
AMPK Activation Serves as a Common Pro-Survival Pathway in Esophageal Adenocarcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Chemicals and Reagents
2.3. Western Blot
2.4. Seahorse Flux Analysis
2.5. siRNA Transfection
2.6. Quantitative Real Time PCR
2.7. Patient-Derived Organoids
2.8. Cytotoxicity Assay
2.9. Statistical Analysis
3. Results
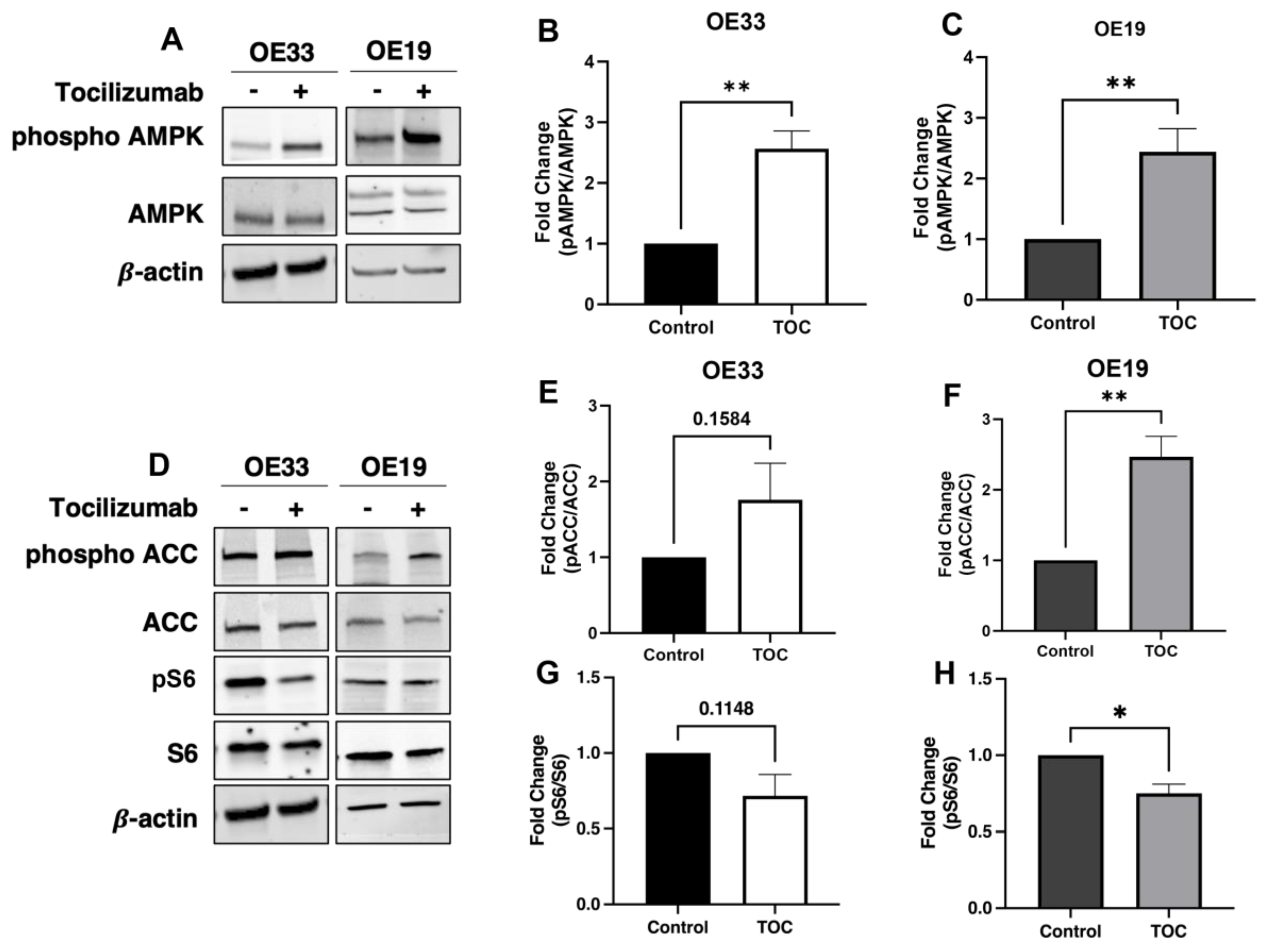
3.1. Il-6 Inhibition with Tocilizumab Activates AMPK and Downstream Targets
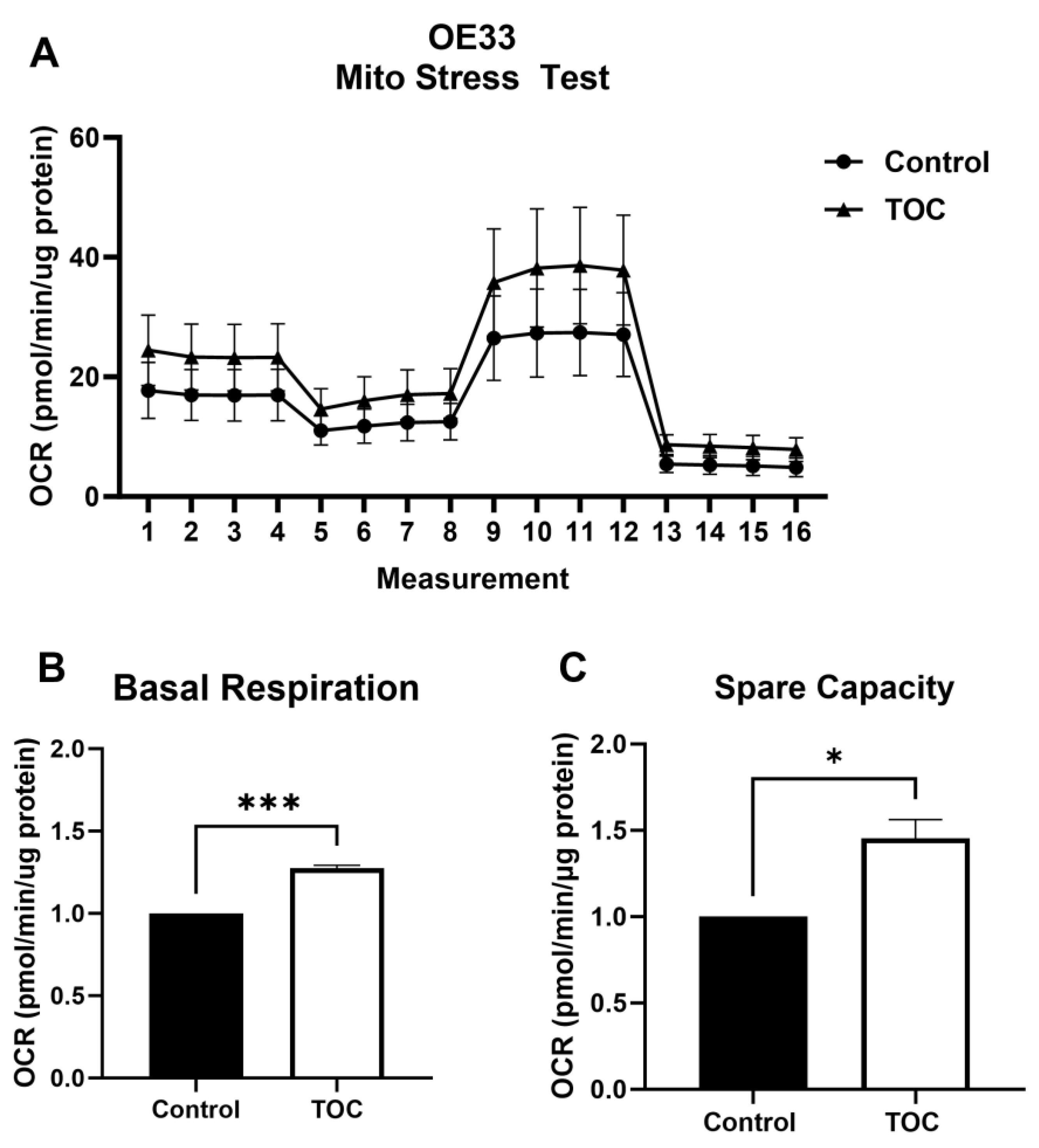
3.2. Il-6 Inhibition with Tocilizumab Increases Oxidative Metabolism
3.3. AMPK Knockdown Decreases Oxidative Metabolism in the Presence of Tocilizumab
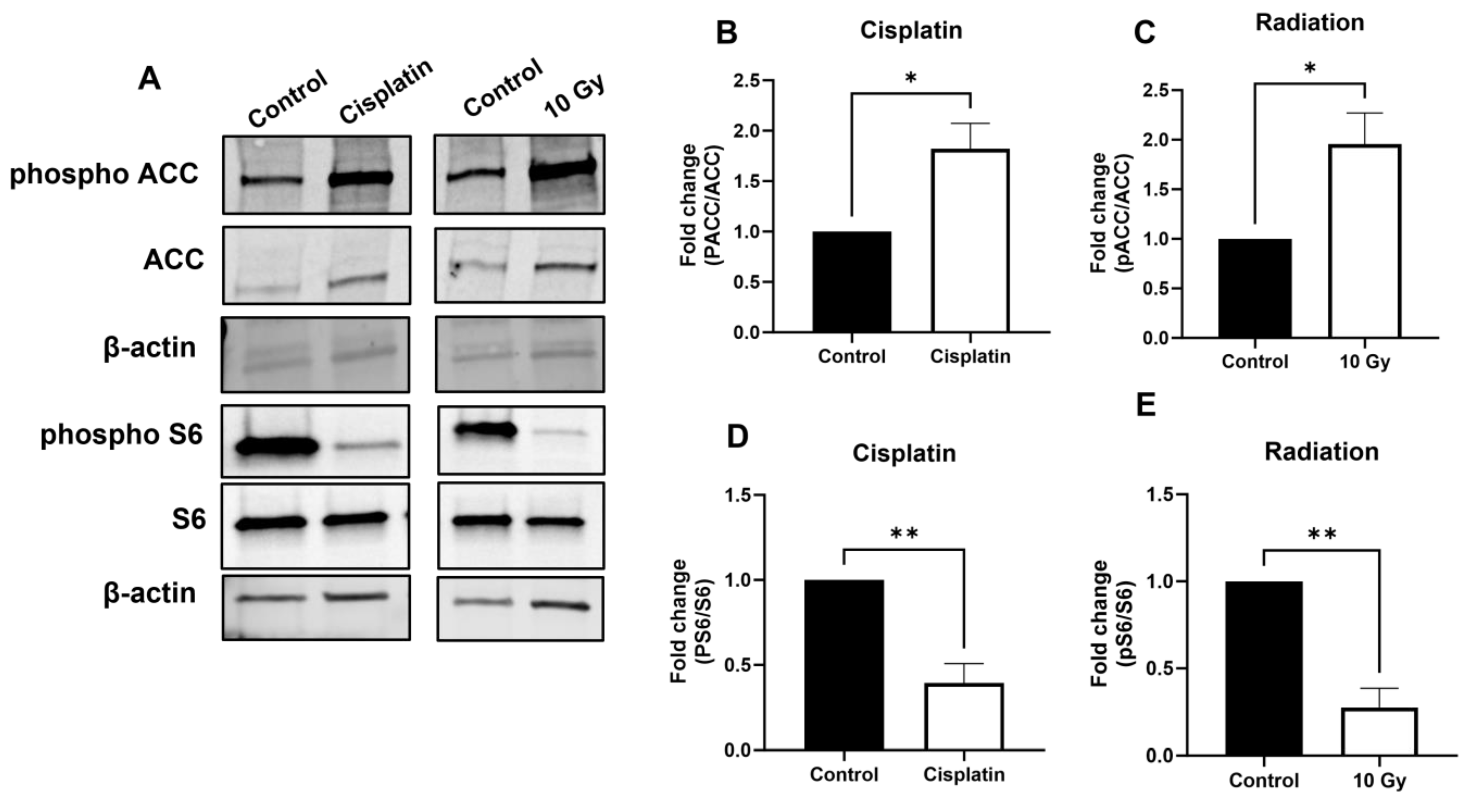
3.4. AMPK Signaling Is Activated upon Treatment with Chemotherapeutics and Radiation
3.5. AMPK Inhibition Promotes the Cytotoxic Effect of Chemotherapies and Radiation in a Synergistic Manner
3.6. Inhibition of AMPK Enhances Cytotoxic Effects of Chemotherapy and Radiation in Patient-Derived EAC Organoids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Penfold, L.; Woods, A.; Pollard, A.E.; Arizanova, J.; Pascual-Navarro, E.; Muckett, P.J.; Dore, M.H.; Montoya, A.; Whilding, C.; Fets, L.; et al. AMPK activation protects against prostate cancer by inducing a catabolic cellular state. Cell Rep. 2023, 42, 112396. [Google Scholar] [CrossRef] [PubMed]
- Vincent, E.E.; Coelho, P.P.; Blagih, J.; Griss, T.; Viollet, B.; Jones, R.G. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 2014, 34, 3627–3639. [Google Scholar] [CrossRef]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth In Vivo. Cell Metab. 2012, 17, 113–124. [Google Scholar] [CrossRef]
- Pan, Y.; Shao, D.; Zhao, Y.; Zhang, F.; Zheng, X.; Tan, Y.; He, K.; Li, J.; Chen, L. Berberine Reverses Hypoxia-induced Chemoresistance in Breast Cancer through the Inhibition of AMPK-HIF-1α. Int. J. Biol. Sci. 2017, 13, 794–803. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef]
- Quante, M.; Graham, T.A.; Jansen, M. Insights into the Pathophysiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 406–420. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef]
- Sheikh, M.; Roshandel, G.; McCormack, V.; Malekzadeh, R. Current Status and Future Prospects for Esophageal Cancer. Cancers 2023, 15, 765. [Google Scholar] [CrossRef]
- van Hagen, P.; Hulshof, M.C.C.M.; Van Lanschot, J.J.B.; Steyerberg, E.W.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.L.; Richel, D.J.; Nieuwenhuijzen, G.A.P.; Hospers, G.A.P.; Bonenkamp, J.J.; et al. Preoperative Chemoradiotherapy for Esophageal or Junctional Cancer. N. Engl. J. Med. 2012, 366, 2074–2084. [Google Scholar] [CrossRef]
- Li, S.; Hoefnagel, S.J.M.; Krishnadath, K.K. Molecular Biology and Clinical Management of Esophageal Adenocarcinoma. Cancers 2023, 15, 5410. [Google Scholar] [CrossRef] [PubMed]
- Hoefnagel, S.J.M.; Boonstra, J.J.; Russchen, M.J.A.M.; Krishnadath, K.K. Towards Personalized Treatment Strategies for Esophageal Adenocarcinoma; A Review on the Molecular Characterization of Esophageal Adenocarcinoma and Current Research Efforts on Individualized Curative Treatment Regimens. Cancers 2021, 13, 4881. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef] [PubMed]
- Karakasheva, T.A.; Kijima, T.; Shimonosono, M.; Maekawa, H.; Sahu, V.; Gabre, J.T.; Cruz-Acuña, R.; Giroux, V.; Sangwan, V.; Whelan, K.A.; et al. Generation and Characterization of Patient-Derived Head and Neck, Oral, and Esophageal Cancer Organoids. Curr. Protoc. Stem Cell Biol. 2020, 53, 109. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Gabre, J.T.; Sachdeva, U.M.; Cruz-Acuña, R.; Lin, E.W.; DeMarshall, M.; Falk, G.W.; Ginsberg, G.G.; Yang, Z.; Kim, M.M.; et al. Patient-derived organoids as a platform for modeling a patient’s response to chemoradiotherapy in esophageal cancer. Sci. Rep. 2021, 11, 21304. [Google Scholar] [CrossRef]
- Chandramouleeswaran, P.M.; Guha, M.; Shimonosono, M.; Whelan, K.A.; Maekawa, H.; Sachdeva, U.M.; Ruthel, G.; Mukherjee, S.; Engel, N.; Gonzalez, M.V.; et al. Autophagy mitigates ethanol-induced mitochondrial dysfunction and oxidative stress in esophageal keratinocytes. PLoS ONE 2020, 15, e0239625. [Google Scholar] [CrossRef]
- Lo, L.; Uchenunu, O.; Botelho, R.J.; Antonescu, C.N.; Karshafian, R. AMPK is required for recovery from metabolic stress induced by ultrasound microbubble treatment. iScience 2023, 26, 105883. [Google Scholar] [CrossRef] [PubMed]
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef]
- Zhao, H.; Zhuang, Y.; Li, R.; Liu, Y.; Mei, Z.; He, Z.; Zhou, F.; Zhou, Y. Effects of different doses of X-ray irradiation on cell apoptosis, cell cycle, DNA damage repair and glycolysis in HeLa cells. Oncol. Lett. 2019, 17, 42–54. [Google Scholar] [CrossRef]
- Han, F.; Li, C.-F.; Cai, Z.; Zhang, X.; Jin, G.; Zhang, W.-N.; Xu, C.; Wang, C.-Y.; Morrow, J.; Zhang, S.; et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat. Commun. 2018, 9, 4728. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, D.-L.; Zhou, M.; Zheng, Z.-S.; He, M.-F.; Huang, S.; Liao, X.-Z.; Zhang, J.-X. Cordycepin enhances the chemosensitivity of esophageal cancer cells to cisplatin by inducing the activation of AMPK and suppressing the AKT signaling pathway. Cell Death Dis. 2020, 11, 866. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, B.; Xu, W.W.; Chan, K.W.; Guan, X.Y.; Qin, Y.R.; Lee, N.P.Y.; Chan, K.T.; Law, S.; Tsao, S.W.; et al. Role of AMPK signaling in mediating the anticancer effects of silibinin in esophageal squamous cell carcinoma. Expert. Opin. Ther. Targets 2015, 20, 7–18. [Google Scholar] [CrossRef]
- He, Q.; Li, J.; Dong, F.; Cai, C.; Zou, X. LKB1 promotes radioresistance in esophageal cancer cells exposed to radiation, by suppression of apoptosis and activation of autophagy via the AMPK pathway. Mol. Med. Rep. 2017, 16, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Park, H.U.; Suy, S.; Danner, M.; Dailey, V.; Zhang, Y.; Li, H.; Hyduke, D.R.; Collins, B.T.; Gagnon, G.; Kallakury, B.; et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol. Cancer Ther. 2009, 8, 733–741. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McNamee, N.; Rajagopalan, P.; Tal-Mason, A.; Roytburd, S.; Sachdeva, U.M. AMPK Activation Serves as a Common Pro-Survival Pathway in Esophageal Adenocarcinoma Cells. Biomolecules 2024, 14, 1115. https://doi.org/10.3390/biom14091115
McNamee N, Rajagopalan P, Tal-Mason A, Roytburd S, Sachdeva UM. AMPK Activation Serves as a Common Pro-Survival Pathway in Esophageal Adenocarcinoma Cells. Biomolecules. 2024; 14(9):1115. https://doi.org/10.3390/biom14091115
Chicago/Turabian StyleMcNamee, Niamh, Pavithra Rajagopalan, Aya Tal-Mason, Samuel Roytburd, and Uma M. Sachdeva. 2024. "AMPK Activation Serves as a Common Pro-Survival Pathway in Esophageal Adenocarcinoma Cells" Biomolecules 14, no. 9: 1115. https://doi.org/10.3390/biom14091115
APA StyleMcNamee, N., Rajagopalan, P., Tal-Mason, A., Roytburd, S., & Sachdeva, U. M. (2024). AMPK Activation Serves as a Common Pro-Survival Pathway in Esophageal Adenocarcinoma Cells. Biomolecules, 14(9), 1115. https://doi.org/10.3390/biom14091115