of quinacrine treatment for prion diseases. J. Virol. 2003, 77, 8462–8469. [Google Scholar] [CrossRef]
- Ghaemmaghami, S.; Ahn, M.; Lessard, P.; Giles, K.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009, 5, e1000673. [Google Scholar] [CrossRef]
- Berry, D.B.; Lu, D.; Geva, M.; Watts, J.C.; Bhardwaj, S.; Oehler, A.; Renslo, A.R.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Drug resistance confounding prion therapeutics. Proc. Natl. Acad. Sci. USA 2013, 110, E4160–E4169. [Google Scholar] [CrossRef]
- Caughey, B.; Race, R.E. Potent inhibition of scrapie-associated PrP accumulation by congo red. J. Neurochem. 1992, 59, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Martino, P.A.; Villa, S.; Carcassola, G.; Giannino, M.L.; Dall’Ara, P.; Pollera, C.; Iussich, S.; Tranquillo, V.M.; Bareggi, S.; et al. Evaluation of anti-prion activity of congo red and its derivatives in experimentally infected hamsters. Arzneimittelforschung 2004, 54, 406–415. [Google Scholar] [CrossRef]
- Rudyk, H.; Vasiljevic, S.; Hennion, R.M.; Birkett, C.R.; Hope, J.; Gilbert, I.H. Screening Congo Red and its analogues for their ability to prevent the formation of PrP-res in scrapie-infected cells. J. Gen. Virol. 2000, 81, 1155–1164. [Google Scholar] [CrossRef]
- Demaimay, R.; Harper, J.; Gordon, H.; Weaver, D.; Chesebro, B.; Caughey, B. Structural aspects of Congo red as an inhibitor of protease-resistant prion protein formation. J. Neurochem. 1998, 71, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Caspi, S.; Halimi, M.; Yanai, A.; Sasson, S.B.; Taraboulos, A.; Gabizon, R. The anti-prion activity of Congo red. Putative Mech. J. Biol. Chem. 1998, 273, 3484–3489. [Google Scholar] [CrossRef]
- Ryou, C.; Legname, G.; Peretz, D.; Craig, J.C.; Baldwin, M.A.; Prusiner, S.B. Differential inhibition of prion propagation by enantiomers of quinacrine. Lab. Investig. 2003, 83, 837–843. [Google Scholar] [CrossRef]
- Korth, C.; May, B.C.; Cohen, F.E.; Prusiner, S.B. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. USA 2001, 98, 9836–9841. [Google Scholar] [CrossRef]
- Collins, S.J.; Lewis, V.; Brazier, M.; Hill, A.F.; Fletcher, A.; Masters, C.L. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann. Neurol. 2002, 52, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, M.D.; Kuo, A.L.; Wong, K.S.; Haman, A.; Devereux, G.; Raudabaugh, B.J.; Johnson, D.Y.; Torres-Chae, C.C.; Finley, R.; Garcia, P.; et al. Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology 2013, 81, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Gorham, M.; Hudson, F.; Kennedy, A.; Keogh, G.; Pal, S.; Rossor, M.; Rudge, P.; Siddique, D.; Spyer, M.; et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study):A patient-preference trial. Lancet Neurol. 2009, 8, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, L.D.; Raymond, G.J.; Maxson, L.; Silveira, J.; Baron, G.S. Inhibition of protease-resistant prion protein accumulation in vitro by curcumin. J. Virol. 2003, 77, 5499–5502. [Google Scholar] [CrossRef] [PubMed]
- Kocisko, D.A.; Baron, G.S.; Rubenstein, R.; Chen, J.; Kuizon, S.; Caughey, B. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J. Virol. 2003, 77, 10288–10294. [Google Scholar] [CrossRef]
- Hyeon, J.W.; Kim, S.Y.; Lee, S.M.; Lee, J.; An, S.S.; Lee, M.K.; Lee, Y.S. Anti-Prion Screening for Acridine, Dextran, and Tannic Acid using Real Time-Quaking Induced Conversion: A Comparison with PrPSc-Infected Cell Screening. PLoS ONE 2017, 12, e0170266. [Google Scholar] [CrossRef]
- Cavaliere, P.; Torrent, J.; Prigent, S.; Granata, V.; Pauwels, K.; Pastore, A.; Rezaei, H.; Zagari, A. Binding of methylene blue to a surface cleft inhibits the oligomerization and fibrillization of prion protein. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 20–28. [Google Scholar] [CrossRef]
- Villa, V.; Thellung, S.; Bajetto, A.; Gatta, E.; Robello, M.; Novelli, F.; Tasso, B.; Tonelli, M.; Florio, T. Novel celecoxib analogues inhibit glial production of prostaglandin E2, nitric oxide, and oxygen radicals reverting the neuroinflammatory responses induced by misfolded prion protein fragment 90-231 or lipopolysaccharide. Pharmacol. Res. 2016, 113, 500–514. [Google Scholar] [CrossRef]
- Pickhardt, M.; Lawatscheck, C.; Borner, H.G.; Mandelkow, E. Inhibition of Tau Protein Aggregation by Rhodanine-based Compounds Solubilized Via Specific Formulation Additives to Improve Bioavailability and Cell Viability. Curr. Alzheimer Res. 2017, 14, 742–752. [Google Scholar] [CrossRef]
- Stincardini, C.; Massignan, T.; Biggi, S.; Elezgarai, S.R.; Sangiovanni, V.; Vanni, I.; Pancher, M.; Adami, V.; Moreno, J.; Stravalaci, M.; et al. An antipsychotic drug exerts anti-prion effects by altering the localization of the cellular prion protein. PLoS ONE 2017, 12, e0182589. [Google Scholar] [CrossRef] [PubMed]
- De, L.A.; Colombo, L.; Diomede, L.; Capobianco, R.; Mangieri, M.; Miccolo, C.; Limido, L.; Forloni, G.; Tagliavini, F.; Salmona, M. The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS ONE 2008, 3, e1888. [Google Scholar]
- Zerr, I. Investigating new treatments for Creutzfeldt-Jakob disease. Lancet Neurol. 2022, 21, 299–300. [Google Scholar] [CrossRef]
- Panegyres, P.K.; Armari, E. Therapies for human prion diseases. Am. J. Neurodegener. Dis. 2013, 2, 176–186. [Google Scholar] [PubMed]
- Charveriat, M.; Reboul, M.; Wang, Q.; Picoli, C.; Lenuzza, N.; Montagnac, A.; Nhiri, N.; Jacquet, E.; Gueritte, F.; Lallemand, J.Y.; et al. New inhibitors of prion replication that target the amyloid precursor. J. Gen. Virol. 2009, 90, 1294–1301. [Google Scholar] [CrossRef]
- Bertsch, U.; Winklhofer, K.F.; Hirschberger, T.; Bieschke, J.; Weber, P.; Hartl, F.U.; Tavan, P.; Tatzelt, J.; Kretzschmar, H.A.; Giese, A. Systematic identification of antiprion drugs by high-throughput screening based on scanning for intensely fluorescent targets. J. Virol. 2005, 79, 7785–7791. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pritzkow, S.; Schauer, I.; Tupaki-Sreepurna, A.; Morales, R.; Soto, C. Screening of Anti-Prion Compounds Using the Protein Misfolding Cyclic Amplification Technology. Biomolecules 2024, 14, 1113. https://doi.org/10.3390/biom14091113
Pritzkow S, Schauer I, Tupaki-Sreepurna A, Morales R, Soto C. Screening of Anti-Prion Compounds Using the Protein Misfolding Cyclic Amplification Technology. Biomolecules. 2024; 14(9):1113. https://doi.org/10.3390/biom14091113
Chicago/Turabian StylePritzkow, Sandra, Isaac Schauer, Ananya Tupaki-Sreepurna, Rodrigo Morales, and Claudio Soto. 2024. "Screening of Anti-Prion Compounds Using the Protein Misfolding Cyclic Amplification Technology" Biomolecules 14, no. 9: 1113. https://doi.org/10.3390/biom14091113