
Extracellular Matrix Components and Mechanosensing Pathways in Health and Disease


 ,
,  , and
, and 
Abstract
:1. Introduction
2. The Interplay between the ECM and Its Specific Component, the Cellular Glycocalyx (GCX)
2.1. Collagens
2.2. Elastin
2.3. PGs
2.3.1. Syndecans and Glypicans
2.3.2. Pericellular PGs
2.4. GAGs
2.5. Glycoproteins
2.6. Cross-Linking Molecules
2.7. The ECM as a Unit
3. Several Critical Mechanical Properties of the ECM Contribute to Its Functionality
3.1. Stiffness/Elasticity
3.2. Viscoelasticity
3.3. Strength and Toughness
3.4. Anisotropy
3.5. Adhesive Properties
3.6. Remodeling and Plasticity
4. Receptors Involved in the Process of Mechanotransduction
4.1. Integrins
4.2. Cadherins
4.3. PIEZO and TRP
4.4. G Protein-Coupled Receptors (GPCRs)
5. Mechanisms of Mechanosensing
5.1. Overview of Mechanosensitive Pathways in Cells
5.2. Role of GAGs and PGs in Mechanotransduction
5.2.1. PGs and Cells
5.2.2. PGs and Cadherin Mechanotransduction Properties
5.2.3. PG Cell–ECM Interactions
5.2.4. PGs Affect the Response to Shear Stress Exerted by Blood Fluid on Endothelial Cells
6. GAG/PG-Mediated Mechanosensing in Cancer
6.1. Tumor Microenvironment and ECM Remodeling
6.2. Dysregulated Mechanosensing in Cancer Progression
6.2.1. ECM Stiffness and Desmoplasia
6.2.2. Fiber Alignment
6.2.3. Cancer Cell Movement
6.2.4. Elasticity, Viscoelasticity, and Plasticity
6.3. Impact of GAGs and PGs on Cancer Cell Behavior
6.3.1. Syndecans
6.3.2. Agrin
6.3.3. Serglycin
6.3.4. Small Leucine-Rich PGs (SLRPs)
6.3.5. Hyaluronan (HA)
6.3.6. Other Chondroitin Sulfate PGs (CSPGs)
7. GAG/PG-Mediated Mechanosensing in Inflammation and Fibrosis
7.1. ECM Remodeling during Inflammation
7.2. GAG/PG Involvement in Immune Cell Recruitment and Activation
7.3. Contribution to Inflammatory Signaling Pathways
7.4. Role in Tissue Repair, Fibrosis, and Resolution of Inflammation
8. Therapeutic Targeting of GAG/PG-Mediated Mechanosensing
8.1. Current Approaches and Challenges in Therapeutic Interventions
8.2. Potential Impact on Cancer, Inflammation, and Other Diseases
9. Future Perspectives
9.1. Emerging Research Directions and Unanswered Questions
9.2. Technological Advancements Enabling Further Understanding of GAG/PG-Mediated Mechanosensing
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABS | actin-binding site |
| AFM | atomic force microscopy |
| AGE | advanced glycosylation end product |
| AI | artificial intelligence |
| Arf6 | ADP-ribosylation factor 6 |
| ARP | actin-related protein |
| BBB | blood–brain barrier |
| CD44 | cluster of differentiation 44 |
| CDH | cadherin |
| CLS | cord-like structures |
| CREB | cAMP response element-binding protein |
| CS | chondroitin sulfate |
| CSPGs | chondroitin sulfate proteoglycans |
| CXCL | chemokine (C-X-C motif) ligand |
| Da | Daltons |
| DAMP | damage-associated molecular pattern |
| DCCM | directional collective cell migration |
| DDR | discoidin domain receptor |
| DS | dermatan sulfate |
| ECM | extracellular matrix |
| eNOS | endothelial nitric oxide synthase |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| FAK | focal adhesion kinase |
| FAT domain | focal adhesion targeting domain |
| FERM | F for protein 4.1, E for ezrin, R for radixin, and M for moesin |
| FSS | fluid shear stress |
| GAGs | glycosaminoglycans |
| Gal | galactose |
| GBM | glioblastoma multiforme |
| GCX | cellular glycocalyx |
| GPCR | G protein-coupled receptors |
| H1Rs | histamine H1 receptors |
| HA | hyaluronan |
| HCC | hepatocarcinoma |
| Hep | heparin |
| HS | heparan sulfate |
| HSPG | heparan sulfate proteoglycan |
| HSCs | hepatic stellate cells |
| IGF-IR | insulin growth factor I receptor |
| IL | interleukin |
| KLF | Krüppel-like factor |
| KS | keratan sulfate |
| LOX | lysyl oxidase |
| LOXLs | lysyl oxidase-like proteins |
| LYVE-1 | lymphatic vessel endothelial hyaluronan receptor 1 |
| MAPK | mitogen-activated protein kinase |
| MMPs | matrix metalloproteinases |
| MT1-MMP | membrane type I matrix metalloproteinase |
| NF-κB | nuclear factor κB |
| NRP | N-rich protein |
| PAMP | pathogen-associated molecular pattern |
| PECAM | platelet and endothelial cell adhesion molecule |
| PGs | proteoglycans |
| PI3K | phosphatidylinositol 3-kinase |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PRELP | proline/arginine-rich end and leucine-rich protein |
| RHAMM | hyaluronan-mediated motility receptor |
| ROCK | Rho-associated protein kinase |
| SLRP | small leucine-rich proteoglycan |
| TAZ | transcriptional co-activator with PDZ-binding motif |
| TEAD | transcriptional enhanced associate domain |
| TGFβ | transforming growth factor beta |
| THD | talin head domain |
| TLRs | Toll-like receptors |
| TNBC | triple-negative breast cancer |
| TNF | tumor necrosis factor |
| TRP | transient receptor potential family |
| TRPV4 | transient receptor potential cation channel, subfamily V, member 4 |
| TSG-6 | tumor necrosis factor-stimulated gene 6 |
| VBSs | vinculin-binding sites |
| VEGF | vascular endothelial growth factor |
| WISPs | WNT1-inducible signaling pathway proteins |
| YAP | yes-associated protein |
References
- Ingber, D.E. Cellular Mechanotransduction: Putting All the Pieces Together Again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Iskratsch, T.; Wolfenson, H.; Sheetz, M.P. Appreciating Force and Shape—The Rise of Mechanotransduction in Cell Biology. Nat. Rev. Mol. Cell Biol. 2014, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Inman, A.; Smutny, M. Feeling the Force: Multiscale Force Sensing and Transduction at the Cell-Cell Interface. Semin. Cell Dev. Biol. 2021, 120, 53–65. [Google Scholar] [CrossRef] [PubMed]
- DuFort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing Forces: Architectural Control of Mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. From Tensegrity to Human Organs-on-Chips: Implications for Mechanobiology and Mechanotherapeutics. Biochem. J. 2023, 480, 243–257. [Google Scholar] [CrossRef]
- Moore, K.H.; Murphy, H.A.; George, E.M. The Glycocalyx: A Central Regulator of Vascular Function. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2021, 320, R508–R518. [Google Scholar] [CrossRef]
- The Machinery of Life|SpringerLink. Available online: https://link.springer.com/book/10.1007/978-0-387-84925-6 (accessed on 4 August 2024).
- Zhou, C.J.; Guo, Y. Mini Review on Collagens in Normal Skin and Pathological Scars: Current Understanding and Future Perspective. Front. Med. 2024, 11, 1449597. [Google Scholar] [CrossRef]
- Wang, K.; Meng, X.; Guo, Z. Elastin Structure, Synthesis, Regulatory Mechanism and Relationship with Cardiovascular Diseases. Front. Cell Dev. Biol. 2021, 9, 596702. [Google Scholar] [CrossRef]
- Couchman, J.R.; Pataki, C.A. An Introduction to Proteoglycans and Their Localization. J. Histochem. Cytochem. 2012, 60, 885–897. [Google Scholar] [CrossRef]
- Fox, C.M. The Role of Heparan Sulfate Proteoglycans during Development of the Zebrafish Lateral Line. Ph.D. Thesis, Johns Hopkins University, Baltimore, MD, USA, 2016. [Google Scholar]
- Shriver, Z.; Capila, I.; Venkataraman, G.; Sasisekharan, R. Heparin and Heparan Sulfate: Analyzing Structure and Microheterogeneity. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 159–176. [Google Scholar] [CrossRef]
- Funderburgh, J.L. Keratan Sulfate Biosynthesis. IUBMB Life 2002, 54, 187–194. [Google Scholar] [CrossRef]
- Lindahl, U.; Couchman, J.; Kimata, K.; Esko, J.D. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Xu, S.; Xu, X.; Wu, R. Deciphering the Properties and Functions of Glycoproteins Using Quantitative Proteomics. J. Proteome Res. 2023, 22, 1571–1588. [Google Scholar] [CrossRef] [PubMed]
- Quantifying Collagen Fibre Architecture in Articular Cartilage Using Small-Angle X-Ra Scattering—IOS Press. Available online: https://content.iospress.com/articles/biomedical-spectroscopy-and-imaging/bsi164 (accessed on 26 July 2024).
- Hulmes, D.J.S. Collagen Diversity, Synthesis and Assembly. In Collagen: Structure and Mechanics; Fratzl, P., Ed.; Springer: Boston, MA, USA, 2008; pp. 15–47. ISBN 978-0-387-73906-9. [Google Scholar]
- CHAPTER 2: Osmotic Properties of Cartilage. Available online: https://www.researchgate.net/publication/312062035_CHAPTER_2_Osmotic_Properties_of_Cartilage (accessed on 26 July 2024).
- Muiznieks, L.D.; Keeley, F.W. Molecular Assembly and Mechanical Properties of the Extracellular Matrix: A Fibrous Protein Perspective. Biochim. Biophys. Acta 2013, 1832, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Lujan, T.J.; Underwood, C.J.; Jacobs, N.T.; Weiss, J.A. Contribution of Glycosaminoglycans to Viscoelastic Tensile Behavior of Human Ligament. J. Appl. Physiol. 2009, 106, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Readioff, R.; Geraghty, B.; Kharaz, Y.A.; Elsheikh, A.; Comerford, E. Proteoglycans Play a Role in the Viscoelastic Behaviour of the Canine Cranial Cruciate Ligament. Front. Bioeng. Biotechnol. 2022, 10, 984224. [Google Scholar] [CrossRef] [PubMed]
- Vidal, C.D.M.P.; Leme-Kraus, A.A.; Rahman, M.; Farina, A.P.; Bedran-Russo, A.K. Role of Proteoglycans on the Biochemical and Biomechanical Properties of Dentin Organic Matrix. Arch. Oral Biol. 2017, 82, 203–208. [Google Scholar] [CrossRef]
- Halper, J.; Kjaer, M. Basic components of connective tissues and extracellular matrix: Elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv Exp Med Biol. 2014, 802, 31–47. [Google Scholar] [CrossRef]
- Xie, W.; Wei, X.; Kang, H.; Jiang, H.; Chu, Z.; Lin, Y.; Hou, Y.; Wei, Q. Static and Dynamic: Evolving Biomaterial Mechanical Properties to Control Cellular Mechanotransduction. Adv. Sci. 2023, 10, e2204594. [Google Scholar] [CrossRef]
- Cieśluk, M.; Pogoda, K.; Piktel, E.; Wnorowska, U.; Deptuła, P.; Bucki, R. Mechanical Properties of the Extracellular Environment of Human Brain Cells Drive the Effectiveness of Drugs in Fighting Central Nervous System Cancers. Brain Sci. 2022, 12, 927. [Google Scholar] [CrossRef]
- Vittum, Z.; Cocchiaro, S.; Mensah, S.A. Basal Endothelial Glycocalyx’s Response to Shear Stress: A Review of Structure, Function, and Clinical Implications. Front. Cell Dev. Biol. 2024, 12, 1371769. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Kadry, Y.A.; Calderwood, D.A. Chapter 22: Structural and Signaling Functions of Integrins. Biochim. Biophys. Acta-Biomembranes 2020, 1862, 183206. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-Mediated Mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical Regulation of a Molecular Clutch Defines Force Transmission and Transduction in Response to Matrix Rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Campbell, I.D.; Critchley, D.R. Talins and Kindlins: Partners in Integrin-Mediated Adhesion. Nat. Rev. Mol. Cell Biol. 2013, 14, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Tseng, H.-Y.; Tan, S.; Senger, F.; Kurzawa, L.; Dedden, D.; Mizuno, N.; Wasik, A.A.; Thery, M.; Dunn, A.R.; et al. Kank2 Activates Talin, Reduces Force Transduction across Integrins and Induces Central Adhesion Formation. Nat. Cell Biol. 2016, 18, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Deng, Y.; Sun, K.; Yang, H.; Liu, J.; Wang, M.; Zhang, Z.; Lin, J.; Wu, C.; Wei, Z.; et al. Structural Basis of Kindlin-Mediated Integrin Recognition and Activation. Proc. Natl. Acad. Sci. USA 2017, 114, 9349–9354. [Google Scholar] [CrossRef] [PubMed]
- Bledzka, K.; Liu, J.; Xu, Z.; Perera, H.D.; Yadav, S.P.; Bialkowska, K.; Qin, J.; Ma, Y.-Q.; Plow, E.F. Spatial Coordination of Kindlin-2 with Talin Head Domain in Interaction with Integrin β Cytoplasmic Tails. J. Biol. Chem. 2012, 287, 24585–24594. [Google Scholar] [CrossRef]
- Niessen, C.M.; Gottardi, C.J. Molecular Components of the Adherens Junction. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 562–571. [Google Scholar] [CrossRef]
- Leckband, D.E.; de Rooij, J. Cadherin Adhesion and Mechanotransduction. Annu. Rev. Cell Dev. Biol. 2014, 30, 291–315. [Google Scholar] [CrossRef]
- Shapiro, L.; Weis, W.I. Structure and Biochemistry of Cadherins and Catenins. Cold Spring Harb. Perspect. Biol. 2009, 1, a003053. [Google Scholar] [CrossRef]
- Yap, A.S.; Kovacs, E.M. Direct Cadherin-Activated Cell Signaling: A View from the Plasma Membrane. J. Cell Biol. 2002, 160, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Sarpal, R.; Ishiyama, N.; Pellikka, M.; Ikura, M.; Tepass, U. Monomeric α-Catenin Links Cadherin to the Actin Cytoskeleton. Nat. Cell Biol. 2013, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, S. A Mechanism of Mechanotransduction at the Cell-Cell Interface. BioEssays 2011, 33, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Tang, V.W.; Brieher, W.M. α-Actinin-4/FSGS1 Is Required for Arp2/3-Dependent Actin Assembly at the Adherens Junction. J. Cell Biol. 2012, 196, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef]
- Piezo1 Channels as Force Sensors in Mechanical Force-Related Chronic Inflammation—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/35154133/ (accessed on 27 July 2024).
- Nilius, B.; Szallasi, A. Transient Receptor Potential Channels as Drug Targets: From the Science of Basic Research to the Art of Medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef]
- Ludwig, M.-G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-Sensing G-Protein-Coupled Receptors. Nature 2003, 425, 93–98. [Google Scholar] [CrossRef]
- Erdogmus, S.; Storch, U.; Danner, L.; Becker, J.; Winter, M.; Ziegler, N.; Wirth, A.; Offermanns, S.; Hoffmann, C.; Gudermann, T.; et al. Helix 8 Is the Essential Structural Motif of Mechanosensitive GPCRs. Nat. Commun. 2019, 10, 5784. [Google Scholar] [CrossRef]
- Geiger, B.; Yamada, K.M. Molecular Architecture and Function of Matrix Adhesions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005033. [Google Scholar] [CrossRef]
- Burridge, K. Focal Adhesions: A Personal Perspective on a Half Century of Progress. FEBS J. 2017, 284, 3355–3361. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Calderwood, D.A. Organization, dynamics and mechanoregulation of integrin-mediated cell-ECM adhesions. Nat. Rev. Mol. Cell Biol. 2023, 24, 142–161. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and Their Regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Klapholz, B.; Brown, N.H. Talin—The Master of Integrin Adhesions. J. Cell Sci. 2017, 130, 2435–2446. [Google Scholar] [CrossRef] [PubMed]
- Lietha, D.; Cai, X.; Ceccarelli, D.F.J.; Li, Y.; Schaller, M.D.; Eck, M.J. Structural Basis for the Autoinhibition of Focal Adhesion Kinase. Cell 2007, 129, 1177–1187. [Google Scholar] [CrossRef]
- Humphries, J.D.; Paul, N.R.; Humphries, M.J.; Morgan, M.R. Emerging Properties of Adhesion Complexes: What Are They and What Do They Do? Trends Cell Biol. 2015, 25, 388–397. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional Atlas of the Integrin Adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in Cancer: Mechanistic Findings and Clinical Applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Lundin, V.; Sugden, W.W.; Theodore, L.N.; Sousa, P.M.; Han, A.; Chou, S.; Wrighton, P.J.; Cox, A.G.; Ingber, D.E.; Goessling, W.; et al. YAP Regulates Hematopoietic Stem Cell Formation in Response to the Biomechanical Forces of Blood Flow. Dev. Cell 2020, 52, 446–460.e5. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ Drives Fibroblast Activation and Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef]
- Kahle, E.R.; Han, B.; Chandrasekaran, P.; Phillips, E.R.; Mulcahey, M.K.; Lu, X.L.; Marcolongo, M.S.; Han, L. Molecular Engineering of Pericellular Microniche via Biomimetic Proteoglycans Modulates Cell Mechanobiology. ACS Nano 2022, 16, 1220–1230. [Google Scholar] [CrossRef]
- Möckl, L. The Emerging Role of the Mammalian Glycocalyx in Functional Membrane Organization and Immune System Regulation. Front. Cell Dev. Biol. 2020, 8, 253. [Google Scholar] [CrossRef] [PubMed]
- Henrich-Noack, P.; Nikitovic, D.; Neagu, M.; Docea, A.O.; Engin, A.B.; Gelperina, S.; Shtilman, M.; Mitsias, P.; Tzanakakis, G.; Gozes, I.; et al. The Blood–Brain Barrier and beyond: Nano-Based Neuropharmacology and the Role of Extracellular Matrix. Nanomed. Nanotechnol. Biol. Med. 2019, 17, 359–379. [Google Scholar] [CrossRef] [PubMed]
- Afratis, N.A.; Nikitovic, D.; Multhaupt, H.A.B.; Theocharis, A.D.; Couchman, J.R.; Karamanos, N.K. Syndecans—Key Regulators of Cell Signaling and Biological Functions. FEBS J. 2017, 284, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, A.N.; Multhaupt, H.A.B.; Couchman, J.R. Syndecans in Wound Healing, Inflammation and Vascular Biology. Int. J. Biochem. Cell Biol. 2007, 39, 505–528. [Google Scholar] [CrossRef]
- Gopal, S.; Arokiasamy, S.; Pataki, C.; Whiteford, J.R.; Couchman, J.R. Syndecan Receptors: Pericellular Regulators in Development and Inflammatory Disease. Open Biol. 2021, 11, 200377. [Google Scholar] [CrossRef]
- Lekka, M.; Herman, K.; Zemła, J.; Bodek, Ł.; Pyka-Fościak, G.; Gil, D.; Dulińska-Litewka, J.; Ptak, A.; Laidler, P. Probing the Recognition Specificity of αVβ1 Integrin and Syndecan-4 Using Force Spectroscopy. Micron 2020, 137, 102888. [Google Scholar] [CrossRef]
- Kennelly, T.M.; Li, Y.; Cao, Y.; Qwarnstrom, E.E.; Geoghegan, M. Distinct Binding Interactions of A5β1-Integrin and Proteoglycans with Fibronectin. Biophys. J. 2019, 117, 688–695. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V.; Róg, T.; Lee, D.A.; Hytönen, V.P.; Del Río Hernández, A.E. Syndecan-4 Tunes Cell Mechanics by Activating the Kindlin-Integrin-RhoA Pathway. Nat. Mater. 2020, 19, 669–678. [Google Scholar] [CrossRef]
- Burgos-Bravo, F.; Martínez-Meza, S.; Quest, A.F.G.; Wilson, C.A.M.; Leyton, L. Application of Force to a Syndecan-4 Containing Complex with Thy-1-αVβ3 Integrin Accelerates Neurite Retraction. Front. Mol. Biosci. 2020, 7, 582257. [Google Scholar] [CrossRef]
- Romaine, A.; Melleby, A.O.; Alam, J.; Lobert, V.H.; Lu, N.; Lockwood, F.E.; Hasic, A.; Lunde, I.G.; Sjaastad, I.; Stenmark, H.; et al. Integrin A11β1 and Syndecan-4 Dual Receptor Ablation Attenuate Cardiac Hypertrophy in the Pressure Overloaded Heart. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H1057–H1071. [Google Scholar] [CrossRef]
- Morgan, M.R.; Hamidi, H.; Bass, M.D.; Warwood, S.; Ballestrem, C.; Humphries, M.J. Syndecan-4 Phosphorylation Is a Control Point for Integrin Recycling. Dev. Cell 2013, 24, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Karimi, F.; Thombare, V.J.; Hutton, C.A.; O’Connor, A.J.; Qiao, G.G.; Heath, D.E. Biomaterials Functionalized with Nanoclusters of Integrin- and Syndecan-Binding Ligands Improve Cell Adhesion and Mechanosensing under Shear Flow Conditions. J. Biomed. Mater. Res. Part A 2021, 109, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Sharma, U.; Kasuba, K.C.; Strohmeyer, N.; Müller, D.J. Engineered Biomimetic Fibrillar Fibronectin Matrices Regulate Cell Adhesion Initiation, Migration, and Proliferation via A5β1 Integrin and Syndecan-4 Crosstalk. Adv. Sci. 2023, 10, e2300812. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.M.; Rapraeger, A.C. Syndecan-1-Mediated Cell Spreading Requires Signaling by Avβ3 Integrins in Human Breast Carcinoma Cells. Exp. Cell Res. 2003, 286, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Leavitt, L.; Ramaswamy, R.; Rapraeger, A.C. Interaction of Syndecan and A6β4 Integrin Cytoplasmic Domains. J. Biol. Chem 2010, 285, 13569–13579. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Panitch, A. Proteoglycans and Proteoglycan Mimetics for Tissue Engineering. Am. J. Physiol. Cell Physiol. 2022, 322, C754–C761. [Google Scholar] [CrossRef]
- Pannekoek, W.-J.; de Rooij, J.; Gloerich, M. Force Transduction by Cadherin Adhesions in Morphogenesis. F1000Research 2019, 8, 1044. [Google Scholar] [CrossRef]
- Wang, A.; Dunn, A.R.; Weis, W.I. Mechanism of the Cadherin-Catenin F-Actin Catch Bond Interaction. Elife 2022, 11, e80130. [Google Scholar] [CrossRef]
- Buckley, C.D.; Tan, J.; Anderson, K.L.; Hanein, D.; Volkmann, N.; Weis, W.I.; Nelson, W.J.; Dunn, A.R. The Minimal Cadherin-Catenin Complex Binds to Actin Filaments under Force. Science 2014, 346, 1254211. [Google Scholar] [CrossRef]
- Maruthamuthu, V.; Sabass, B.; Schwarz, U.S.; Gardel, M.L. Cell-ECM Traction Force Modulates Endogenous Tension at Cell-Cell Contacts. Proc. Natl. Acad. Sci. USA 2011, 108, 4708–4713. [Google Scholar] [CrossRef]
- Zuidema, A.; Wang, W.; Sonnenberg, A. Crosstalk between Cell Adhesion Complexes in Regulation of Mechanotransduction. Bioessays 2020, 42, e2000119. [Google Scholar] [CrossRef] [PubMed]
- Plutoni, C.; Bazellieres, E.; Le Borgne-Rochet, M.; Comunale, F.; Brugues, A.; Séveno, M.; Planchon, D.; Thuault, S.; Morin, N.; Bodin, S.; et al. P-Cadherin Promotes Collective Cell Migration via a Cdc42-Mediated Increase in Mechanical Forces. J. Cell Biol. 2016, 212, 199–217. [Google Scholar] [CrossRef] [PubMed]
- Bazellières, E.; Conte, V.; Elosegui-Artola, A.; Serra-Picamal, X.; Bintanel-Morcillo, M.; Roca-Cusachs, P.; Muñoz, J.J.; Sales-Pardo, M.; Guimerà, R.; Trepat, X. Control of Cell-Cell Forces and Collective Cell Dynamics by the Intercellular Adhesome. Nat. Cell Biol. 2015, 17, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne-Rochet, M.; Angevin, L.; Bazellières, E.; Ordas, L.; Comunale, F.; Denisov, E.V.; Tashireva, L.A.; Perelmuter, V.M.; Bièche, I.; Vacher, S.; et al. P-Cadherin-Induced Decorin Secretion Is Required for Collagen Fiber Alignment and Directional Collective Cell Migration. J. Cell Sci. 2019, 132, jcs233189. [Google Scholar] [CrossRef] [PubMed]
- Barua, D.; Nagel, M.; Winklbauer, R. Cell-Cell Contact Landscapes in Xenopus Gastrula Tissues. Proc. Natl. Acad. Sci. USA 2021, 118, e2107953118. [Google Scholar] [CrossRef]
- Davaapil, H.; Hopkins, J.; Bonnin, N.; Papadaki, V.; Leung, A.; Kosuge, H.; Tashima, T.; Nakakido, M.; Sekido, R.; Tsumoto, K.; et al. PRELP Secreted from Mural Cells Protects the Function of Blood Brain Barrier through Regulation of Endothelial Cell-Cell Integrity. Front. Cell Dev. Biol. 2023, 11, 1147625. [Google Scholar] [CrossRef]
- Krakowski, P.; Rejniak, A.; Sobczyk, J.; Karpiński, R. Cartilage Integrity: A Review of Mechanical and Frictional Properties and Repair Approaches in Osteoarthritis. Healthcare 2024, 12, 1648. [Google Scholar] [CrossRef]
- Ateshian, G.A. The Role of Interstitial Fluid Pressurization in Articular Cartilage Lubrication. J. Biomech. 2009, 42, 1163–1176. [Google Scholar] [CrossRef]
- Reye, G.; Huang, X.; Haupt, L.M.; Murphy, R.J.; Northey, J.J.; Thompson, E.W.; Momot, K.I.; Hugo, H.J. Mechanical Pressure Driving Proteoglycan Expression in Mammographic Density: A Self-Perpetuating Cycle? J. Mammary Gland Biol. Neoplasia 2021, 26, 277–296. [Google Scholar] [CrossRef]
- McConnell, J.C.; O’Connell, O.V.; Brennan, K.; Weiping, L.; Howe, M.; Joseph, L.; Knight, D.; O’Cualain, R.; Lim, Y.; Leek, A.; et al. Increased Peri-Ductal Collagen Micro-Organization May Contribute to Raised Mammographic Density. Breast Cancer Res. 2016, 18, 5. [Google Scholar] [CrossRef]
- Vuoriluoto, K.; Jokinen, J.; Kallio, K.; Salmivirta, M.; Heino, J.; Ivaska, J. Syndecan-1 Supports Integrin A2β1-Mediated Adhesion to Collagen. Exp. Cell Res. 2008, 314, 3369–3381. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Friedl, A. Syndecan-1-Induced ECM Fiber Alignment Requires Integrin Avβ3 and Syndecan-1 Ectodomain and Heparan Sulfate Chains. PLoS ONE 2016, 11, e0150132. [Google Scholar] [CrossRef] [PubMed]
- Northey, J.J.; Barrett, A.S.; Acerbi, I.; Hayward, M.-K.; Talamantes, S.; Dean, I.S.; Mouw, J.K.; Ponik, S.M.; Lakins, J.N.; Huang, P.-J.; et al. Stiff Stroma Increases Breast Cancer Risk by Inducing the Oncogene ZNF217. J. Clin. Investig. 2020, 130, 5721–5737. [Google Scholar] [CrossRef] [PubMed]
- Han, Y. Analysis of the Role of the Hippo Pathway in Cancer. J. Transl. Med. 2019, 17, 116. [Google Scholar] [CrossRef]
- Meinhardt, G.; Haider, S.; Kunihs, V.; Saleh, L.; Pollheimer, J.; Fiala, C.; Hetey, S.; Feher, Z.; Szilagyi, A.; Than, N.G.; et al. Pivotal Role of the Transcriptional Co-Activator YAP in Trophoblast Stemness of the Developing Human Placenta. Proc. Natl. Acad. Sci. USA 2020, 117, 13562–13570. [Google Scholar] [CrossRef]
- Lee, W.; Leddy, H.A.; Chen, Y.; Lee, S.H.; Zelenski, N.A.; McNulty, A.L.; Wu, J.; Beicker, K.N.; Coles, J.; Zauscher, S.; et al. Synergy between Piezo1 and Piezo2 Channels Confers High-Strain Mechanosensitivity to Articular Cartilage. Proc. Natl. Acad. Sci. USA 2014, 111, E5114–E5122. [Google Scholar] [CrossRef]
- Alcaide-Ruggiero, L.; Cugat, R.; Domínguez, J.M. Proteoglycans in Articular Cartilage and Their Contribution to Chondral Injury and Repair Mechanisms. Int. J. Mol. Sci. 2023, 24, 10824. [Google Scholar] [CrossRef]
- Savadipour, A.; Nims, R.J.; Rashidi, N.; Garcia-Castorena, J.M.; Tang, R.; Marushack, G.K.; Oswald, S.J.; Liedtke, W.B.; Guilak, F. Membrane Stretch as the Mechanism of Activation of PIEZO1 Ion Channels in Chondrocytes. Proc. Natl. Acad. Sci. USA 2023, 120, e2221958120. [Google Scholar] [CrossRef]
- Di, X.; Gao, X.; Peng, L.; Ai, J.; Jin, X.; Qi, S.; Li, H.; Wang, K.; Luo, D. Cellular Mechanotransduction in Health and Diseases: From Molecular Mechanism to Therapeutic Targets. Signal. Transduct. Target. Ther. 2023, 8, 282. [Google Scholar] [CrossRef]
- Kutikhin, A.G.; Sinitsky, M.Y.; Yuzhalin, A.E.; Velikanova, E.A. Shear Stress: An Essential Driver of Endothelial Progenitor Cells. J. Mol. Cell. Cardiol. 2018, 118, 46–69. [Google Scholar] [CrossRef]
- Askari, H.; Sadeghinejad, M.; Fancher, I.S. Chapter Three—Mechanotransduction and the Endothelial Glycocalyx: Interactions with Membrane and Cytoskeletal Proteins to Transduce Force. In Current Topics in Membranes; Fancher, I.S., Chignalia, A.Z., Eds.; The Cardiovascular Glycocalyx in Health and Disease; Academic Press: Cambridge, MA, USA, 2023; Volume 91, pp. 43–60. [Google Scholar]
- Zeng, Y. Endothelial Glycocalyx as a Critical Signalling Platform Integrating the Extracellular Haemodynamic Forces and Chemical Signalling. J. Cell. Mol. Med. 2017, 21, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Le, V.; Mei, L.; Voyvodic, P.L.; Zhao, C.; Busch, D.J.; Stachowiak, J.C.; Baker, A.B. Molecular Tension in Syndecan-1 Is Regulated by Extracellular Mechanical Cues and Fluidic Shear Stress. Biomaterials 2021, 275, 120947. [Google Scholar] [CrossRef] [PubMed]
- Voyvodic, P.L.; Min, D.; Liu, R.; Williams, E.; Chitalia, V.; Dunn, A.K.; Baker, A.B. Loss of Syndecan-1 Induces a pro-Inflammatory Phenotype in Endothelial Cells with a Dysregulated Response to Atheroprotective Flow. J. Biol. Chem. 2014, 289, 9547–9559. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, H.; Beauvais, D.M.; Rapraeger, A.C. Cytoplasmic Domain Interactions of Syndecan-1 and Syndecan-4 with A6β4 Integrin Mediate Human Epidermal Growth Factor Receptor (HER1 and HER2)-Dependent Motility and Survival. J. Biol. Chem. 2014, 289, 30318–30332. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.Z.; Luo, K.H.; Ventikos, Y. Principal Mode of Syndecan-4 Mechanotransduction for the Endothelial Glycocalyx Is a Scissor-like Dimer Motion. Acta Physiol. 2020, 228, e13376. [Google Scholar] [CrossRef]
- Guilluy, C.; Dolega, M.E. Syndecan-4 forces integrins to cooperate. Nat. Mater. 2020, 19, 587–588. [Google Scholar] [CrossRef]
- Foolen, J.; Janssen-van den Broek, M.W.J.T.; Baaijens, F.P.T. Synergy between Rho Signaling and Matrix Density in Cyclic Stretch-Induced Stress Fiber Organization. Acta Biomater. 2014, 10, 1876–1885. [Google Scholar] [CrossRef]
- Elfenbein, A.; Rhodes, J.M.; Meller, J.; Schwartz, M.A.; Matsuda, M.; Simons, M. Suppression of RhoG Activity Is Mediated by a Syndecan 4-Synectin-RhoGDI1 Complex and Is Reversed by PKCα in a Rac1 Activation Pathway. J. Cell Biol. 2009, 186, 75–83. [Google Scholar] [CrossRef]
- Baeyens, N.; Mulligan-Kehoe, M.J.; Corti, F.; Simon, D.D.; Ross, T.D.; Rhodes, J.M.; Wang, T.Z.; Mejean, C.O.; Simons, M.; Humphrey, J.; et al. Syndecan 4 Is Required for Endothelial Alignment in Flow and Atheroprotective Signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 17308–17313. [Google Scholar] [CrossRef]
- Dong, C.; Choi, Y.K.; Lee, J.; Zhang, X.F.; Honerkamp-Smith, A.; Widmalm, G.; Lowe-Krentz, L.J.; Im, W. Structure, Dynamics, and Interactions of GPI-Anchored Human Glypican-1 with Heparan Sulfates in a Membrane. Glycobiology 2021, 31, 593–602. [Google Scholar] [CrossRef]
- Bartosch, A.M.W.; Mathews, R.; Mahmoud, M.M.; Cancel, L.M.; Haq, Z.S.; Tarbell, J.M. Heparan Sulfate Proteoglycan Glypican-1 and PECAM-1 Cooperate in Shear-Induced Endothelial Nitric Oxide Production. Sci. Rep. 2021, 11, 11386. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Mayer, M.; Cancel, L.M.; Bartosch, A.M.; Mathews, R.; Tarbell, J.M. The Glycocalyx Core Protein Glypican 1 Protects Vessel Wall Endothelial Cells from Stiffness-Mediated Dysfunction and Disease. Cardiovasc. Res. 2021, 117, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Baghy, K.; Ladányi, A.; Reszegi, A.; Kovalszky, I. Insights into the Tumor Microenvironment-Components, Functions and Therapeutics. Int. J. Mol. Sci. 2023, 24, 17536. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Berdiaki, A.; Giatagana, E.-M.; Tzanakakis, G.; Nikitovic, D. The Landscape of Small Leucine-Rich Proteoglycan Impact on Cancer Pathogenesis with a Focus on Biglycan and Lumican. Cancers 2023, 15, 3549. [Google Scholar] [CrossRef]
- Sorensen, H.T.; Friis, S.; Olsen, J.H.; Thulstrup, A.M.; Mellemkjaer, L.; Linet, M.; Trichopoulos, D.; Vilstrup, H.; Olsen, J. Risk of Liver and Other Types of Cancer in Patients with Cirrhosis: A Nationwide Cohort Study in Denmark. Hepatology 1998, 28, 921–925. [Google Scholar] [CrossRef]
- Morris, B.A.; Burkel, B.; Ponik, S.M.; Fan, J.; Condeelis, J.S.; Aguirre-Ghiso, J.A.; Castracane, J.; Denu, J.M.; Keely, P.J. Collagen Matrix Density Drives the Metabolic Shift in Breast Cancer Cells. EBioMedicine 2016, 13, 146–156. [Google Scholar] [CrossRef]
- Schaefer, L.; Schaefer, R.M. Proteoglycans: From Structural Compounds to Signaling Molecules. Cell Tissue Res. 2010, 339, 237–246. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan Form and Function: A Comprehensive Nomenclature of Proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Neagu, M.; Tsatsakis, A.; Nikitovic, D. Proteoglycans and Immunobiology of Cancer—Therapeutic Implications. Front. Immunol. 2019, 10, 875. [Google Scholar] [CrossRef]
- Nikitovic, D.; Berdiaki, A.; Spyridaki, I.; Krasanakis, T.; Tsatsakis, A.; Tzanakakis, G.N. Proteoglycans-Biomarkers and Targets in Cancer Therapy. Front. Endocrinol. 2018, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Yip, G.W.; Smollich, M.; Götte, M. Therapeutic Value of Glycosaminoglycans in Cancer. Mol. Cancer Ther. 2006, 5, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Wieboldt, R.; Läubli, H. Glycosaminoglycans in Cancer Therapy. Am. J. Physiol.-Cell Physiol. 2022, 322, C1187–C1200. [Google Scholar] [CrossRef] [PubMed]
- Vigetti, D.; Karousou, E.; Viola, M.; Deleonibus, S.; De Luca, G.; Passi, A. HA: Biosynthesis and Signaling. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, C.; Ween, M.P.; Lokman, N.A.; Tan, I.A.; Pyragius, C.E.; Oehler, M.K. Chemotherapy-Induced HA Production: A Novel Chemoresistance Mechanism in Ovarian Cancer. BMC Cancer 2013, 13, 476. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Chiodoni, C.; Tripodo, C.; Colombo, M.P. The Good and Bad of Targeting Cancer-Associated Extracellular Matrix. Curr. Opin. Pharmacol. 2017, 35, 75–82. [Google Scholar] [CrossRef]
- Long, Y.; Niu, Y.; Liang, K.; Du, Y. Mechanical Communication in Fibrosis Progression. Trends Cell Biol. 2022, 32, 70–90. [Google Scholar] [CrossRef]
- Burgess, J.K.; Mauad, T.; Tjin, G.; Karlsson, J.C.; Westergren-Thorsson, G. The Extracellular Matrix—The under-Recognized Element in Lung Disease? J. Pathol. 2016, 240, 397–409. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Singh, A.P.; Moniaux, N.; Senapati, S.; Chakraborty, S.; Meza, J.L.; Batra, S.K. MUC4 Mucin Potentiates Pancreatic Tumor Cell Proliferation, Survival, and Invasive Properties and Interferes with Its Interaction to Extracellular Matrix Proteins. Mol. Cancer Res. 2007, 5, 309–320. [Google Scholar] [CrossRef]
- Purushothaman, A.; Mohajeri, M.; Lele, T.P. The Role of Glycans in the Mechanobiology of Cancer. J. Biol. Chem. 2023, 299, 102935. [Google Scholar] [CrossRef]
- Suresh, S. Biomechanics and Biophysics of Cancer Cells. Acta Biomater. 2007, 3, 413–438. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Weaver, V.M. Mechanics, Malignancy, and Metastasis: The Force Journey of a Tumor Cell. Cancer Metastasis Rev. 2009, 28, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The Physics of Cancer: The Role of Physical Interactions and Mechanical Forces in Metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.I.; Kang, I.; You, W.-K.; McDonald, D.M.; Weaver, V.M. In Situ Force Mapping of Mammary Gland Transformation. Integr. Biol. 2011, 3, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Torrino, S.; Grasset, E.M.; Audebert, S.; Belhadj, I.; Lacoux, C.; Haynes, M.; Pisano, S.; Abélanet, S.; Brau, F.; Chan, S.Y.; et al. Mechano-Induced Cell Metabolism Promotes Microtubule Glutamylation to Force Metastasis. Cell Metab. 2021, 33, 1342–1357.e10. [Google Scholar] [CrossRef]
- Moeendarbary, E.; Harris, A.R. Cell Mechanics: Principles, Practices, and Prospects. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 6, 371–388. [Google Scholar] [CrossRef]
- Malandrino, A.; Kamm, R.D.; Moeendarbary, E. In Vitro Modeling of Mechanics in Cancer Metastasis. ACS Biomater. Sci. Eng. 2018, 4, 294–301. [Google Scholar] [CrossRef]
- Guo, Q.; Sun, D.; Barrett, A.S.; Jindal, S.; Pennock, N.D.; Conklin, M.W.; Xia, Z.; Mitchell, E.; Samatham, R.; Mirza, N.; et al. Mammary Collagen Is under Reproductive Control with Implications for Breast Cancer. Matrix Biol. 2022, 105, 104–126. [Google Scholar] [CrossRef]
- Pach, E.; Brinckmann, J.; Rübsam, M.; Kümper, M.; Mauch, C.; Zigrino, P. Fibroblast MMP14-Dependent Collagen Processing Is Necessary for Melanoma Growth. Cancers 2021, 13, 1984. [Google Scholar] [CrossRef]
- Yang, N.; Mosher, R.; Seo, S.; Beebe, D.; Friedl, A. Syndecan-1 in breast cancer stroma fibroblasts regulates extracellular matrix fiber organization and carcinoma cell motility. Am. J. Pathol. 2011, 178, 325–335. [Google Scholar] [CrossRef]
- Mechanosensing during Directed Cell Migration Requires Dynamic Actin Polymerization at Focal Adhesions—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/31594807/ (accessed on 1 August 2024).
- Yang, H.; Guan, L.; Li, S.; Jiang, Y.; Xiong, N.; Li, L.; Wu, C.; Zeng, H.; Liu, Y. Mechanosensitive Caveolin-1 Activation-Induced PI3K/Akt/mTOR Signaling Pathway Promotes Breast Cancer Motility, Invadopodia Formation and Metastasis in Vivo. Oncotarget 2016, 7, 16227–16247. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.C.; Ray, A.-M.; Ramolu, L.; Macabre, C.; Simon, F.; Noulet, F.; Blandin, A.-F.; Renner, G.; Lehmann, M.; Choulier, L.; et al. Caveolin-1-Negative Head and Neck Squamous Cell Carcinoma Primary Tumors Display Increased Epithelial to Mesenchymal Transition and Prometastatic Properties. Oncotarget 2015, 6, 41884–41901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Ma, Y.; Yang, L.; Wang, T.; Meng, X.; Zong, Z.; Sun, X.; Hua, X.; Li, H. Yes-Associated Protein (YAP) Binds to HIF-1α and Sustains HIF-1α Protein Stability to Promote Hepatocellular Carcinoma Cell Glycolysis under Hypoxic Stress. J. Exp. Clin. Cancer Res. 2018, 37, 216. [Google Scholar] [CrossRef]
- Wei, C.; Wang, Y.; Li, X. The Role of Hippo Signal Pathway in Breast Cancer Metastasis. OncoTargets Ther. 2018, 11, 2185–2193. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stroma Cells in Tumor Microenvironment and Breast Cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Cancer-Associated Fibroblasts: An Emerging Target of Anti-Cancer Immunotherapy—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/31462327/ (accessed on 1 August 2024).
- Ohshio, Y.; Teramoto, K.; Hanaoka, J.; Tezuka, N.; Itoh, Y.; Asai, T.; Daigo, Y.; Ogasawara, K. Cancer-Associated Fibroblast-Targeted Strategy Enhances Antitumor Immune Responses in Dendritic Cell-Based Vaccine. Cancer Sci. 2015, 106, 134–142. [Google Scholar] [CrossRef]
- Khalilgharibi, N.; Mao, Y. To Form and Function: On the Role of Basement Membrane Mechanics in Tissue Development, Homeostasis and Disease. Open Biol. 2021, 11, 200360. [Google Scholar] [CrossRef]
- Zhang, J.; Reinhart-King, C.A. Targeting Tissue Stiffness in Metastasis: Mechanomedicine Improves Cancer Therapy. Cancer Cell 2020, 37, 754–755. [Google Scholar] [CrossRef]
- Willumsen, N.; Bager, C.L.; Leeming, D.J.; Smith, V.; Karsdal, M.A.; Dornan, D.; Bay-Jensen, A.-C. Extracellular Matrix Specific Protein Fingerprints Measured in Serum Can Separate Pancreatic Cancer Patients from Healthy Controls. BMC Cancer 2013, 13, 554. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Shields, M.A.; Dangi-Garimella, S.; Redig, A.J.; Munshi, H.G. Biochemical Role of the Collagen-Rich Tumour Microenvironment in Pancreatic Cancer Progression. Biochem. J. 2012, 441, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Delvin, E.; Christiansen, C. Protein Fingerprints—Relying on and Understanding the Information of Serological Protein Measurements. Clin. Biochem. 2011, 44, 1278–1279. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of Pancreatic Ductal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef] [PubMed]
- Tenti, P.; Vannucci, L. Lysyl Oxidases: Linking Structures and Immunity in the Tumor Microenvironment. Cancer Immunol. Immunother. 2020, 69, 223–235. [Google Scholar] [CrossRef]
- Mammoto, T.; Jiang, E.; Jiang, A.; Mammoto, A. Extracellular Matrix Structure and Tissue Stiffness Control Postnatal Lung Development through the Lipoprotein Receptor-Related Protein 5/Tie2 Signaling System. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1009–1018. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Zhang, X.; Feng, M.; Ma, J.; Li, J.; Yang, X.; Fang, F.; Xia, Q.; Zhang, Z.; et al. Exosome-Mediated Secretion of LOXL4 Promotes Hepatocellular Carcinoma Cell Invasion and Metastasis. Mol. Cancer 2019, 18, 18. [Google Scholar] [CrossRef]
- Ghasemi, H.; Mousavibahar, S.H.; Hashemnia, M.; Karimi, J.; Khodadadi, I.; Mirzaei, F.; Tavilani, H. Tissue Stiffness Contributes to YAP Activation in Bladder Cancer Patients Undergoing Transurethral Resection. Ann. N. Y. Acad. Sci. 2020, 1473, 48–61. [Google Scholar] [CrossRef]
- Yu, M.; Shen, W.; Shi, X.; Wang, Q.; Zhu, L.; Xu, X.; Yu, J.; Liu, L.; Yu, M.; Shen, W.; et al. Upregulated LOX and Increased Collagen Content Associated with Aggressive Clinicopathological Features and Unfavorable Outcome in Oral Squamous Cell Carcinoma. J. Cell. Biochem. 2019, 120, 14348–14359. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, L.; Li, C.; Yang, C.; Li, L.; Song, S.; Wu, H.; Liu, F.; Wang, L.; Gu, J. LOX-1 Is a Poor Prognostic Indicator and Induces Epithelial-Mesenchymal Transition and Metastasis in Pancreatic Cancer Patients. Cell. Oncol. 2018, 41, 73–84. [Google Scholar] [CrossRef]
- Yamauchi, M.; Gibbons, D.L.; Zong, C.; Fradette, J.J.; Bota-Rabassedas, N.; Kurie, J.M. Fibroblast Heterogeneity and Its Impact on Extracellular Matrix and Immune Landscape Remodeling in Cancer. Matrix Biol. 2020, 91–92, 8–18. [Google Scholar] [CrossRef]
- Nicolas-Boluda, A.; Vaquero, J.; Vimeux, L.; Guilbert, T.; Barrin, S.; Kantari-Mimoun, C.; Ponzo, M.; Renault, G.; Deptula, P.; Pogoda, K.; et al. Tumor Stiffening Reversion through Collagen Crosslinking Inhibition Improves T Cell Migration and Anti-PD-1 Treatment. Elife 2021, 10, e58688. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Lee, D.; Lee, J.E.; Park, H.S.; Jung, S.S.; Park, D.; Kang, D.H.; Lee, S.-I.; Woo, S.-D.; Chung, C. The Matrix Stiffness Coordinates the Cell Proliferation and PD-L1 Expression via YAP in Lung Adenocarcinoma. Cancers 2024, 16, 598. [Google Scholar] [CrossRef] [PubMed]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The Receptor RAGE: Bridging Inflammation and Cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Oh, S.W.; Lee, Y.; Kim, J.Y.; Ji, E.S.; Kim, P. Targeting Extracellular Matrix Glycation to Attenuate Fibroblast Activation. Acta Biomater. 2022, 141, 255–263. [Google Scholar] [CrossRef]
- Rodriguez-Teja, M.; Gronau, J.H.; Breit, C.; Zhang, Y.Z.; Minamidate, A.; Caley, M.P.; McCarthy, A.; Cox, T.R.; Erler, J.T.; Gaughan, L.; et al. AGE-Modified Basement Membrane Cooperates with Endo180 to Promote Epithelial Cell Invasiveness and Decrease Prostate Cancer Survival. J. Pathol. 2014, 141, 255–263. [Google Scholar] [CrossRef]
- Leeming, D.J.; Bay-Jensen, A.C.; Vassiliadis, E.; Larsen, M.R.; Henriksen, K.; Karsdal, M.A. Post-Translational Modifications of the Extracellular Matrix Are Key Events in Cancer Progression: Opportunities for Biochemical Marker Development. Biomarkers 2011, 16, 193–205. [Google Scholar] [CrossRef]
- Pogoda, K.; Bucki, R.; Byfield, F.J.; Cruz, K.; Lee, T.; Marcinkiewicz, C.; Janmey, P.A. Soft Substrates Containing Hyaluronan Mimic the Effects of Increased Stiffness on Morphology, Motility, and Proliferation of Glioma Cells. Biomacromolecules 2017, 18, 3040–3051. [Google Scholar] [CrossRef]
- Rehfeldt, F.; Brown, A.E.X.; Raab, M.; Cai, S.; Zajac, A.L.; Zemel, A.; Discher, D.E. Hyaluronic Acid Matrices Show Matrix Stiffness in 2D and 3D Dictates Cytoskeletal Order and Myosin-II Phosphorylation within Stem Cells. Integr. Biol. 2012, 4, 422–430. [Google Scholar] [CrossRef]
- Hammer, A.M.; Sizemore, G.M.; Shukla, V.C.; Avendano, A.; Sizemore, S.T.; Chang, J.J.; Kladney, R.D.; Cuitiño, M.C.; Thies, K.A.; Verfurth, Q.; et al. Stromal PDGFR-α Activation Enhances Matrix Stiffness, Impedes Mammary Ductal Development, and Accelerates Tumor Growth. Neoplasia 2017, 19, 496–508. [Google Scholar] [CrossRef]
- Spada, S.; Tocci, A.; Di Modugno, F.; Nisticò, P. Fibronectin as a Multiregulatory Molecule Crucial in Tumor Matrisome: From Structural and Functional Features to Clinical Practice in Oncology. J. Exp. Clin. Cancer Res. 2021, 40, 102. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Gentile, M.T.; Pentimalli, F.; Cortellino, S.; Grieco, M.; Giordano, A. Multiple Aspects of Matrix Stiffness in Cancer Progression. Front. Oncol. 2024, 14, 1406644. [Google Scholar] [CrossRef] [PubMed]
- Drifka, C.R.; Loeffler, A.G.; Mathewson, K.; Keikhosravi, A.; Eickhoff, J.C.; Liu, Y.; Weber, S.M.; Kao, W.J.; Eliceiri, K.W. Highly Aligned Stromal Collagen Is a Negative Prognostic Factor Following Pancreatic Ductal Adenocarcinoma Resection. Oncotarget 2016, 7, 76197–76213. [Google Scholar] [CrossRef] [PubMed]
- Brett, E.A.; Sauter, M.A.; Machens, H.-G.; Duscher, D. Tumor-Associated Collagen Signatures: Pushing Tumor Boundaries. Cancer Metab. 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Beunk, L.; Bakker, G.-J.; van Ens, D.; Bugter, J.; Gal, F.; Svoren, M.; Friedl, P.; Wolf, K. Actomyosin Contractility Requirements and Reciprocal Cell–Tissue Mechanics for Cancer Cell Invasion through Collagen-Based Channels. Eur. Phys. J. E 2022, 45, 48. [Google Scholar] [CrossRef]
- Hanley, C.J.; Noble, F.; Ward, M.; Bullock, M.; Drifka, C.; Mellone, M.; Manousopoulou, A.; Johnston, H.E.; Hayden, A.; Thirdborough, S.; et al. A Subset of Myofibroblastic Cancer-Associated Fibroblasts Regulate Collagen Fiber Elongation, Which Is Prognostic in Multiple Cancers. Oncotarget 2016, 7, 6159–6174. [Google Scholar] [CrossRef]
- Garrison, C.M.; Schwarzbauer, J.E. Fibronectin Fibril Alignment Is Established upon Initiation of Extracellular Matrix Assembly. Mol. Biol. Cell 2021, 32, 739–752. [Google Scholar] [CrossRef]
- Chute, C.; Yang, X.; Meyer, K.; Yang, N.; O’Neil, K.; Kasza, I.; Eliceiri, K.; Alexander, C.; Friedl, A. Syndecan-1 Induction in Lung Microenvironment Supports the Establishment of Breast Tumor Metastases. Breast Cancer Res. 2018, 20, 66. [Google Scholar] [CrossRef]
- Madsen, D.H.; Jürgensen, H.J.; Siersbæk, M.S.; Kuczek, D.E.; Cloud, L.G.; Liu, S.; Behrendt, N.; Grøntved, L.; Weigert, R.; Bugge, T.H. Tumor-Associated Macrophages Derived from Circulating Inflammatory Monocytes Degrade Collagen through Cellular Uptake. Cell Rep. 2017, 21, 3662–3671. [Google Scholar] [CrossRef]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef]
- Wisdom, K.M.; Indana, D.; Chou, P.-E.; Desai, R.; Kim, T.; Chaudhuri, O. Covalent Cross-Linking of Basement Membrane-like Matrices Physically Restricts Invasive Protrusions in Breast Cancer Cells. Matrix Biol. 2020, 85–86, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-P.; Alisafaei, F.; Adebawale, K.; Chang, J.; Shenoy, V.B.; Chaudhuri, O. The Nuclear Piston Activates Mechanosensitive Ion Channels to Generate Cell Migration Paths in Confining Microenvironments. Sci. Adv. 2021, 7, eabd4058. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, J.; Jiang, K.; Fernandez, J.G.; Lim, C.T. Extracellular Matrix Mechanobiology in Cancer Cell Migration. Acta Biomater. 2023, 163, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, D.-H.; Kim, H.-N.; Wang, C.J.; Kwak, M.K.; Hur, E.; Suh, K.-Y.; An, S.S.; Levchenko, A. Directed Migration of Cancer Cells guided by the Graded Texture of the Underlying Matrix. Nat. Mater. 2016, 15, 792–801. [Google Scholar] [CrossRef]
- Kushiro, K.; Yaginuma, T.; Ryo, A.; Takai, M. Differences in Three-Dimensional Geometric Recognition by Non-Cancerous and Cancerous Epithelial Cells on Microgroove-Based Topography. Sci. Rep. 2017, 7, 4244. [Google Scholar] [CrossRef]
- Pieuchot, L.; Marteau, J.; Guignandon, A.; Dos Santos, T.; Brigaud, I.; Chauvy, P.-F.; Cloatre, T.; Ponche, A.; Petithory, T.; Rougerie, P.; et al. Curvotaxis Directs Cell Migration through Cell-Scale Curvature Landscapes. Nat. Commun. 2018, 9, 3995. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Le Berre, M.; Lautenschlaeger, F.; Maiuri, P.; Callan-Jones, A.; Heuzé, M.; Takaki, T.; Voituriez, R.; Piel, M. Confinement and Low Adhesion Induce Fast Amoeboid Migration of Slow Mesenchymal Cells. Cell 2015, 160, 659–672. [Google Scholar] [CrossRef]
- Das, A.; Barai, A.; Monteiro, M.; Kumar, S.; Sen, S. Nuclear Softening Is Essential for Protease-Independent Migration. Matrix Biol. 2019, 82, 4–19. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer. 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Isomursu, A.; Park, K.-Y.; Hou, J.; Cheng, B.; Mathieu, M.; Shamsan, G.A.; Fuller, B.; Kasim, J.; Mahmoodi, M.M.; Lu, T.J.; et al. Directed Cell Migration towards Softer Environments. Nat. Mater. 2022, 21, 1081–1090. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, Z.; Chen, Y.; Li, S.; Jiang, Y.; Yang, H.; Wu, C.; You, F.; Zheng, C.; Zhu, J.; et al. ROCK Isoforms Differentially Modulate Cancer Cell Motility by Mechanosensing the Substrate Stiffness. Acta Biomater. 2019, 88, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Kollmannsberger, P.; Bidan, C.M.; Dunlop, J.W.C.; Fratzl, P.; Vogel, V. Tensile Forces Drive a Reversible Fibroblast-to-Myofibroblast Transition during Tissue Growth in Engineered Clefts. Sci. Adv. 2018, 4, eaao4881. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human Breast Cancer Invasion and Aggression Correlates with ECM Stiffening and Immune Cell Infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.K.; Halbrook, C.J.; Kerk, S.A.; Radyk, M.; Wisner, S.; Kremer, D.M.; Sajjakulnukit, P.; Andren, A.; Hou, S.W.; Trivedi, A.; et al. Hyaluronic Acid Fuels Pancreatic Cancer Cell Growth. Elife 2021, 10, e62645. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-W.E.; Pedron, S.; Harley, B.A.C. The Combined Influence of Hydrogel Stiffness and Matrix-Bound Hyaluronic Acid Content on Glioblastoma Invasion. Macromol. Biosci. 2017, 17, 1700018. [Google Scholar] [CrossRef]
- Grolman, J.M.; Weinand, P.; Mooney, D.J. Extracellular Matrix Plasticity as a Driver of Cell Spreading. Proc. Natl. Acad. Sci. USA 2020, 117, 25999–26007. [Google Scholar] [CrossRef]
- Martinez-Garcia, F.D.; de Hilster, R.H.J.; Sharma, P.K.; Borghuis, T.; Hylkema, M.N.; Burgess, J.K.; Harmsen, M.C. Architecture and Composition Dictate Viscoelastic Properties of Organ-Derived Extracellular Matrix Hydrogels. Polymers 2021, 13, 3113. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Gupta, A.; Najibi, A.J.; Seo, B.R.; Garry, R.; Tringides, C.M.; de Lázaro, I.; Darnell, M.; Gu, W.; Zhou, Q.; et al. Matrix Viscoelasticity Controls Spatiotemporal Tissue Organization. Nat. Mater. 2023, 22, 117–127. [Google Scholar] [CrossRef]
- Deng, H.; Wang, Y.; Yin, Y.; Shu, J.; Zhang, J.; Shu, X.; Wu, F.; He, J. Effects of Matrix Viscoelasticity on Cell–Matrix Interaction, Actin Cytoskeleton Organization, and Apoptosis of Osteosarcoma MG-63 Cells. J. Mater. Chem. B 2024, 12, 222–232. [Google Scholar] [CrossRef]
- Fan, Y.; Sun, Q.; Li, X.; Feng, J.; Ao, Z.; Li, X.; Wang, J. Substrate Stiffness Modulates the Growth, Phenotype, and Chemoresistance of Ovarian Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 718834. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Vivès, R.R.; Schaefer, L.; Götte, M.; Merline, R.; Passi, A.; Heldin, P.; Magalhães, A.; Reis, C.A.; Skandalis, S.S.; et al. A Biological Guide to Glycosaminoglycans: Current Perspectives and Pending Questions. FEBS J. 2024, 291, 3331–3366. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-P.; Martin, L.J.; Hanna, W.; Banerjee, D.; Miller, N.; Fishell, E.; Khokha, R.; Boyd, N.F. Growth Factors and Stromal Matrix Proteins Associated with Mammographic Densities. Cancer Epidemiol. Biomark. Prev. 2001, 10, 243–248. [Google Scholar]
- Andrlová, H.; Mastroianni, J.; Madl, J.; Kern, J.S.; Melchinger, W.; Dierbach, H.; Wernet, F.; Follo, M.; Technau-Hafsi, K.; Has, C.; et al. Biglycan Expression in the Melanoma Microenvironment Promotes Invasiveness via Increased Tissue Stiffness Inducing Integrin-Β1 Expression. Oncotarget 2017, 8, 42901–42916. [Google Scholar] [CrossRef] [PubMed]
- Szarvas, T.; Reis, H.; Kramer, G.; Shariat, S.F.; vom Dorp, F.; Tschirdewahn, S.; Schmid, K.W.; Kovalszky, I.; Rübben, H. Enhanced Stromal Syndecan-1 Expression Is an Independent Risk Factor for Poor Survival in Bladder Cancer. Hum. Pathol. 2014, 45, 674–682. [Google Scholar] [CrossRef]
- Szarvas, T.; Reis, H.; Vom Dorp, F.; Tschirdewahn, S.; Niedworok, C.; Nyirady, P.; Schmid, K.W.; Rübben, H.; Kovalszky, I. Soluble Syndecan-1 (SDC1) Serum Level as an Independent Pre-Operative Predictor of Cancer-Specific Survival in Prostate Cancer. Prostate 2016, 76, 977–985. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Karamanos, N.K. Proteoglycans Remodeling in Cancer: Underlying Molecular Mechanisms. Matrix Biol. 2019, 75–76, 220–259. [Google Scholar] [CrossRef]
- Mytilinaiou, M.; Nikitovic, D.; Berdiaki, A.; Kostouras, A.; Papoutsidakis, A.; Tsatsakis, A.M.; Tzanakakis, G.N. Emerging Roles of Syndecan 2 in Epithelial and Mesenchymal Cancer Progression. IUBMB Life 2017, 69, 824–833. [Google Scholar] [CrossRef]
- Mytilinaiou, M.; Bano, A.; Nikitovic, D.; Berdiaki, A.; Voudouri, K.; Kalogeraki, A.; Karamanos, N.K.; Tzanakakis, G.N. Syndecan-2 Is a Key Regulator of Transforming Growth Factor Beta 2/Smad2-Mediated Adhesion in Fibrosarcoma Cells. IUBMB Life 2013, 65, 134–143. [Google Scholar] [CrossRef]
- Chalkiadaki, G.; Nikitovic, D.; Berdiaki, A.; Sifaki, M.; Krasagakis, K.; Katonis, P.; Karamanos, N.K.; Tzanakakis, G.N. Fibroblast Growth Factor-2 Modulates Melanoma Adhesion and Migration through a Syndecan-4-Dependent Mechanism. Int. J. Biochem. Cell. Biol. 2009, 41, 1323–1331. [Google Scholar] [CrossRef]
- Bellin, R.M.; Kubicek, J.D.; Frigault, M.J.; Kamien, A.J.; Steward, R.L.; Barnes, H.M.; DiGiacomo, M.B.; Duncan, L.J.; Edgerly, C.K.; Morse, E.M.; et al. Defining the Role of Syndecan-4 in Mechanotransduction Using Surface-Modification Approaches. Proc. Natl. Acad. Sci. USA 2009, 106, 22102–22107. [Google Scholar] [CrossRef]
- Woods, A.; Couchman, J.R. Syndecan-4 and Focal Adhesion Function. Curr. Opin. Cell Biol. 2001, 13, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Saoncella, S.; Echtermeyer, F.; Denhez, F.; Nowlen, J.K.; Mosher, D.F.; Robinson, S.D.; Hynes, R.O.; Goetinck, P.F. Syndecan-4 Signals Cooperatively with Integrins in a Rho-Dependent Manner in the Assembly of Focal Adhesions and Actin Stress Fibers. Proc. Natl. Acad. Sci. USA 1999, 96, 2805–2810. [Google Scholar] [CrossRef] [PubMed]
- Fiore, V.F.; Ju, L.; Chen, Y.; Zhu, C.; Barker, T.H. Dynamic Catch of a Thy-1-A5β1+syndecan-4 Trimolecular Complex. Nat. Commun. 2014, 5, 4886. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S.; Oka, Y.; Fujiki, F.; Morimoto, S.; Nakajima, H.; Nakae, Y.; Nakata, J.; Nishida, S.; Hosen, N.; Tatsumi, N.; et al. Syndecan-4 as a Biomarker to Predict Clinical Outcome for Glioblastoma Multiforme Treated with WT1 Peptide Vaccine. Futur. Sci. OA 2016, 2, FSO96. [Google Scholar] [CrossRef] [PubMed]
- Na, K.Y.; Bacchini, P.; Bertoni, F.; Kim, Y.W.; Park, Y.-K. Syndecan-4 and Fibronectin in Osteosarcoma. Pathology 2012, 44, 325–330. [Google Scholar] [CrossRef]
- Burgess, R.W.; Skarnes, W.C.; Sanes, J.R. Agrin Isoforms with Distinct Amino Termini: Differential Expression, Localization, and Function. J. Cell Biol. 2000, 151, 41–52. [Google Scholar] [CrossRef]
- Rivera, C.; Zandonadi, F.S.; Sánchez-Romero, C.; Soares, C.D.; Granato, D.C.; González-Arriagada, W.A.; Leme, A.F.P. Agrin Has a Pathological Role in the Progression of Oral Cancer. Br. J. Cancer 2018, 118, 1628–1638. [Google Scholar] [CrossRef]
- Chakraborty, S.; Lakshmanan, M.; Swa, H.L.F.; Chen, J.; Zhang, X.; Ong, Y.S.; Loo, L.S.; Aklncllar, S.C.; Gunaratne, J.; Tergaonkar, V.; et al. An Oncogenic Role of Agrin in Regulating Focal Adhesion Integrity in Hepatocellular Carcinoma. Nat. Commun. 2015, 6, 6184. [Google Scholar] [CrossRef]
- Chakraborty, S.; Njah, K.; Pobbati, A.V.; Lim, Y.B.; Raju, A.; Lakshmanan, M.; Tergaonkar, V.; Lim, C.T.; Hong, W. Agrin as a Mechanotransduction Signal Regulating YAP through the Hippo Pathway. Cell Rep. 2017, 18, 2464–2479. [Google Scholar] [CrossRef]
- Kolset, S.O.; Tveit, H. Serglycin—Structure and Biology. Cell. Mol. Life Sci. 2008, 65, 1073–1085. [Google Scholar] [CrossRef]
- Purushothaman, A.; Bandari, S.K.; Chandrashekar, D.S.; Jones, R.J.; Lee, H.C.; Weber, D.M.; Orlowski, R.Z. Chondroitin Sulfate Proteoglycan Serglycin Influences Protein Cargo Loading and Functions of Tumor-Derived Exosomes. Oncotarget 2017, 8, 73723–73732. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Seidel, C.; Borset, M.; Dobra, K.; Baykov, V.; Labropoulou, V.; Kanakis, I.; Dalas, E.; Karamanos, N.K.; Sundan, A.; et al. Serglycin Constitutively Secreted by Myeloma Plasma Cells Is a Potent Inhibitor of Bone Mineralization in Vitro. J. Biol. Chem. 2006, 281, 35116–35128. [Google Scholar] [CrossRef] [PubMed]
- Korpetinou, A.; Papachristou, D.J.; Lampropoulou, A.; Bouris, P.; Labropoulou, V.T.; Noulas, A.; Karamanos, N.K.; Theocharis, A.D. Increased Expression of Serglycin in Specific Carcinomas and Aggressive Cancer Cell Lines. Biomed. Res. Int. 2015, 2015, 690721. [Google Scholar] [CrossRef] [PubMed]
- Baghy, K.; Tátrai, P.; Regős, E.; Kovalszky, I. Proteoglycans in Liver Cancer. World J. Gastroenterol. 2016, 22, 379. [Google Scholar] [CrossRef]
- Zhang, Z.; Qiu, N.; Yin, J.; Zhang, J.; Liu, H.; Guo, W.; Liu, M.; Liu, T.; Chen, D.; Luo, K.; et al. SRGN Crosstalks with YAP to Maintain Chemoresistance and Stemness in Breast Cancer Cells by Modulating HDAC2 Expression. Theranostics 2020, 10, 4290–4307. [Google Scholar] [CrossRef]
- Wang, H.-B.; Dembo, M.; Hanks, S.K.; Wang, Y.-L. Focal Adhesion Kinase Is Involved in Mechanosensing during Fibroblast Migration. Proc. Natl. Acad. Sci. USA 2001, 98, 11295–11300. [Google Scholar] [CrossRef]
- Robinson, K.A.; Sun, M.; Barnum, C.E.; Weiss, S.N.; Huegel, J.; Shetye, S.S.; Lin, L.; Saez, D.; Adams, S.M.; Iozzo, R.V.; et al. Decorin and Biglycan Are Necessary for Maintaining Collagen Fibril Structure, Fiber Realignment, and Mechanical Properties of Mature Tendons. Matrix Biol. 2017, 64, 81–93. [Google Scholar] [CrossRef]
- Lewis, J.L.; Krawczak, D.A.; Oegema, T.R.; Westendorf, J.J. Effect of Decorin and Dermatan Sulfate on the Mechanical Properties of a Neocartilage. Connect Tissue Res. 2010, 51, 159–170. [Google Scholar] [CrossRef]
- Rühland, C.; Schönherr, E.; Robenek, H.; Hansen, U.; Iozzo, R.V.; Bruckner, P.; Seidler, D.G. The Glycosaminoglycan Chain of Decorin Plays an Important Role in Collagen Fibril Formation at the Early Stages of Fibrillogenesis. FEBS J. 2007, 274, 4246–4255. [Google Scholar] [CrossRef]
- Hu, L.; Duan, Y.; Li, J.; Su, L.; Yan, M.; Zhu, Z.; Liu, B.; Yang, Q. Biglycan Enhances Gastric Cancer Invasion by Activating FAK Signaling Pathway. Oncotarget 2014, 5, 1885–1896. [Google Scholar] [CrossRef]
- Manupati, K.; Paul, R.; Hao, M.; Haas, M.; Bian, Z.C.; Holm, T.M.; Guan, J.-L.; Yeo, S.K. Biglycan Promotes Cancer Stem Cell Properties, NFκB Signaling and Metastatic Potential in Breast Cancer Cells. Cancers 2022, 14, 455. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Thomas, V.J.; Coulson-Thomas, Y.M.; Gesteira, T.F.; de Paula, C.A.A.; Mader, A.M.; Waisberg, J.; Pinhal, M.A.; Friedl, A.; Toma, L.; Nader, H.B. Colorectal Cancer Desmoplastic Reaction Up-Regulates Collagen Synthesis and Restricts Cancer Cell Invasion. Cell Tissue Res. 2011, 346, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Aggelidakis, J.; Berdiaki, A.; Nikitovic, D.; Papoutsidakis, A.; Papachristou, D.J.; Tsatsakis, A.M.; Tzanakakis, G.N. Biglycan Regulates MG63 Osteosarcoma Cell Growth Through a LPR6/β-Catenin/IGFR-IR Signaling Axis. Front. Oncol. 2018, 8, 470. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Thomas, V.J.; Coulson-Thomas, Y.M.; Gesteira, T.F.; de Paula, C.A.A.; Carneiro, C.R.W.; Ortiz, V.; Toma, L.; Kao, W.W.-Y.; Nader, H.B. Lumican Expression, Localization and Antitumor Activity in Prostate Cancer. Exp. Cell Res. 2013, 319, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Radwanska, A.; Baczynska, D.; Nowak, D.; Brézillon, S.; Popow, A.; Maquart, F.-X.; Wegrowski, Y.; Malicka-Blaszkiewicz, M. Lumican Affects Actin Cytoskeletal Organization in Human Melanoma A375 Cells. Life Sci. 2008, 83, 651–660. [Google Scholar] [CrossRef]
- Zeltz, C.; Brézillon, S.; Käpylä, J.; Eble, J.A.; Bobichon, H.; Terryn, C.; Perreau, C.; Franz, C.M.; Heino, J.; Maquart, F.-X.; et al. Lumican Inhibits Cell Migration through A2β1 Integrin. Exp. Cell Res. 2010, 316, 2922–2931. [Google Scholar] [CrossRef]
- Karamanou, K.; Franchi, M.; Proult, I.; Rivet, R.; Vynios, D.; Brézillon, S. Lumican Inhibits In Vivo Melanoma Metastasis by Altering Matrix-Effectors and Invadopodia Markers. Cells 2021, 10, 841. [Google Scholar] [CrossRef]
- Karamanou, K.; Franchi, M.; Piperigkou, Z.; Perreau, C.; Maquart, F.-X.; Vynios, D.H.; Brézillon, S. Lumican Effectively Regulates the Estrogen Receptors-Associated Functional Properties of Breast Cancer Cells, Expression of Matrix Effectors and Epithelial-to-Mesenchymal Transition. Sci. Rep. 2017, 7, 45138. [Google Scholar] [CrossRef]
- Nikitovic, D.; Papoutsidakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican affects tumor cell functions, tumor-ECM interactions, angiogenesis and inflammatory response. Matrix Biol. 2014, 35, 206–214. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Q.; Yu, Z.; Wu, X.; Chen, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; Liu, B.; et al. Cancer-Associated Fibroblast-Derived Lumican Promotes Gastric Cancer Progression via the Integrin Β1-FAK Signaling Pathway. Int. J. Cancer 2017, 141, 998–1010. [Google Scholar] [CrossRef]
- Papoutsidakis, A.; Giatagana, E.M.; Berdiaki, A.; Spyridaki, I.; Spandidos, D.A.; Tsatsakis, A.; Tzanakakis, G.N.; Nikitovic, D. Lumican Mediates HTB94 Chondrosarcoma Cell Growth via an IGF-IR/Erk1/2 Axis. Int. J. Oncol. 2020, 57, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Katonis, P.; Tsatsakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican, a Small Leucine-Rich Proteoglycan. IUBMB Life 2008, 60, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Berdiaki, A.; Zafiropoulos, A.; Katonis, P.; Tsatsakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican Expression Is Positively Correlated with the Differentiation and Negatively with the Growth of Human Osteosarcoma Cells. FEBS J. 2008, 275, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Tzardi, M.; Berdiaki, A.; Tsatsakis, A.; Tzanakakis, G.N. Cancer Microenvironment and Inflammation: Role of HA. Front. Immunol. 2015, 6, 169. [Google Scholar] [CrossRef]
- Caon, I.; Bartolini, B.; Parnigoni, A.; Caravà, E.; Moretto, P.; Viola, M.; Karousou, E.; Vigetti, D.; Passi, A. Revisiting the Hallmarks of Cancer: The Role of HA. Semin. Cancer Biol. 2020, 62, 9–19. [Google Scholar] [CrossRef]
- Tammi, R.H.; Kultti, A.; Kosma, V.-M.; Pirinen, R.; Auvinen, P.; Tammi, M.I. HA in Human Tumors: Pathobiological and Prognostic Messages from Cell-Associated and Stromal HA. Semin. Cancer Biol. 2008, 18, 288–295. [Google Scholar] [CrossRef]
- Toole, B.P. HA Promotes the Malignant Phenotype. Glycobiology 2002, 12, 37R–42R. [Google Scholar] [CrossRef]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.I.; Reis, G.F.; McKnight, T.R.; et al. Tissue Mechanics Promote IDH1-Dependent HIF1α-Tenascin C Feedback to Regulate Glioblastoma Aggression. Nat. Cell. Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef]
- Voutouri, C.; Stylianopoulos, T. Accumulation of Mechanical Forces in Tumors Is Related to HA Content and Tissue Stiffness. PLoS ONE 2018, 13, e0193801. [Google Scholar] [CrossRef]
- Kim, Y.; Kumar, S. CD44-Mediated Adhesion to Hyaluronic Acid Contributes to Mechanosensing and Invasive Motility. Mol. Cancer Res. 2014, 12, 1416–1429. [Google Scholar] [CrossRef]
- Khoonkari, M.; Liang, D.; Kamperman, M.; Kruyt, F.A.E.; van Rijn, P. Physics of Brain Cancer: Multiscale Alterations of Glioblastoma Cells under Extracellular Matrix Stiffening. Pharmaceutics 2022, 14, 1031. [Google Scholar] [CrossRef] [PubMed]
- Pranda, M.A.; Gray, K.M.; DeCastro, A.J.L.; Dawson, G.M.; Jung, J.W.; Stroka, K.M. Tumor Cell Mechanosensing During Incorporation into the Brain Microvascular Endothelium. Cell Mol. Bioeng. 2019, 12, 455–480. [Google Scholar] [CrossRef] [PubMed]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Tsolakis, I.; Tzanakakis, G.N.; Karamanos, N.K. Chondroitin Sulfate as a Key Molecule in the Development of Atherosclerosis and Cancer Progression. In Advances in Pharmacology; Chondroitin Sulfate: Structure, Role and Pharmacological Activity; Academic Press: Cambridge, MA, USA, 2006; Volume 53, pp. 281–295. [Google Scholar]
- Labropoulou, V.T.; Theocharis, A.D.; Ravazoula, P.; Perimenis, P.; Hjerpe, A.; Karamanos, N.K.; Kalofonos, H.P. Versican but Not Decorin Accumulation Is Related to Metastatic Potential and Neovascularization in Testicular Germ Cell Tumours. Histopathology 2006, 49, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Umehara, Y.; Higashiyama, S.; Itoh, N.; Sugahara, K. Specific Molecular Interactions of Oversulfated Chondroitin Sulfate E with Various Heparin-Binding Growth Factors: Implications as a Physiological Binding Partner in the Brain and Other Tissues. J. Biol. Chem. 2002, 277, 43707–43716. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ten Dam, G.B.; Murugan, S.; Yamada, S.; Hashiguchi, T.; Mizumoto, S.; Oguri, K.; Okayama, M.; van Kuppevelt, T.H.; Sugahara, K. Involvement of Highly Sulfated Chondroitin Sulfate in the Metastasis of the Lewis Lung Carcinoma Cells. J. Biol. Chem. 2008, 283, 34294–34304. [Google Scholar] [CrossRef]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of Matrix Metalloproteinases in Cancer Progression and Their Pharmacological Targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef]
- Yang, J.; Price, M.A.; Li, G.Y.; Bar-Eli, M.; Salgia, R.; Jagedeeswaran, R.; Carlson, J.H.; Ferrone, S.; Turley, E.A.; McCarthy, J.B. Melanoma Proteoglycan Modifies Gene Expression to Stimulate Tumor Cell Motility, Growth, and Epithelial-to-Mesenchymal Transition. Cancer Res. 2009, 69, 7538–7547. [Google Scholar] [CrossRef]
- Bret, C.; Hose, D.; Reme, T.; Sprynski, A.-C.; Mahtouk, K.; Schved, J.-F.; Quittet, P.; Rossi, J.-F.; Goldschmidt, H.; Klein, B. Expression of Genes Encoding for Proteins Involved in Heparan Sulphate and Chondroitin Sulphate Chain Synthesis and Modification in Normal and Malignant Plasma Cells. Br. J. Haematol. 2009, 145, 350–368. [Google Scholar] [CrossRef]
- Taylor, K.R.; Gallo, R.L. Glycosaminoglycans and Their Proteoglycans: Host-Associated Molecular Patterns for Initiation and Modulation of Inflammation. FASEB J. 2006, 20, 9–22. [Google Scholar] [CrossRef]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C Is an Endogenous Activator of Toll-like Receptor 4 That Is Essential for Maintaining Inflammation in Arthritic Joint Disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Babelova, A.; Kiss, E.; Hausser, H.-J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Götte, M.; et al. The Matrix Component Biglycan Is Proinflammatory and Signals through Toll-like Receptors 4 and 2 in Macrophages. J. Clin. Investig. 2005, 115, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Breaking Down Chronic Inflammatory Diseases: The Role of Biglycan in Promoting a Switch between Inflammation and Au-tophagy—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30776184/ (accessed on 28 July 2024).
- Appunni, S.; Rubens, M.; Ramamoorthy, V.; Anand, V.; Khandelwal, M.; Sharma, A. Biglycan: An Emerging Small Leucine-Rich Proteoglycan (SLRP) Marker and Its Clinicopathological Significance. Mol. Cell Biochem. 2021, 476, 3935–3950. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.; Asari, A.A.; Sugahara, K.N. HA Fragments: An Information-Rich System. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef]
- Berdiaki, A.; Neagu, M.; Spyridaki, I.; Kuskov, A.; Perez, S.; Nikitovic, D. HA and Reactive Oxygen Species Signaling-Novel Cues from the Matrix? Antioxidants 2023, 12, 824. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of Lung Injury and Repair by Toll-like Receptors and HA. Nat. Med. 2005, 11, 1173–1179. [Google Scholar] [CrossRef]
- Noble, P.W. HA and Its Catabolic Products in Tissue Injury and Repair. Matrix Biol. 2002, 21, 25–29. [Google Scholar] [CrossRef]
- McKee, C.M.; Penno, M.B.; Cowman, M.; Burdick, M.D.; Strieter, R.M.; Bao, C.; Noble, P.W. HA (HA) Fragments Induce Chemokine Gene Expression in Alveolar Macrophages. The Role of HA Size and CD44. J. Clin. Investig. 1996, 98, 2403–2413. [Google Scholar] [CrossRef]
- Mummert, M.E.; Mummert, D.; Edelbaum, D.; Hui, F.; Matsue, H.; Takashima, A. Synthesis and Surface Expression of HA by Dendritic Cells and Its Potential Role in Antigen Presentation. J. Immunol. 2002, 169, 4322–4331. [Google Scholar] [CrossRef]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J.C. Oligosaccharides of HA Activate Dendritic Cells via Toll-like Receptor 4. J. Exp. Med. 2002, 195, 99–111. [Google Scholar] [CrossRef]
- Monslow, J.; Govindaraju, P.; Puré, E. HA—A Functional and Structural Sweet Spot in the Tissue Microenvironment. Front. Immunol. 2015, 6, 231. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; oude Egbrink, M.G.A. The Endothelial Glycocalyx: Composition, Functions, and Visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Katakam, S.K.; Urbanowitz, A.-K.; Gotte, M. Heparan Sulphate as a Regulator of Leukocyte Recruitment in Inflammation. Curr. Protein. Pept. Sci. 2015, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, M.A.; Genemaras, K.; Dailey, H.L.; Jedlicka, S.; Zhang, X.F. Dual Regulation of L-Selectin-Mediated Leukocyte Adhesion by Endothelial Surface Glycocalyx. Cell Mol. Bioeng. 2017, 10, 102–113. [Google Scholar] [CrossRef]
- Norgard-Sumnicht, K.; Varki, A. Endothelial Heparan Sulfate Proteoglycans That Bind to L-Selectin Have Glucosamine Residues with Unsubstituted Amino Groups. J. Biol. Chem. 1995, 270, 12012–12024. [Google Scholar] [CrossRef]
- Falanga, V. Wound Healing and Its Impairment in the Diabetic Foot. Lancet 2005, 366, 1736–1743. [Google Scholar] [CrossRef]
- Evanko, S.P.; Tammi, M.I.; Tammi, R.H.; Wight, T.N. HA-Dependent Pericellular Matrix. Adv. Drug. Deliv. Rev. 2007, 59, 1351–1365. [Google Scholar] [CrossRef]
- Schneider, A.; Francius, G.; Obeid, R.; Schwinté, P.; Hemmerlé, J.; Frisch, B.; Schaaf, P.; Voegel, J.-C.; Senger, B.; Picart, C. Polyelectrolyte Multilayers with a Tunable Young’s Modulus: Influence of Film Stiffness on Cell Adhesion. Langmuir 2006, 22, 1193–1200. [Google Scholar] [CrossRef]
- Fan, F.; Su, B.; Kolodychak, A.; Ekwueme, E.; Alderfer, L.; Saha, S.; Webber, M.J.; Hanjaya-Putra, D. Hyaluronic Acid Hydrogels with Phototunable Supramolecular Cross-Linking for Spatially Controlled Lymphatic Tube Formation. ACS Appl. Mater. Interfaces 2023, 15, 58181–58195. [Google Scholar] [CrossRef]
- Rinaldi, E.; Baggi, F. LYVE-1 Is “on Stage” Now: An Emerging Player in Dendritic Cell Docking to Lymphatic Endothelial Cells. Cell. Mol. Immunol. 2018, 15, 663–665. [Google Scholar] [CrossRef]
- Kieu, T.Q.; Tazawa, K.; Kawashima, N.; Noda, S.; Fujii, M.; Nara, K.; Hashimoto, K.; Han, P.; Okiji, T. Kinetics of LYVE-1-Positive M2-like Macrophages in Developing and Repairing Dental Pulp in Vivo and Their pro-Angiogenic Activity in Vitro. Sci. Rep. 2022, 12, 5176. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.D.; Shworak, N.W.; Liu, J.; Schwartz, J.J.; Zhang, L. Heparan Sulfate Proteoglycans of the Cardiovascular System. Specific Structures Emerge but How Is Synthesis Regulated? J. Clin. Investig. 1997, 99, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Tarbell, J.M.; Simon, S.I.; Curry, F.-R.E. Mechanosensing at the Vascular Interface. Annu. Rev. Biomed. Eng. 2014, 16, 505–532. [Google Scholar] [CrossRef] [PubMed]
- Florian, J.A.; Kosky, J.R.; Ainslie, K.; Pang, Z.; Dull, R.O.; Tarbell, J.M. Heparan Sulfate Proteoglycan Is a Mechanosensor on Endothelial Cells. Circ. Res. 2003, 93, e136–e142. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Rabodzey, A.; Dewey, C.F. Glycocalyx Modulates the Motility and Proliferative Response of Vascular Endothelium to Fluid Shear Stress. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1023–H1030. [Google Scholar] [CrossRef]
- Giantsos-Adams, K.M.; Koo, A.J.-A.; Song, S.; Sakai, J.; Sankaran, J.; Shin, J.H.; Garcia-Cardena, G.; Dewey, C.F. Heparan Sulfate Regrowth Profiles Under Laminar Shear Flow Following Enzymatic Degradation. Cell. Mol. Bioeng. 2013, 6, 160–174. [Google Scholar] [CrossRef]
- Maiti, G.; Ashworth, S.; Choi, T.; Chakravarti, S. Molecular Cues for Immune Cells from Small Leucine-Rich Repeat Proteoglycans in Their Extracellular Matrix-Associated and Free Forms. Matrix Biol. 2023, 123, 48–58. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The Biology and Role of CD44 in Cancer Progression: Therapeutic Implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Jariyal, H.; Srivastava, A. Hyaluronic Acid: More than a Carrier, Having an Overpowering Extracellular and Intracellular Impact on Cancer. Carbohydr. Polym. 2023, 317, 121081. [Google Scholar] [CrossRef]
- Amorim, S.; da Costa, D.S.; Mereiter, S.; Pashkuleva, I.; Reis, C.A.; Reis, R.L.; Pires, R.A. Multilayer Platform to Model the Bioactivity of Hyaluronic Acid in Gastric Cancer. Mater. Sci. Eng. C 2021, 119, 111616. [Google Scholar] [CrossRef]
- Chen, J.; Meng, J.; Li, X.; Li, X.; Liu, Y.; Jin, C.; Zhang, L.; Hao, Z.; Chen, X.; Zhang, M.; et al. HA/CD44 Regulates the T Helper 1 Cells Differentiation by Activating Annexin A1/Akt/mTOR Signaling to Drive the Pathogenesis of EAP. Front. Immunol. 2022, 13, 875412. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican—A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Hashiguchi, T.; Kitagawa, H. Aberrant Glycosaminoglycan Biosynthesis by Tumor Suppressor EXTL2 Deficiency Promotes Liver Inflammation and Tumorigenesis through Toll-like 4 Receptor Signaling. FASEB J. 2020, 34, 8385–8401. [Google Scholar] [CrossRef] [PubMed]
- Hanoux, V.; Eguida, J.; Fleurot, E.; Levallet, J.; Bonnamy, P.-J. Increase in Hyaluronic Acid Degradation Decreases the Expression of Estrogen Receptor Alpha in MCF7 Breast Cancer Cell Line. Mol. Cell. Endocrinol. 2018, 476, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Jariyal, H.; Gupta, C.; Srivastava, A. Hyaluronic Acid Induction on Breast Cancer Stem Cells Unfolds Subtype Specific Variations in Stemness and Epithelial-to-Mesenchymal Transition. Int. J. Biol. Macromol. 2020, 160, 1078–1089. [Google Scholar] [CrossRef]
- Li, L.; Qi, L.; Liang, Z.; Song, W.; Liu, Y.; Wang, Y.; Sun, B.; Zhang, B.; Cao, W. Transforming Growth Factor-Β1 Induces EMT by the Transactivation of Epidermal Growth Factor Signaling through HA/CD44 in Lung and Breast Cancer Cells. Int. J. Mol. Med. 2015, 36, 113–122. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W. Matrix HA Promotes Specific MicroRNA Upregulation Leading to Drug Resistance and Tumor Progression. Int. J. Mol. Sci. 2016, 17, 517. [Google Scholar] [CrossRef]
- Afroz, R.; Zhou, Y.; Little, P.J.; Xu, S.; Mohamed, R.; Stow, J.; Kamato, D. Toll-like Receptor 4 Stimulates Gene Expression via Smad2 Linker Region Phosphorylation in Vascular Smooth Muscle Cells. ACS Pharmacol. Transl. Sci. 2020, 3, 524–534. [Google Scholar] [CrossRef]
- Leppert, P.C.; Jayes, F.L.; Segars, J.H. The Extracellular Matrix Contributes to Mechanotransduction in Uterine Fibroids. Obstet. Gynecol. Int. 2014, 2014, 783289. [Google Scholar] [CrossRef]
- Remodelling the Extracellular Matrix in Development and Disease—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/25415508/ (accessed on 28 July 2024).
- Zhu, L.; Liu, L.; Wang, A.; Liu, J.; Huang, X.; Zan, T. Positive Feedback Loops between Fibroblasts and the Mechanical Environment Contribute to Dermal Fibrosis. Matrix Biol. 2023, 121, 1–21. [Google Scholar] [CrossRef]
- Li, S.; Li, C.; Zhang, Y.; He, X.; Chen, X.; Zeng, X.; Liu, F.; Chen, Y.; Chen, J. Targeting Mechanics-Induced Fibroblast Activation through CD44-RhoA-YAP Pathway Ameliorates Crystalline Silica-Induced Silicosis. Theranostics 2019, 9, 4993–5008. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-Q.; Deng, X.-W.; Xu, G.-Q.; Lin, J.; Lu, H.-Z.; Chen, J. Mechanical Homeostasis Imbalance in Hepatic Stellate Cells Activation and Hepatic Fibrosis. Front. Mol. Biosci. 2023, 10, 1183808. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Toyoki, Y.; Hakamada, K.; Yoshihara, S.; Sasaki, M. Characterization of Heparan Sulfate on Hepatocytes in Regenerating Rat Liver. J. Hepato-Biliary-Pancreat. Surg. 2008, 15, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Mead, T.J.; Bhutada, S.; Martin, D.R.; Apte, S.S. Proteolysis: A Key Post-Translational Modification Regulating Proteoglycans. Am. J. Physiol. Cell. Physiol. 2022, 323, C651–C665. [Google Scholar] [CrossRef] [PubMed]
- Force-Dependent Breaching of the Basement Membrane—PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5328923/ (accessed on 28 July 2024).
- Lachowski, D.; Cortes, E.; Rice, A.; Pinato, D.; Rombouts, K.; del Rio Hernandez, A. Matrix Stiffness Modulates the Activity of MMP-9 and TIMP-1 in Hepatic Stellate Cells to Perpetuate Fibrosis. Sci. Rep. 2019, 9, 7299. [Google Scholar] [CrossRef]
- Kim, J.; Seki, E. HA in Liver Fibrosis: Basic Mechanisms, Clinical Implications, and Therapeutic Targets. Hepatol. Commun. 2023, 7, e0083. [Google Scholar] [CrossRef]
- Masola, V.; Greco, N.; Gambaro, G.; Franchi, M.; Onisto, M. Heparanase as Active Player in Endothelial Glycocalyx Remodeling. Matrix Biol. Plus 2021, 13, 100097. [Google Scholar] [CrossRef]
- Berdiaki, A.; Neagu, M.; Giatagana, E.-M.; Kuskov, A.; Tsatsakis, A.M.; Tzanakakis, G.N.; Nikitovic, D. Glycosaminoglycans: Carriers and Targets for Tailored Anti-Cancer Therapy. Biomolecules 2021, 11, 395. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Giatagana, E.-M.; Kuskov, A.; Berdiaki, A.; Tsatsakis, A.; Neagu, M.; Nikitovic, D. Proteoglycans in the Pathogenesis of Hormone-Dependent Cancers: Mediators and Effectors. Cancers 2020, 12, 2401. [Google Scholar] [CrossRef]
- Espinoza-Sánchez, N.A.; Götte, M. Role of Cell Surface Proteoglycans in Cancer Immunotherapy. Semin. Cancer Biol. 2020, 62, 48–67. [Google Scholar] [CrossRef]
- Dong, Y.; Zheng, Q.; Wang, Z.; Lin, X.; You, Y.; Wu, S.; Wang, Y.; Hu, C.; Xie, X.; Chen, J.; et al. Higher Matrix Stiffness as an Independent Initiator Triggers Epithelial-Mesenchymal Transition and Facilitates HCC Metastasis. J. Hematol. Oncol. 2019, 12, 112. [Google Scholar] [CrossRef] [PubMed]
- Pankova, D.; Chen, Y.; Terajima, M.; Schliekelman, M.J.; Baird, B.N.; Fahrenholtz, M.; Sun, L.; Gill, B.J.; Vadakkan, T.J.; Kim, M.P.; et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-Link Switch in Tumor Stroma. Mol. Cancer Res. 2016, 14, 287–295. [Google Scholar] [CrossRef] [PubMed]
- De Felice, D.; Alaimo, A. Mechanosensitive Piezo Channels in Cancer: Focus on Altered Calcium Signaling in Cancer Cells and in Tumor Progression. Cancers 2020, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Kalli, M.; Poskus, M.D.; Stylianopoulos, T.; Zervantonakis, I.K. Beyond Matrix Stiffness: Targeting Force-Induced Cancer Drug Resistance. Trends Cancer 2023, 9, 937–954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Jia, Y.; Yu, Y.; Zhang, B.; Xu, F.; Guo, H. Targeting the Tumor Biophysical Microenvironment to Reduce Resistance to Immunotherapy. Adv. Drug Deliv. Rev. 2022, 186, 114319. [Google Scholar] [CrossRef]
- Tilsed, C.M.; Fisher, S.A.; Nowak, A.K.; Lake, R.A.; Lesterhuis, W.J. Cancer Chemotherapy: Insights into Cellular and Tumor Microenvironmental Mechanisms of Action. Front. Oncol. 2022, 12, 960317. [Google Scholar] [CrossRef]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Epithelial Mesenchymal Transition and Cancer Stem Cell-like Phenotypes Facilitate Chemoresistance in Recurrent Ovarian Cancer. Curr. Cancer Drug Targets 2010, 10, 268–278. [Google Scholar] [CrossRef]
- Darvishi, B.; Eisavand, M.R.; Majidzadeh-A, K.; Farahmand, L. Matrix Stiffening and Acquired Resistance to Chemotherapy: Concepts and Clinical Significance. Br. J. Cancer 2022, 126, 1253–1263. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-Mediated Drug Resistance: A Major Contributor to Minimal Residual Disease. Nat. Rev. Cancer. 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Deng, M.; Lin, J.; Nowsheen, S.; Liu, T.; Zhao, Y.; Villalta, P.W.; Sicard, D.; Tschumperlin, D.J.; Lee, S.; Kim, J.; et al. Extracellular Matrix Stiffness Determines DNA Repair Efficiency and Cellular Sensitivity to Genotoxic Agents. Sci. Adv. 2020, 6, eabb2630. [Google Scholar] [CrossRef]
- Baltes, F.; Pfeifer, V.; Silbermann, K.; Caspers, J.; Wantoch von Rekowski, K.; Schlesinger, M.; Bendas, G. β1-Integrin Binding to Collagen Type 1 Transmits Breast Cancer Cells into Chemoresistance by Activating ABC Efflux Transporters. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118663. [Google Scholar] [CrossRef] [PubMed]
- Bell-McGuinn, K.M.; Matthews, C.M.; Ho, S.N.; Barve, M.; Gilbert, L.; Penson, R.T.; Lengyel, E.; Palaparthy, R.; Gilder, K.; Vassos, A.; et al. A Phase II, Single-Arm Study of the Anti-A5β1 Integrin Antibody Volociximab as Monotherapy in Patients with Platinum-Resistant Advanced Epithelial Ovarian or Primary Peritoneal Cancer. Gynecol. Oncol. 2011, 121, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Haddad, T.; Qin, R.; Lupu, R.; Satele, D.; Eadens, M.; Goetz, M.P.; Erlichman, C.; Molina, J. A Phase I Study of Cilengitide and Paclitaxel in Patients with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2017, 79, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- McNeel, D.G.; Eickhoff, J.; Lee, F.T.; King, D.M.; Alberti, D.; Thomas, J.P.; Friedl, A.; Kolesar, J.; Marnocha, R.; Volkman, J.; et al. Phase I Trial of a Monoclonal Antibody Specific for αvβ3 Integrin (MEDI-522) in Patients with Advanced Malignancies, Including an Assessment of Effect on Tumor Perfusion. Clin. Cancer Res. 2005, 11, 7851–7860. [Google Scholar] [CrossRef] [PubMed]
- Tedla, Y.G.; Yano, Y.; Carnethon, M.; Greenland, P. Association Between Long-Term Blood Pressure Variability and 10-Year Progression in Arterial Stiffness: The Multiethnic Study of Atherosclerosis. Hypertension 2017, 69, 118–127. [Google Scholar] [CrossRef]
- Goswami, R.; Cohen, J.; Sharma, S.; Zhang, D.X.; Lafyatis, R.; Bhawan, J.; Rahaman, S.O. TRPV4 ION Channel Is Associated with Scleroderma. J. Investig. Dermatol. 2017, 137, 962–965. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Grove, L.M.; Paruchuri, S.; Southern, B.D.; Abraham, S.; Niese, K.A.; Scheraga, R.G.; Ghosh, S.; Thodeti, C.K.; Zhang, D.X.; et al. TRPV4 Mediates Myofibroblast Differentiation and Pulmonary Fibrosis in Mice. J. Clin. Investig. 2014, 124, 5225–5238. [Google Scholar] [CrossRef]
- Tang, W.; Fan, Y. SIRT6 as a Potential Target for Treating Insulin Resistance. Life Sci. 2019, 231, 116558. [Google Scholar] [CrossRef]
- Li, L.; Wang, F.; Wei, X.; Liang, Y.; Cui, Y.; Gao, F.; Zhong, J.; Pu, Y.; Zhao, Y.; Yan, Z.; et al. Transient Receptor Potential Vanilloid 1 Activation by Dietary Capsaicin Promotes Urinary Sodium Excretion by Inhibiting Epithelial Sodium Channel α Subunit-Mediated Sodium Reabsorption. Hypertension 2014, 64, 397–404. [Google Scholar] [CrossRef]
- Ma, L.; Zhong, J.; Zhao, Z.; Luo, Z.; Ma, S.; Sun, J.; He, H.; Zhu, T.; Liu, D.; Zhu, Z.; et al. Activation of TRPV1 Reduces Vascular Lipid Accumulation and Attenuates Atherosclerosis. Cardiovasc. Res. 2011, 92, 504–513. [Google Scholar] [CrossRef]
- Xu, S.; Liu, B.; Yin, M.; Koroleva, M.; Mastrangelo, M.; Ture, S.; Morrell, C.N.; Zhang, D.X.; Fisher, E.A.; Jin, Z.G. A Novel TRPV4-Specific Agonist Inhibits Monocyte Adhesion and Atherosclerosis. Oncotarget 2016, 7, 37622–37635. [Google Scholar] [CrossRef] [PubMed]
- Thorneloe, K.S.; Cheung, M.; Bao, W.; Alsaid, H.; Lenhard, S.; Jian, M.-Y.; Costell, M.; Maniscalco-Hauk, K.; Krawiec, J.A.; Olzinski, A.; et al. An Orally Active TRPV4 Channel Blocker Prevents and Resolves Pulmonary Edema Induced by Heart Failure. Sci. Transl. Med. 2012, 4, 159ra148. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Skrdla, P.; Schroyer, R.; Kumar, S.; Fernando, D.; Oughton, A.; Norton, N.; Sprecher, D.L.; Cheriyan, J. Clinical Pharmacokinetics, Safety, and Tolerability of a Novel, First-in-Class TRPV4 Ion Channel Inhibitor, GSK2798745, in Healthy and Heart Failure Subjects. Am. J. Cardiovasc. Drugs 2019, 19, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Rahaman, S.G.; Goswami, R.; Dutta, B.; Mahanty, M.; Rahaman, S.O. Role of Mechanosensitive Channels/Receptors in Atherosclerosis. Am. J. Physiol. Cell Physiol. 2022, 322, C927–C938. [Google Scholar] [CrossRef] [PubMed]
- Puech, P.-H.; Bongrand, P. Mechanotransduction as a Major Driver of Cell Behaviour: Mechanisms, and Relevance to Cell Organization and Future Research. Open Biol. 2021, 11, 210256. [Google Scholar] [CrossRef] [PubMed]
- Bongrand, P. Understanding How Cells Probe the World: A Preliminary Step towards Modeling Cell Behavior? Int. J. Mol. Sci. 2023, 24, 2266. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep Learning. Nature. 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Caicedo, J.C.; Cooper, S.; Heigwer, F.; Warchal, S.; Qiu, P.; Molnar, C.; Vasilevich, A.S.; Barry, J.D.; Bansal, H.S.; Kraus, O.; et al. Data-Analysis Strategies for Image-Based Cell Profiling. Nat. Methods 2017, 14, 849–863. [Google Scholar] [CrossRef]
- Das, A.; Fischer, R.S.; Pan, D.; Waterman, C.M. YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-Dependent, Myosin II- and Phospho-YAP-Independent Pathway during Extracellular Matrix Mechanosensing. J. Biol. Chem. 2016, 291, 6096–6110. [Google Scholar] [CrossRef]
- Bonnevie, E.D.; Ashinsky, B.G.; Dekky, B.; Volk, S.W.; Smith, H.E.; Mauck, R.L. Cell Morphology and Mechanosensing Can Be Decoupled in Fibrous Microenvironments and Identified Using Artificial Neural Networks. Sci. Rep. 2021, 11, 5950. [Google Scholar] [CrossRef]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a Distance: Mechanically Coupling the Extracellular Matrix with the Nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Gann, P.H.; Deaton, R.; Amatya, A.; Mohnani, M.; Rueter, E.E.; Yang, Y.; Ananthanarayanan, V. Development of a Nuclear Morphometric Signature for Prostate Cancer Risk in Negative Biopsies. PLoS ONE 2013, 8, e69457. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.Y.; Wu, Y.; Goh, D.; Tan, V.; Ng, C.W.; Lim, J.C.T.; Lau, M.C.; Yeong, J.P.S. Application of Artificial Intelligence to In Vitro Tumor Modeling and Characterization of the Tumor Microenvironment. Adv. Healthc. Mater. 2023, 12, e2202457. [Google Scholar] [CrossRef] [PubMed]
- Binas, D.A.; Tzanakakis, P.; Economopoulos, T.L.; Konidari, M.; Bourgioti, C.; Moulopoulos, L.A.; Matsopoulos, G.K. A Novel Approach for Estimating Ovarian Cancer Tissue Heterogeneity through the Application of Image Processing Techniques and Artificial Intelligence. Cancers 2023, 15, 1058. [Google Scholar] [CrossRef] [PubMed]
- SubramanianBalachandar, V.; Islam, M.M.; Steward, R.L. A Machine Learning Approach to Predict Cellular Mechanical Stresses in Response to Chemical Perturbation. Biophys. J. 2023, 122, 3413–3424. [Google Scholar] [CrossRef] [PubMed]

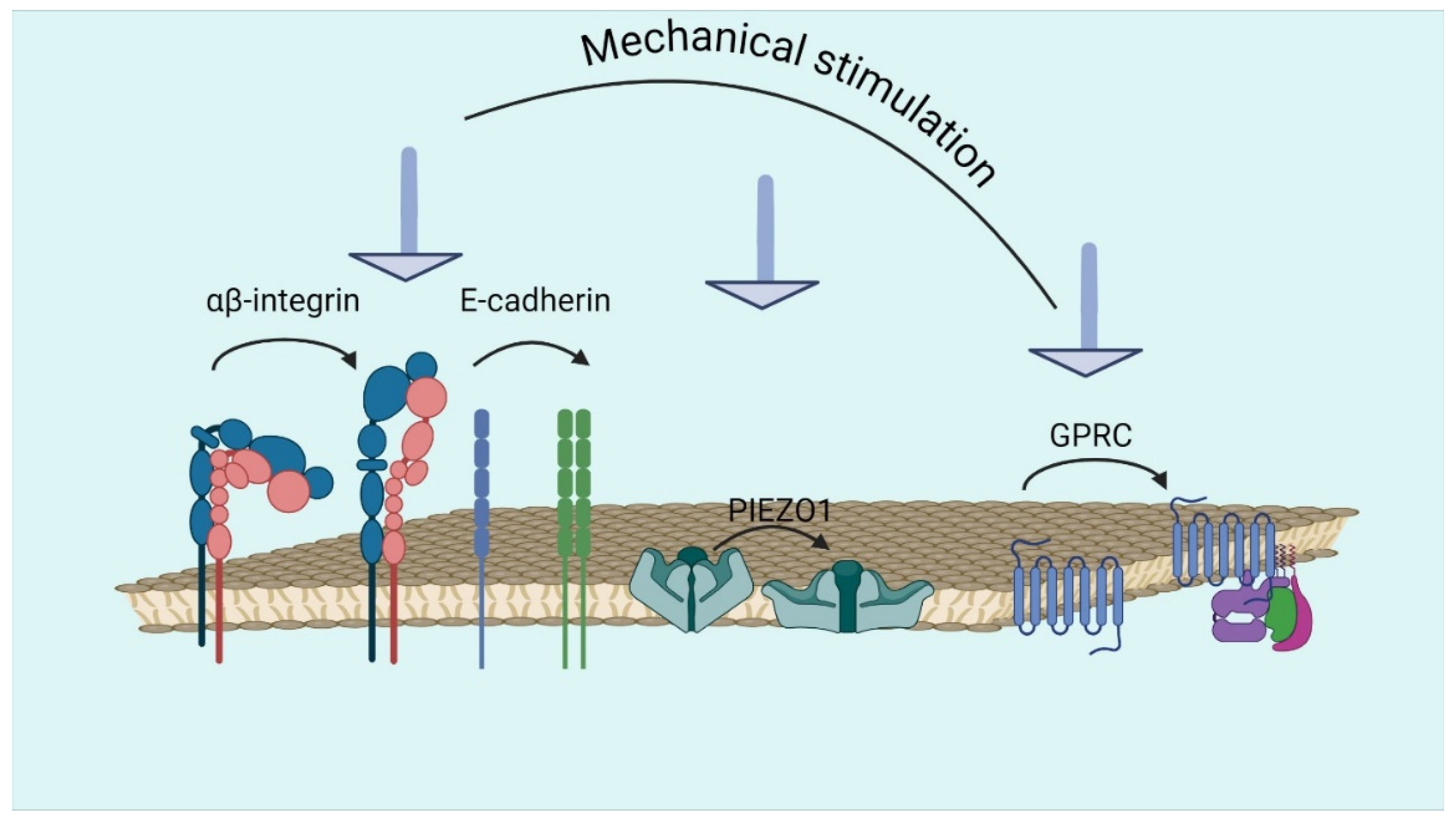


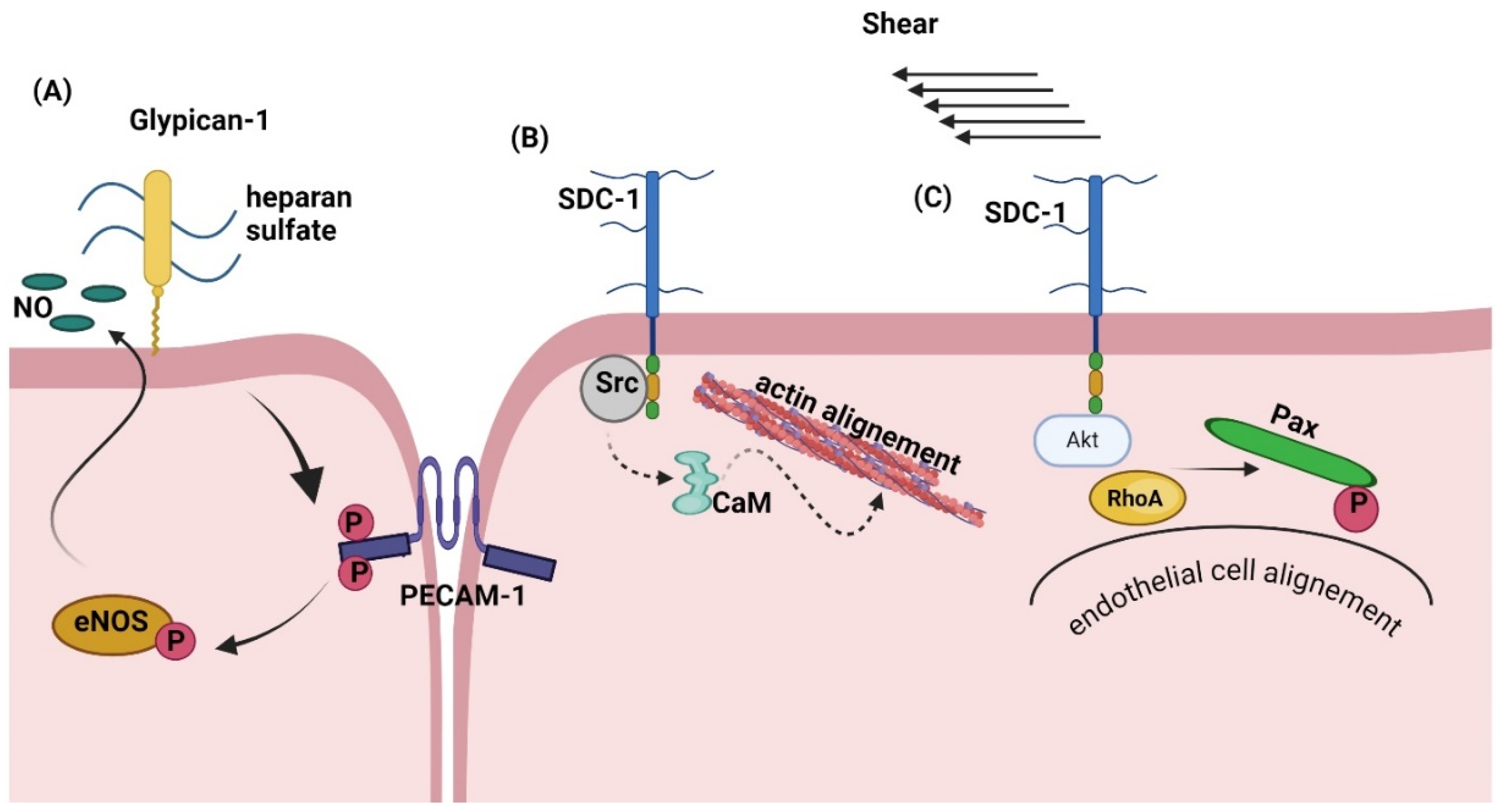
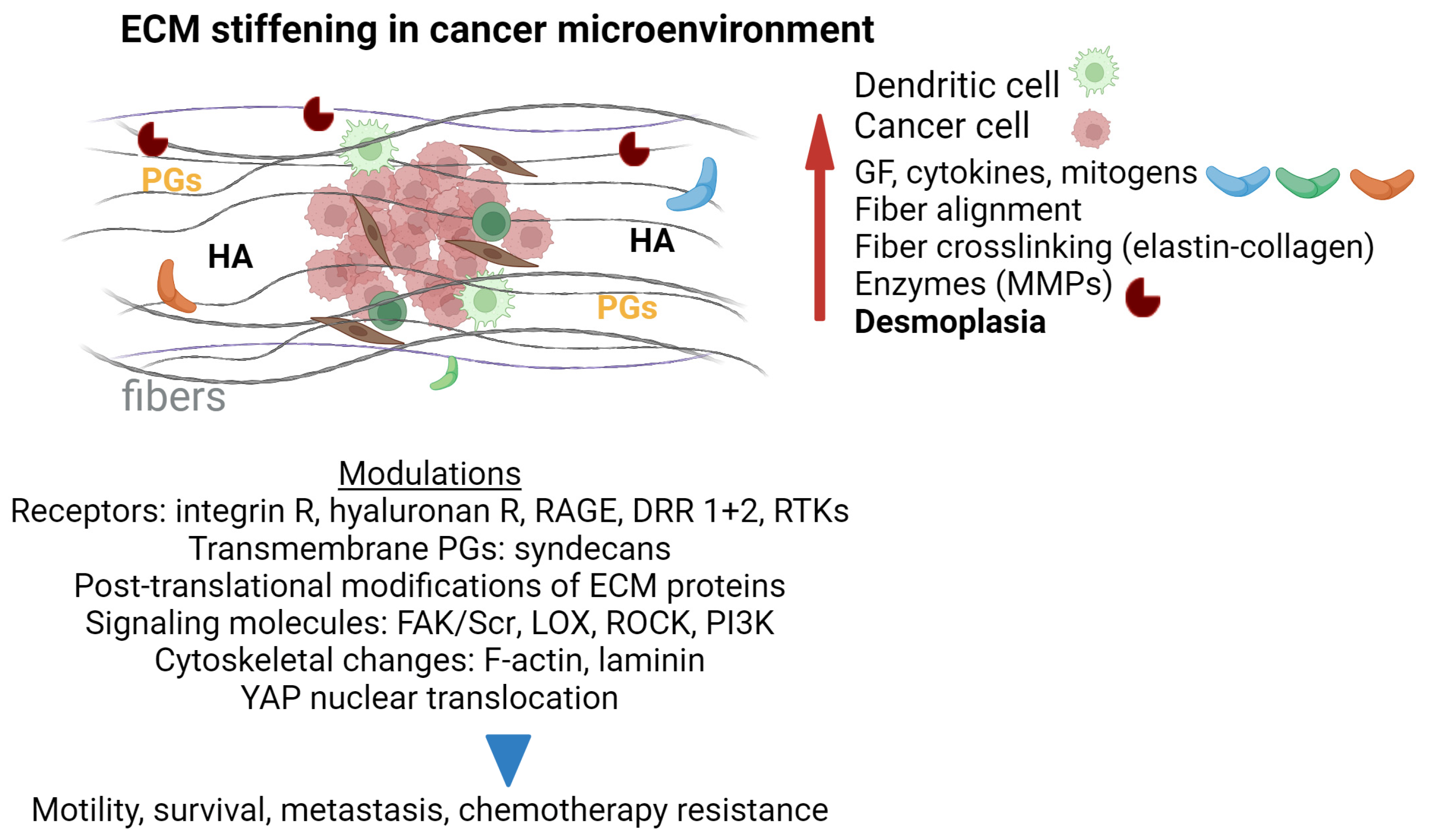
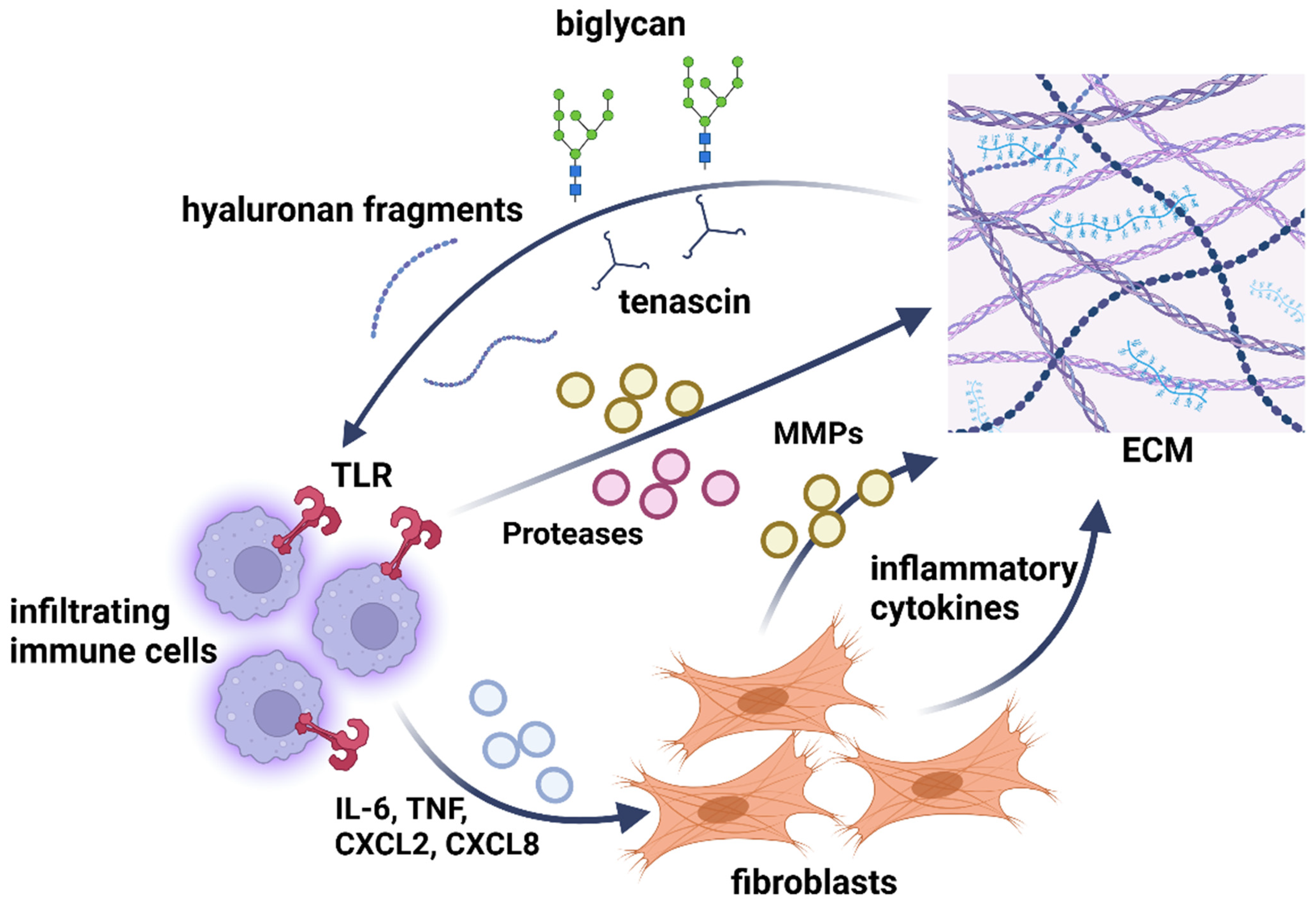
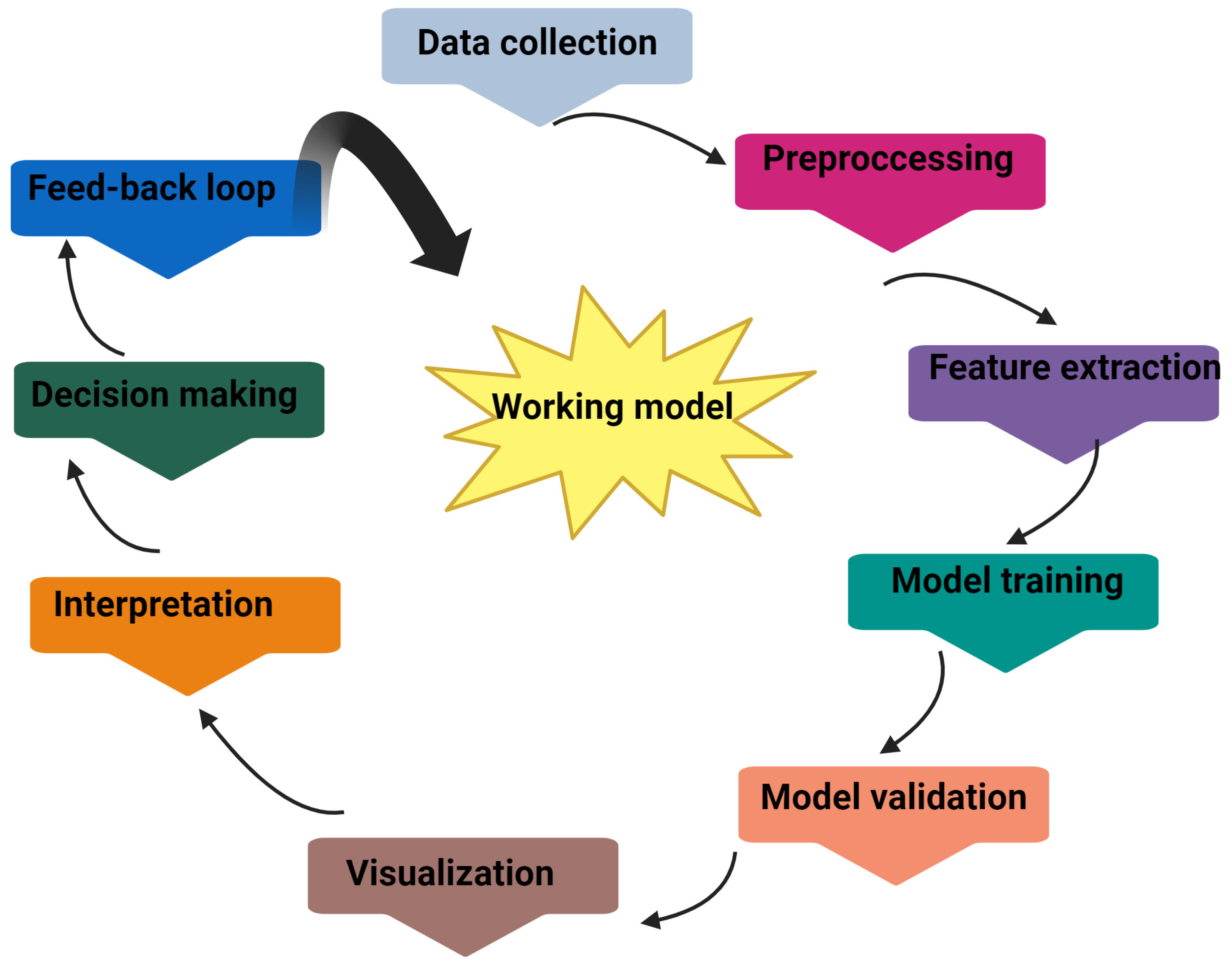

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stiffness/Elasticity | Viscoelasticity | Strength & Toughness | Anisotropy | Adhesive Properties | Remodeling & Plasticity |
|---|---|---|---|---|---|
| Collagen | Collagen | Collagen | Collagen | Collagen | Cell-matrix adhesion receptors |
| Elastin | Elastin | Elastin | Elastin | PGs | Cytokines & growth factors |
| Glycoproteins | Glycoproteins | Glycoproteins | Glycoproteins | Glycoproteins tenascin | TGF-b |
| Crosslinking molecules | Crosslinking molecules | Crosslinking molecules | Crosslinking molecules | Fibronectin | Fibroblasts & myofibroblasts |
| PGs | PGs | PGs | PGs | Laminin | Crosslinking enzymes |
| Water | Water | Water | Water | Integrins | Matrix metalloprorteinases |
| Matrix metalloproteinases | GAGs | GAGs | Tissue inhibitors of MMPs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berdiaki, A.; Neagu, M.; Tzanakakis, P.; Spyridaki, I.; Pérez, S.; Nikitovic, D. Extracellular Matrix Components and Mechanosensing Pathways in Health and Disease. Biomolecules 2024, 14, 1186. https://doi.org/10.3390/biom14091186
Berdiaki A, Neagu M, Tzanakakis P, Spyridaki I, Pérez S, Nikitovic D. Extracellular Matrix Components and Mechanosensing Pathways in Health and Disease. Biomolecules. 2024; 14(9):1186. https://doi.org/10.3390/biom14091186
Chicago/Turabian StyleBerdiaki, Aikaterini, Monica Neagu, Petros Tzanakakis, Ioanna Spyridaki, Serge Pérez, and Dragana Nikitovic. 2024. "Extracellular Matrix Components and Mechanosensing Pathways in Health and Disease" Biomolecules 14, no. 9: 1186. https://doi.org/10.3390/biom14091186
APA StyleBerdiaki, A., Neagu, M., Tzanakakis, P., Spyridaki, I., Pérez, S., & Nikitovic, D. (2024). Extracellular Matrix Components and Mechanosensing Pathways in Health and Disease. Biomolecules, 14(9), 1186. https://doi.org/10.3390/biom14091186