
VDAC1-Based Peptides as Potential Modulators of VDAC1 Interactions with Its Partners and as a Therapeutic for Cancer, NASH, and Diabetes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Overview
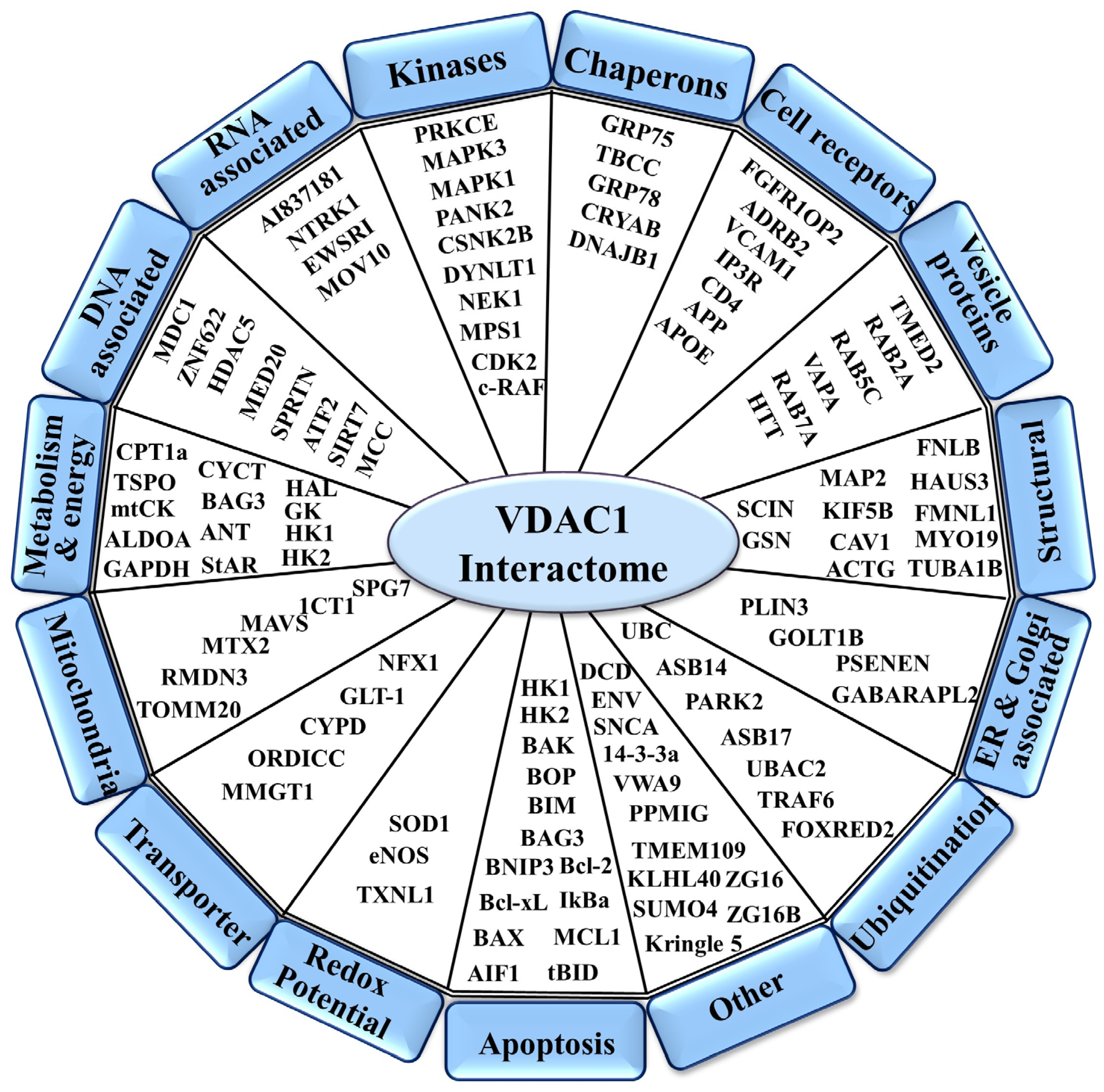
2. VDAC1 as a Hub Protein—Modulation of Its Interactome to Target Apoptosis, Metabolism, and Inflammation
2.1. VDAC1 Interaction with Metabolism-Related Proteins
2.2. VDAC1 Interaction with Apoptosis-Related Proteins
2.3. VDAC1 Interaction with Cytoskeletal Proteins
2.4. VDAC1 Interactions with Signaling Proteins
3. Development of Cell-Penetrating, Stable, and Effective VDAC1-Based Peptides
3.1. The VDAC1 N-Terminal-Derived Peptide
3.2. The VDAC1-Derived Peptide R-Tf-D-LP4
4. Proposed Mode of Action of VDAC1-Based Peptides
4.1. Impairment of Cell Metabolism and Energy Homeostasis
4.2. Inhibiting the Anti-Apoptotic Activities of HK, Bcl-2, and Bcl-xL and Promoting Apoptosis
4.3. Elimination of Cancer Stem Cells (CSCs)
4.4. Inducing Senescence
5. VDAC1-Based Peptides Targeting Diseases
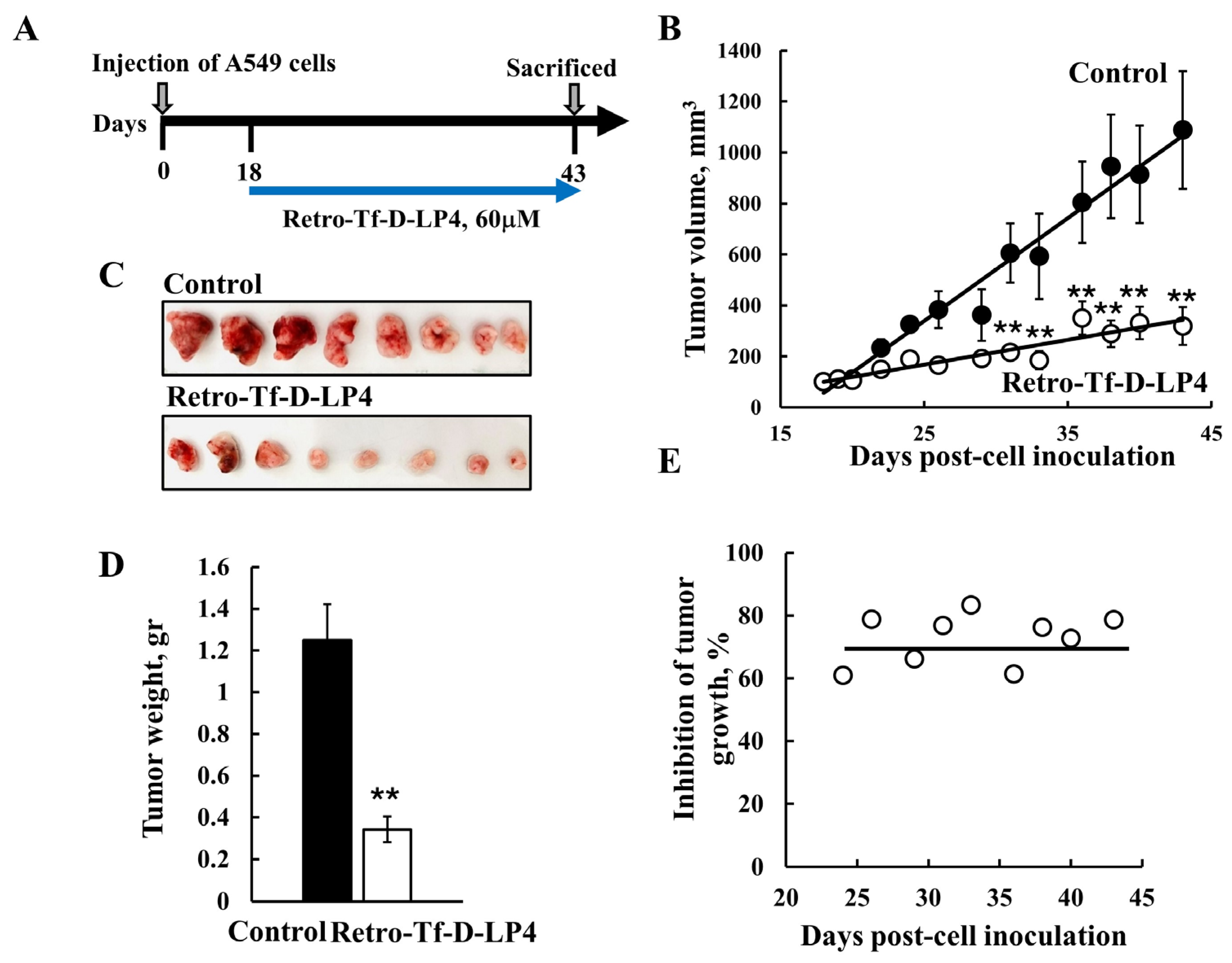
5.1. Targeting Cancer with VDAC1-Derived Peptides
5.2. Targeting NASH with the VDAC1-Derived Peptide R-Tf-D-LP4
5.3. Targeting Diabetes Mellitus with the VDAC1-Derived Peptide R-Tf-D-LP4
5.4. VDAC1-Based Peptide Targeting Levels of Testosterone
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Shteinfer-Kuzmine, A.; Sela, G.; Santhanam, M.; Gottschalk, B.; Boujemaa-Paterski, R.; Medalia, O.; Graier, W.F.; Shoshan-Barmatz, V. The Multicellular Effects of VDAC1 N-Terminal-Derived Peptide. Biomolecules 2022, 12, 1387. [Google Scholar] [CrossRef]
- Prezma, T.; Shteinfer, A.; Admoni, L.; Raviv, Z.; Sela, I.; Levi, I.; Shoshan-Barmatz, V. VDAC1-based peptides: Novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia. Cell Death Dis. 2013, 4, e809. [Google Scholar] [CrossRef] [PubMed]
- Shteinfer-Kuzmine, A.; Amsalem, Z.; Arif, T.; Zooravlov, A.; Shoshan-Barmatz, V. Selective induction of cancer cell death by VDAC1-based peptides and their potential use in cancer therapy. Mol. Oncol. 2018, 12, 1077–1103. [Google Scholar] [CrossRef] [PubMed]
- Shteinfer-Kuzmine, A.; Argueti, S.; Gupta, R.; Shvil, N.; Abu-Hamad, S.; Gropper, Y.; Hoeber, J.; Magri, A.; Messina, A.; Kozlova, E.N.; et al. A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS. Front. Cell Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef]
- Shteinfer-Kuzmine, A.; Arif, T.; Krelin, Y.; Tripathi, S.S.; Paul, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1-based peptides: Attacking oncogenic properties in glioblastoma. Oncotarget 2017, 8, 31329–31346. [Google Scholar] [CrossRef]
- Kuzmanov, U.; Emili, A. Protein-protein interaction networks: Probing disease mechanisms using model systems. Genome Med. 2013, 5, 37. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Petta, I.; Lievens, S.; Libert, C.; Tavernier, J.; De Bosscher, K. Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics. Mol. Ther. 2016, 24, 707–718. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef]
- Pittala, S.; Krelin, Y.; Kuperman, Y.; Shoshan-Barmatz, V. A Mitochondrial VDAC1-Based Peptide Greatly Suppresses Steatosis and NASH-Associated Pathologies in a Mouse Model. Mol. Ther. 2019, 27, 1848–1862. [Google Scholar] [CrossRef]
- Miyata, N.; Fujii, S.; Kuge, O. Porin proteins have critical functions in mitochondrial phospholipid metabolism in yeast. J. Biol. Chem. 2018, 293, 17593–17605. [Google Scholar] [CrossRef]
- Davis, J.; Meyer, T.; Smolnig, M.; Smethurst, D.G.J.; Neuhaus, L.; Heyden, J.; Broeskamp, F.; Edrich, E.S.M.; Knittelfelder, O.; Kolb, D.; et al. A dynamic actin cytoskeleton is required to prevent constitutive VDAC-dependent MAPK signalling and aberrant lipid homeostasis. iScience 2023, 26, 107539. [Google Scholar] [CrossRef]
- Arbel, N.; Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef]
- Arzoine, L.; Zilberberg, N.; Ben-Romano, R.; Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J. Biol. Chem. 2009, 284, 3946–3955. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Mizrachi, D.; Keinan, N. Oligomerization of the mitochondrial protein VDAC1: From structure to function and cancer therapy. Prog. Mol. Biol. Transl. Sci. 2013, 117, 303–334. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Kang, K.; Zhu, X.; Park, S.-J.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, X.; Eno, C.O.; Zhao, G.; Li, C.; White, C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J. Biol. Chem. 2013, 288, 19870–19881. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, J.; Weng, C.; Chen, R.; Zheng, Y.; Chen, Q.; Tang, H. Identification of the protein-protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem. Biophys. Res. Commun. 2003, 305, 989–996. [Google Scholar] [CrossRef]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef]
- Goldin, N.; Arzoine, L.; Heyfets, A.; Israelson, A.; Zaslavsky, Z.; Bravman, T.; Bronner, V.; Notcovich, A.; Shoshan-Barmatz, V.; Flescher, E. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene 2008, 27, 4636–4643. [Google Scholar] [CrossRef]
- Simamura, E.; Shimada, H.; Hatta, T.; Hirai, K.I. Mitochondrial voltage-dependent anion channels (VDACs) as novel pharmacological targets for anti-cancer agents. J. Bioenerg. Biomembr. 2008, 40, 213–217. [Google Scholar] [CrossRef]
- Pernemalm, M.; De Petris, L.; Branca, R.M.; Forshed, J.; Kanter, L.; Soria, J.C.; Girard, P.; Validire, P.; Pawitan, Y.; van den Oord, J.; et al. Quantitative proteomics profiling of primary lung adenocarcinoma tumors reveals functional perturbations in tumor metabolism. J. Proteome Res. 2013, 12, 3934–3943. [Google Scholar] [CrossRef] [PubMed]
- Vyssokikh, M.; Zorova, L.; Zorov, D.; Heimlich, G.; Jurgensmeier, J.; Schreiner, D.; Brdiczka, D. The intra-mitochondrial cytochrome c distribution varies correlated to the formation of a complex between VDAC and the adenine nucleotide translocase: This affects Bax-dependent cytochrome c release. Biochim. Biophys. Acta 2004, 1644, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.L.; van Rossum, D.B.; Kaplin, A.I.; Barrow, R.K.; Snyder, S.H. Inositol 1,4,5-trisphosphate receptor/GAPDH complex augments Ca2+ release via locally derived NADH. Proc. Natl. Acad. Sci. USA 2005, 102, 1357–1359. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 2006, 1762, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef]
- Neumann, D.; Buckers, J.; Kastrup, L.; Hell, S.W.; Jakobs, S. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys. 2010, 3, 4. [Google Scholar] [CrossRef]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Mathupala, S.; Rempel, A.; Geschwind, J.F.; Ko, Y.H. Mitochondrial bound type II hexokinase: A key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. Biophys. Acta 2002, 1555, 14–20. [Google Scholar] [CrossRef]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [PubMed]
- Pittala, S.; Krelin, Y.; Shoshan-Barmatz, V. Targeting Liver Cancer and Associated Pathologies in Mice with a Mitochondrial VDAC1-Based Peptide. Neoplasia 2018, 20, 594–609. [Google Scholar] [CrossRef]
- Fraser, F.; Corstorphine, C.G.; Zammit, V.A. Topology of carnitine palmitoyltransferase I in the mitochondrial outer membrane. Biochem. J. 1997, 323 Pt 3, 711–718. [Google Scholar] [CrossRef]
- Gustafsdottir, S.M.; Schallmeiner, E.; Fredriksson, S.; Gullberg, M.; Söderberg, O.; Jarvius, M.; Jarvius, J.; Howell, M.; Landegren, U. Proximity ligation assays for sensitive and specific protein analyses. Anal. Biochem. 2005, 345, 2–9. [Google Scholar] [CrossRef]
- Joo, H.K.; Lee, Y.R.; Lim, S.Y.; Lee, E.J.; Choi, S.; Cho, E.J.; Park, M.S.; Ryoo, S.; Park, J.B.; Jeon, B.H. Peripheral benzodiazepine receptor regulates vascular endothelial activations via suppression of the voltage-dependent anion channel-1. FEBS Lett. 2012, 586, 1349–1355. [Google Scholar] [CrossRef]
- Veenman, L.; Shandalov, Y.; Gavish, M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr. 2008, 40, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Azarashvili, T.; Krestinina, O.; Baburina, Y.; Odinokova, I.; Grachev, D.; Papadopoulos, V.; Akatov, V.; Lemasters, J.J.; Reiser, G. Combined effect of G3139 and TSPO ligands on Ca2+-induced permeability transition in rat brain mitochondria. Arch. Biochem. Biophys. 2015, 587, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Leschiner, S.; Spanier, I.; Weisinger, G.; Weizman, A.; Gavish, M. PK 11195 attenuates kainic acid-induced seizures and alterations in peripheral-type benzodiazepine receptor (PBR) protein components in the rat brain. J. Neurochem. 2002, 80, 917–927. [Google Scholar] [CrossRef]
- Veenman, L.; Papadopoulos, V.; Gavish, M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 2007, 13, 2385–2405. [Google Scholar] [CrossRef]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Ben-Hail, D.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012, 444, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Ide, T.; Yanagida, T.; Tsujimoto, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000, 275, 12321–12325. [Google Scholar] [CrossRef]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Yamagata, H.; Shimizu, S.; Nishida, Y.; Watanabe, Y.; Craigen, W.J.; Tsujimoto, Y. Requirement of voltage-dependent anion channel 2 for pro-apoptotic activity of Bax. Oncogene 2009, 28, 3563–3572. [Google Scholar] [CrossRef]
- Sugiyama, T.; Shimizu, S.; Matsuoka, Y.; Yoneda, Y.; Tsujimoto, Y. Activation of mitochondrial voltage-dependent anion channel by apro-apoptotic BH3-only protein Bim. Oncogene 2002, 21, 4944–4956. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. The voltage-dependent anion channel: An essential player in apoptosis. Biochimie 2002, 84, 187–193. [Google Scholar] [CrossRef]
- Huang, H.; Shah, K.; Bradbury, N.A.; Li, C.; White, C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 2014, 5, e1482. [Google Scholar] [CrossRef]
- Zhang, X.; Bian, X.; Kong, J. The proapoptotic protein BNIP3 interacts with VDAC to induce mitochondrial release of endonuclease G. PLoS ONE 2014, 9, e113642. [Google Scholar] [CrossRef]
- Takehara, T.; Liu, X.; Fujimoto, J.; Friedman, S.L.; Takahashi, H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001, 34, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Yang, X.; Pater, A.; Tang, S.C. Resistance to apoptosis is correlated with the reduced caspase-3 activation and enhanced expression of antiapoptotic proteins in human cervical multidrug-resistant cells. Biochem. Biophys. Res. Commun. 2000, 270, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Grobholz, R.; Zentgraf, H.; Kohrmann, K.U.; Bleyl, U. Bax, Bcl-2, fas and Fas-L antigen expression in human seminoma: Correlation with the apoptotic index. Apmis 2002, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Moss, S.F.; Krajewski, S.; Song, K.; Holt, P.R.; Reed, J.C. Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas. Cancer Res. 1996, 56, 2422–2427. [Google Scholar] [PubMed]
- Pedersen, P.L. Voltage dependent anion channels (VDACs): A brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J. Bioenerg. Biomembr. 2008, 40, 123–126. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the “Warburg effect” and a pivotal target for effective therapy. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2008. [Google Scholar]
- Kusano, H.; Shimizu, S.; Koya, R.C.; Fujita, H.; Kamada, S.; Kuzumaki, N.; Tsujimoto, Y. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene 2000, 19, 4807–4814. [Google Scholar] [CrossRef]
- Qiao, H.; McMillan, J.R. Gelsolin segment 5 inhibits HIV-induced T-cell apoptosis via Vpr-binding to VDAC. FEBS Lett. 2007, 581, 535–540. [Google Scholar] [CrossRef]
- Carre, M.; Andre, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef]
- Gurnev, P.A.; Rostovtseva, T.K.; Bezrukov, S.M. Tubulin-blocked state of VDAC studied by polymer and ATP partitioning. FEBS Lett. 2011, 585, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Puurand, M.; Tepp, K.; Timohhina, N.; Aid, J.; Shevchuk, I.; Chekulayev, V.; Kaambre, T. Tubulin beta II and beta III Isoforms as the Regulators of VDAC Channel Permeability in Health and Disease. Cells 2019, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Lemasters, J.J. Warburg revisited: Regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J. Pharmacol. Exp. Ther. 2012, 342, 637–641. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Bezrukov, S.M. VDAC inhibition by tubulin and its physiological implications. Biochim. Biophys. Acta 2012, 1818, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Saks, V.; Guzun, R.; Timohhina, N.; Tepp, K.; Varikmaa, M.; Monge, C.; Beraud, N.; Kaambre, T.; Kuznetsov, A.; Kadaja, L.; et al. Structure-function relationships in feedback regulation of energy fluxes in vivo in health and disease: Mitochondrial interactosome. Biochim. Biophys. Acta 2010, 1797, 678–697. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Patnaik, J.; Mullins, M.R.; Lemasters, J.J. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010, 70, 10192–10201. [Google Scholar] [CrossRef]
- Roman, I.; Figys, J.; Steurs, G.; Zizi, M. Direct measurement of VDAC-actin interaction by surface plasmon resonance. Biochim. Biophys. Acta 2006, 1758, 479–486. [Google Scholar] [CrossRef]
- Xu, X.; Forbes, J.G.; Colombini, M. Actin modulates the gating of Neurospora crassa VDAC. J. Membr. Biol. 2001, 180, 73–81. [Google Scholar] [CrossRef]
- Linden, M.; Karlsson, G. Identification of porin as a binding site for MAP2. Biochem. Biophys. Res. Commun. 1996, 218, 833–836. [Google Scholar] [CrossRef]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef]
- Viola, H.M.; Adams, A.M.; Davies, S.M.; Fletcher, S.; Filipovska, A.; Hool, L.C. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. USA 2014, 111, E2905–E2914. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef]
- Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brannstrom, T.; Rehnmark, A.; Marklund, S.L. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J. Neurosci. 2006, 26, 4147–4154. [Google Scholar] [CrossRef]
- Deng, H.X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.Y.; Bigio, E.H.; et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef]
- Tan, W.; Naniche, N.; Bogush, A.; Pedrini, S.; Trotti, D.; Pasinelli, P. Small peptides against the mutant SOD1/Bcl-2 toxic mitochondrial complex restore mitochondrial function and cell viability in mutant SOD1-mediated ALS. J. Neurosci. 2013, 33, 11588–11598. [Google Scholar] [CrossRef]
- Sun, J.; Liao, J.K. Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2002, 99, 13108–13113. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Guan, K.; Zheng, Z.; Song, T.; He, X.; Xu, C.; Zhang, Y.; Ma, S.; Wang, Y.; Xu, Q.; Cao, Y.; et al. MAVS regulates apoptotic cell death by decreasing K48-linked ubiquitination of voltage-dependent anion channel 1. Mol. Cell Biol. 2013, 33, 3137–3149. [Google Scholar] [CrossRef]
- Trishna, S.; Lavon, A.; Shteinfer-Kuzmine, A.; Dafa-Berger, A.; Shoshan-Barmatz, V. Overexpression of the mitochondrial anti-viral signaling protein, MAVS, in cancers is associated with cell survival and inflammation. Mol. Ther. Nucleic Acids 2023, 33, 713–732. [Google Scholar] [CrossRef]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The Voltage-dependent Anion Channel 1 Mediates Amyloid beta Toxicity and Represents a Potential Target for Alzheimer Disease Therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Smith, S.O. Mechanism of penetration of Antp(43-58) into membrane bilayers. Biochemistry 2005, 44, 10110–10118. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.K.; Subramaniam, S.; Senes, A. A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Calpha-H hydrogen bonds. Proc. Natl. Acad. Sci. USA 2014, 111, E888–E895. [Google Scholar] [CrossRef] [PubMed]
- Brooks, H.; Lebleu, B.; Vives, E. Tat peptide-mediated cellular delivery: Back to basics. Adv. Drug Deliv. Rev. 2005, 57, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Bernabeu, E.; Rodriguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta 2012, 1820, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Xie, Z.; Xie, C.; Zhan, C.; Hu, X.; Shen, Q.; Wei, X.; Su, B.; Wang, J.; et al. D-Peptides as Recognition Molecules and Therapeutic Agents. Chem. Rec. 2016, 16, 1772–1786. [Google Scholar] [CrossRef]
- Goodman, M.; Chorev, M. On the concept of linear modified retro-peptide structures. Acc. Chem. Res. 1979, 12, 1–7. [Google Scholar] [CrossRef]
- Chorev, M.; Goodman, M. Recent developments in retro peptides and proteins—An ongoing topochemical exploration. Trends Biotechnol. 1995, 13, 438–445. [Google Scholar] [CrossRef]
- Pittala, S.; Levy, I.; De, S.; Kumar Pandey, S.; Melnikov, N.; Hyman, T.; Shoshan-Barmatz, V. The VDAC1-based R-Tf-D-LP4 Peptide as a Potential Treatment for Diabetes Mellitus. Cells 2020, 9, 481. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Thompson, C.B. Q&A: Cancer: Clues from cell metabolism. Nature 2010, 465, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef]
- Huang, L.; Han, J.; Ben-Hail, D.; He, L.; Li, B.; Chen, Z.; Wang, Y.; Yang, Y.; Liu, L.; Zhu, Y.; et al. A New Fungal Diterpene Induces VDAC1-dependent Apoptosis in Bax/Bak-deficient Cells. J. Biol. Chem. 2015, 290, 23563–23578. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Pahima, H.; Ben-Hail, D.; Shoshan-Barmatz, V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2013, 1833, 1745–1754. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Chaptal, V.; Ping, P.; Abramson, J. Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation. Channels 2009, 3, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Weisthal, S.; Keinan, N.; Ben-Hail, D.; Arif, T.; Shoshan-Barmatz, V. Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta 2014, 1843, 2270–2281. [Google Scholar] [CrossRef]
- Zalk, R.; Israelson, A.; Garty, E.S.; Azoulay-Zohar, H.; Shoshan-Barmatz, V. Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J. 2005, 386, 73–83. [Google Scholar] [CrossRef]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef]
- Lee, J.T.; Herlyn, M. Old disease, new culprit: Tumor stem cells in cancer. J. Cell. Physiol. 2007, 213, 603–609. [Google Scholar] [CrossRef]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Kissel, K.; Hamm, S.; Schulz, M.; Vecchi, A.; Garlanda, C.; Engelhardt, B. Immunohistochemical localization of the murine transferrin receptor (TfR) on blood-tissue barriers using a novel anti-TfR monoclonal antibody. Histochem. Cell Biol. 1998, 110, 63–72. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Leite, N.C.; Salles, G.F.; Araujo, A.L.; Villela-Nogueira, C.A.; Cardoso, C.R. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009, 29, 113–119. [Google Scholar] [CrossRef]
- Lomonaco, R.; Sunny, N.E.; Bril, F.; Cusi, K. Nonalcoholic fatty liver disease: Current issues and novel treatment approaches. Drugs 2013, 73, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Law, K.; Brunt, E.M. Nonalcoholic fatty liver disease. Clin. Liver Dis. 2010, 14, 591–604. [Google Scholar] [CrossRef]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. S1), S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Kharroubi, A.T.; Darwish, H.M. Diabetes mellitus: The epidemic of the century. World J. Diabetes 2015, 6, 850–867. [Google Scholar] [CrossRef] [PubMed]
- Galtier, F. Definition, epidemiology, risk factors. Diabetes Metab. 2010, 36, 628–651. [Google Scholar] [CrossRef] [PubMed]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. beta-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Manco, M. Insulin Resistance and NAFLD: A Dangerous Liaison beyond the Genetics. Children 2017, 4, 74. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Baulies, A.; Mari, M.; Garcia-Roves, P.M.; Fernandez-Checa, J.C. Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance: Cause or consequence? Free Radic. Res. 2013, 47, 854–868. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. StAR search—What we know about how the steroidogenic acute regulatory protein mediates mitochondrial cholesterol import. Mol. Endocrinol. 2007, 21, 589–601. [Google Scholar] [CrossRef]
- Bose, M.; Whittal, R.M.; Miller, W.L.; Bose, H.S. Steroidogenic activity of StAR requires contact with mitochondrial VDAC1 and phosphate carrier protein. J. Biol. Chem. 2008, 283, 8837–8845. [Google Scholar] [CrossRef]
- Hauet, T.; Yao, Z.X.; Bose, H.S.; Wall, C.T.; Han, Z.; Li, W.; Hales, D.B.; Miller, W.L.; Culty, M.; Papadopoulos, V. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol. Endocrinol. 2005, 19, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, Y.; Martinez-Arguelles, D.B.; Fan, J.; Culty, M.; Papadopoulos, V. Induction of androgen formation in the male by a TAT-VDAC1 fusion peptide blocking 14-3-3varepsilon protein adaptor and mitochondrial VDAC1 interactions. Mol. Ther. 2014, 22, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arguelles, D.B.; Nedow, J.W.; Gukasyan, H.J.; Papadopoulos, V. Oral administration of VDAC1-derived small molecule peptides increases circulating testosterone levels in male rats. Front. Endocrinol. 2022, 13, 1003017. [Google Scholar] [CrossRef]
- Smith, M.C.; Gestwicki, J.E. Features of protein-protein interactions that translate into potent inhibitors: Topology, surface area and affinity. Expert. Rev. Mol. Med. 2012, 14, e16. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, U. Novel cell-penetrating peptide targeting mitochondria. FASEB J. 2015, 29, 4589–4599. [Google Scholar] [CrossRef]
- Puckett, C.A.; Barton, J.K. Targeting a ruthenium complex to the nucleus with short peptides. Bioorg. Med. Chem. 2010, 18, 3564–3569. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Santhanam, M.; Pandey, S.K.; Shteinfer-Kuzmine, A.; Paul, A.; Abusiam, N.; Zalk, R.; Shoshan-Barmatz, V. Interaction of SMAC with a survivin-derived peptide alters essential cancer hallmarks: Tumor growth, inflammation, and immunosuppression. Mol. Ther. 2024, 32, 1934–1955. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shteinfer-Kuzmine, A.; Santhanam, M.; Shoshan-Barmatz, V. VDAC1-Based Peptides as Potential Modulators of VDAC1 Interactions with Its Partners and as a Therapeutic for Cancer, NASH, and Diabetes. Biomolecules 2024, 14, 1139. https://doi.org/10.3390/biom14091139
Shteinfer-Kuzmine A, Santhanam M, Shoshan-Barmatz V. VDAC1-Based Peptides as Potential Modulators of VDAC1 Interactions with Its Partners and as a Therapeutic for Cancer, NASH, and Diabetes. Biomolecules. 2024; 14(9):1139. https://doi.org/10.3390/biom14091139
Chicago/Turabian StyleShteinfer-Kuzmine, Anna, Manikandan Santhanam, and Varda Shoshan-Barmatz. 2024. "VDAC1-Based Peptides as Potential Modulators of VDAC1 Interactions with Its Partners and as a Therapeutic for Cancer, NASH, and Diabetes" Biomolecules 14, no. 9: 1139. https://doi.org/10.3390/biom14091139