

SEPT9_i1 and Septin Dynamics in Oncogenesis and Cancer Treatment

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Septin 9 and SEPT9_i1—Structure as Basis of Protein Interactions
3. SEPT9_i1 Contributes to Cancer Formation, Malignancy, and Resistance to Treatment
3.1. SEPT9_i1 Prevents Degradation, Facilitates Transport and Scaffolds for Transcription Factors
3.2. SEPT9_i1 Is Crucial for Integrating Septin Dynamics with Other Components of the Cytoskeleton
3.2.1. SEPT9_i1 Is a Crucial Factor in Cell Invasiveness
3.2.2. SEPT9_i1 Interaction with Microtubules Contributes to Taxane Resistance
3.3. Competition and Synergism between SEPT9_i1 and Its Isoforms and Their Relevance to Cancer Biology
4. SEPT9_i1 and Other Septins as Novel Therapeutic Targets in Cancer
4.1. Forchlorfenuron—Mechanism and Effects of Action
4.2. Septin Chemotherapy beyond Cytotoxicity
4.2.1. Septin Inhibition as a Tool for Overriding Tumor Resistance to Other Forms of Treatment
4.2.2. Septin Inhibitors as Potential Migrastatics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gopal, S.; Sharpless, N.E. Cancer as a Global Health Priority. JAMA 2021, 326, 809. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Zaidi, S.K.; Sengupta, R. AACR Cancer Disparities Progress Report 2022. Cancer Epidemiol. Biomark. Prev. 2022, 31, 1249–1250. [Google Scholar] [CrossRef] [PubMed]
- Biber, G.; Ben-Shmuel, A.; Sabag, B.; Barda-Saad, M. Actin Regulators in Cancer Progression and Metastases: From Structure and Function to Cytoskeletal Dynamics. Int. Rev. Cell Mol. Biol. 2020, 356, 131–196. [Google Scholar] [PubMed]
- Sneeggen, M.; Guadagno, N.A.; Progida, C. Intracellular Transport in Cancer Metabolic Reprogramming. Front. Cell Dev. Biol. 2020, 8, 597608. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. The Cytoskeleton and Cancer. Cancer Metastasis Rev. 2009, 28, 5–14. [Google Scholar] [CrossRef]
- Li, X.; Wang, J. Mechanical Tumor Microenvironment and Transduction: Cytoskeleton Mediates Cancer Cell Invasion and Metastasis. Int. J. Biol. Sci. 2020, 16, 2014–2028. [Google Scholar] [CrossRef]
- Mostowy, S.; Cossart, P. Septins: The Fourth Component of the Cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 183–194. [Google Scholar] [CrossRef]
- Poüs, C.; Klipfel, L.; Baillet, A. Cancer-Related Functions and Subcellular Localizations of Septins. Front. Cell Dev. Biol. 2016, 4, 126. [Google Scholar] [CrossRef]
- Cavini, I.A.; Leonardo, D.A.; Rosa, H.V.D.; Castro, D.K.S.V.; D’Muniz Pereira, H.; Valadares, N.F.; Araujo, A.P.U.; Garratt, R.C. The Structural Biology of Septins and Their Filaments: An Update. Front. Cell Dev. Biol. 2021, 9, 765085. [Google Scholar] [CrossRef]
- Hartwell, L. Genetic Control of the Cell Division Cycle in Yeast *1IV. Genes Controlling Bud Emergence and Cytokinesis. Exp. Cell Res. 1971, 69, 265–276. [Google Scholar] [CrossRef]
- Schmidt, K.; Nichols, B.J. A Barrier to Lateral Diffusion in the Cleavage Furrow of Dividing Mammalian Cells. Curr. Biol. 2004, 14, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Montagna, C.; Sagie, M.; Zechmeister, J. Mammalian Septins in Health and Disease. Res. Rep. Biochem. 2015, 5, 59–72. [Google Scholar] [CrossRef]
- Weems, A.; McMurray, M. The Step-Wise Pathway of Septin Hetero-Octamer Assembly in Budding Yeast. Elife 2017, 6, e23689. [Google Scholar] [CrossRef] [PubMed]
- Radler, M.R.; Suber, A.; Spiliotis, E.T. Spatial Control of Membrane Traffic in Neuronal Dendrites. Mol. Cell. Neurosci. 2020, 105, 103492. [Google Scholar] [CrossRef]
- Spiliotis, E.T.; Kesisova, I.A. Spatial Regulation of Microtubule-Dependent Transport by Septin GTPases. Trends Cell Biol. 2021, 31, 979–993. [Google Scholar] [CrossRef]
- Spiliotis, E.T.; Nakos, K. Cellular Functions of Actin- and Microtubule-Associated Septins. Curr. Biol. 2021, 31, R651–R666. [Google Scholar] [CrossRef]
- Marttinen, M.; Kurkinen, K.M.; Soininen, H.; Haapasalo, A.; Hiltunen, M. Synaptic Dysfunction and Septin Protein Family Members in Neurodegenerative Diseases. Mol. Neurodegener. 2015, 10, 16. [Google Scholar] [CrossRef]
- Osaka, M.; Rowley, J.D.; Zeleznik-Le, N.J. MSF (MLL Septin-like Fusion), a Fusion Partner Gene of MLL, in a Therapy-Related Acute Myeloid Leukemia with a t(11;17)(Q23;Q25). Proc. Natl. Acad. Sci. USA 1999, 96, 6428–6433. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, M.Y.; Li, Y.W.; Zhang, S.W. Structure and Function of Septin 9 and Its Role in Human Malignant Tumors. World J. Gastrointest. Oncol. 2020, 12, 619–631. [Google Scholar] [CrossRef]
- Min, L.; Chen, J.; Yu, M.; Liu, D. Using Circulating Tumor DNA as a Novel Biomarker to Screen and Diagnose Colorectal Cancer: A Meta-Analysis. J. Clin. Med. 2023, 12, 408. [Google Scholar] [CrossRef]
- Li, B.; Huang, H.; Huang, R.; Zhang, W.; Zhou, G.; Wu, Z.; Lv, C.; Han, X.; Jiang, L.; Li, Y.; et al. SEPT9 Gene Methylation as a Noninvasive Marker for Hepatocellular Carcinoma. Dis. Markers 2020, 2020, 6289063. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Yang, G.; Jiang, H.; Chen, X.; Yang, Y.; Yu, Q.; Pan, B.; Wang, B.; Guo, W.; Yang, W.; et al. Role of Plasma Methylated SEPT9 for Predicting Microvascular Invasion and Tumor Proliferation in Hepatocellular Carcinoma. Technol. Cancer Res. Treat. 2022, 21, 153303382211445. [Google Scholar] [CrossRef] [PubMed]
- Matsui, S.; Kagara, N.; Mishima, C.; Naoi, Y.; Shimoda, M.; Shimomura, A.; Shimazu, K.; Kim, S.J.; Noguchi, S. Methylation of the SEPT9_v2 Promoter as a Novel Marker for the Detection of Circulating Tumor DNA in Breast Cancer Patients. Oncol. Rep. 2016, 36, 2225–2235. [Google Scholar] [CrossRef]
- Krausewitz, P.; Kluemper, N.; Richter, A.-P.; Büttner, T.; Kristiansen, G.; Ritter, M.; Ellinger, J. Early Dynamics of Quantitative SEPT9 and SHOX2 Methylation in Circulating Cell-Free Plasma DNA during Prostate Biopsy for Prostate Cancer Diagnosis. Cancers 2022, 14, 4355. [Google Scholar] [CrossRef] [PubMed]
- Powrózek, T.; Krawczyk, P.; Kucharczyk, T.; Milanowski, J. Septin 9 Promoter Region Methylation in Free Circulating DNA—Potential Role in Noninvasive Diagnosis of Lung Cancer: Preliminary Report. Med. Oncol. 2014, 31, 917. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, M.; Zhang, S.; Liu, Y. Plasma-Methylated SEPT9 for the Noninvasive Diagnosis of Gastric Cancer. J. Clin. Med. 2022, 11, 6399. [Google Scholar] [CrossRef]
- Zeng, Y.; Cao, Y.; Liu, L.; Zhao, J.; Zhang, T.; Xiao, L.; Jia, M.; Tian, Q.; Yu, H.; Chen, S.; et al. SEPT9_i1 Regulates Human Breast Cancer Cell Motility through Cytoskeletal and RhoA/FAK Signaling Pathway Regulation. Cell Death Dis. 2019, 10, 720. [Google Scholar] [CrossRef]
- Connolly, D.; Yang, Z.; Castaldi, M.; Simmons, N.; Oktay, M.H.; Coniglio, S.; Fazzari, M.J.; Verdier-Pinard, P.; Montagna, C. Septin 9 Isoform Expression, Localization and Epigenetic Changes during Human and Mouse Breast Cancer Progression. Breast Cancer Res. 2011, 13, R76. [Google Scholar] [CrossRef]
- Jiao, X.; Zhang, S.; Jiao, J.; Zhang, T.; Qu, W.; Muloye, G.M.; Kong, B.; Zhang, Q.; Cui, B. Promoter Methylation of SEPT9 as a Potential Biomarker for Early Detection of Cervical Cancer and Its Overexpression Predicts Radioresistance. Clin. Epigenetics 2019, 11, 120. [Google Scholar] [CrossRef]
- Stanbery, L.; D’Silva, N.J.; Lee, J.S.; Bradford, C.R.; Carey, T.E.; Prince, M.E.; Wolf, G.T.; Worden, F.P.; Cordell, K.G.; Petty, E.M. High SEPT9_v1 Expression Is Associated with Poor Clinical Outcomes in Head and Neck Squamous Cell Carcinoma. Transl. Oncol. 2010, 3, 239–245. [Google Scholar] [CrossRef]
- Peterson, E.; Petty, E. Conquering the Complex World of Human Septins: Implications for Health and Disease. Clin. Genet. 2010, 77, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Marcus, J.; Bejerano-Sagie, M.; Patterson, N.; Bagchi, S.; Verkhusha, V.V.; Connolly, D.; Goldberg, G.L.; Golden, A.; Sharma, V.P.; Condeelis, J.; et al. Septin 9 Isoforms Promote Tumorigenesis in Mammary Epithelial Cells by Increasing Migration and ECM Degradation through Metalloproteinase Secretion at Focal Adhesions. Oncogene 2019, 38, 5839–5859. [Google Scholar] [CrossRef] [PubMed]
- Iv, F.; Martins, C.S.; Castro-Linares, G.; Taveneau, C.; Barbier, P.; Verdier-Pinard, P.; Camoin, L.; Audebert, S.; Tsai, F.-C.; Ramond, L.; et al. Insights into Animal Septins Using Recombinant Human Septin Octamers with Distinct SEPT9 Isoforms. J. Cell Sci. 2021, 134, jcs258484. [Google Scholar] [CrossRef] [PubMed]
- Zuvanov, L.; Mota, D.M.D.; Araujo, A.P.U.; DeMarco, R. A Blueprint of Septin Expression in Human Tissues. Funct. Integr. Genom. 2019, 19, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Gönczi, M.; Dienes, B.; Dobrosi, N.; Fodor, J.; Balogh, N.; Oláh, T.; Csernoch, L. Septins, a Cytoskeletal Protein Family, with Emerging Role in Striated Muscle. J. Muscle Res. Cell Motil. 2021, 42, 251–265. [Google Scholar] [CrossRef]
- Füchtbauer, A.; Lassen, L.B.; Jensen, A.B.; Howard, J.; Quiroga, A.d.S.; Warming, S.; Sørensen, A.B.; Pedersen, F.S.; Füchtbauer, E.-M. Septin9 Is Involved in Septin Filament Formation and Cellular Stability. Biol. Chem. 2011, 392, 769–777. [Google Scholar] [CrossRef]
- Dolat, L.; Hu, Q.; Spiliotis, E.T. Septin Functions in Organ System Physiology and Pathology. Biol. Chem. 2014, 395, 123–141. [Google Scholar] [CrossRef]
- Ono, R.; Ihara, M.; Nakajima, H.; Ozaki, K.; Kataoka-Fujiwara, Y.; Taki, T.; Nagata, K.; Inagaki, M.; Yoshida, N.; Kitamura, T.; et al. Disruption of Sept6, a Fusion Partner Gene of MLL, Does Not Affect Ontogeny, Leukemogenesis Induced by MLL-SEPT6, or Phenotype Induced by the Loss of Sept4. Mol. Cell Biol. 2005, 25, 10965–10978. [Google Scholar] [CrossRef]
- Suber, Y.; Alam, M.N.A.; Nakos, K.; Bhakt, P.; Spiliotis, E.T. Microtubule-Associated Septin Complexes Modulate Kinesin and Dynein Motility with Differential Specificities. J. Biol. Chem. 2023, 299, 105084. [Google Scholar] [CrossRef]
- Angelis, D.; Spiliotis, E.T. Septin Mutations in Human Cancers. Front. Cell Dev. Biol. 2016, 4, 122. [Google Scholar] [CrossRef]
- Auxier, B.; Dee, J.; Berbee, M.L.; Momany, M. Diversity of Opisthokont Septin Proteins Reveals Structural Constraints and Conserved Motifs. BMC Evol. Biol. 2019, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Shuman, B.; Momany, M. Septins From Protists to People. Front. Cell Dev. Biol. 2022, 9, 824850. [Google Scholar] [CrossRef] [PubMed]
- Grupp, B.; Denkhaus, L.; Gerhardt, S.; Vögele, M.; Johnsson, N.; Gronemeyer, T. The Structure of a Tetrameric Septin Complex Reveals a Hydrophobic Element Essential for NC-Interface Integrity. Commun. Biol. 2024, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, D.C.; Macedo, J.N.; Guimarães, S.L.; Barroso da Silva, F.L.; Cassago, A.; Garratt, R.C.; Portugal, R.V.; Araujo, A.P.U. A Revised Order of Subunits in Mammalian Septin Complexes. Cytoskeleton 2019, 76, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Curtis, B.N.; Vogt, E.J.D.; Cannon, K.S.; Gladfelter, A.S. Reconstitution of Septin Assembly at Membranes to Study Biophysical Properties and Functions. J. Vis. Exp. 2022, 185, e64090. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, Y.; Momany, M. Posttranslational Modifications and Assembly of Septin Heteropolymers and Higher-Order Structures. Curr. Opin. Microbiol. 2012, 15, 660–668. [Google Scholar] [CrossRef]
- Castro, D.K.S.V.; Rosa, H.V.D.; Mendonça, D.C.; Cavini, I.A.; Araujo, A.P.U.; Garratt, R.C. Dissecting the Binding Interface of the Septin Polymerization Enhancer Borg BD3. J. Mol. Biol. 2023, 435, 168132. [Google Scholar] [CrossRef]
- Szuba, A.; Bano, F.; Castro-Linares, G.; Iv, F.; Mavrakis, M.; Richter, R.P.; Bertin, A.; Koenderink, G.H. Membrane Binding Controls Ordered Self-Assembly of Animal Septins. Elife 2021, 10, e63349. [Google Scholar] [CrossRef]
- Benoit, B.; Poüs, C.; Baillet, A. Septins as Membrane Influencers: Direct Play or in Association with Other Cytoskeleton Partners. Front. Cell Dev. Biol. 2023, 11, 1112319. [Google Scholar] [CrossRef]
- McIlhatton, M.A.; Burrows, J.F.; Donaghy, P.G.; Chanduloy, S.; Johnston, P.G.; Russell, S.E.H. Genomic Organization, Complex Splicing Pattern and Expression of a Human Septin Gene on Chromosome 17q25.3. Oncogene 2001, 20, 5930–5939. [Google Scholar] [CrossRef]
- Connolly, D.; Hoang, H.G.; Adler, E.; Tazearslan, C.; Simmons, N.; Bernard, V.V.; Castaldi, M.; Oktay, M.H.; Montagna, C. Septin 9 Amplification and Isoform-Specific Expression in Peritumoral and Tumor Breast Tissue. Biol. Chem. 2014, 395, 157–167. [Google Scholar] [CrossRef]
- Smith, C.; Dolat, L.; Angelis, D.; Forgacs, E.; Spiliotis, E.T.; Galkin, V.E. Septin 9 Exhibits Polymorphic Binding to F-Actin and Inhibits Myosin and Cofilin Activity. J. Mol. Biol. 2015, 427, 3273–3284. [Google Scholar] [CrossRef]
- Dolat, L.; Hunyara, J.L.; Bowen, J.R.; Karasmanis, E.P.; Elgawly, M.; Galkin, V.E.; Spiliotis, E.T. Septins Promote Stress Fiber–Mediated Maturation of Focal Adhesions and Renal Epithelial Motility. J. Cell Biol. 2014, 207, 225–235. [Google Scholar] [CrossRef]
- Kesisova, I.A.; Robinson, B.P.; Spiliotis, E.T. A Septin GTPase Scaffold of Dynein–Dynactin Motors Triggers Retrograde Lysosome Transport. J. Cell Biol. 2021, 220, e202005219. [Google Scholar] [CrossRef]
- Omrane, M.; Camara, A.S.; Taveneau, C.; Benzoubir, N.; Tubiana, T.; Yu, J.; Guérois, R.; Samuel, D.; Goud, B.; Poüs, C.; et al. Septin 9 Has Two Polybasic Domains Critical to Septin Filament Assembly and Golgi Integrity. iScience 2019, 13, 138–153. [Google Scholar] [CrossRef]
- Martins, C.S.; Taveneau, C.; Castro-Linares, G.; Baibakov, M.; Buzhinsky, N.; Eroles, M.; Milanović, V.; Omi, S.; Pedelacq, J.-D.; Iv, F.; et al. Human Septins Organize as Octamer-Based Filaments and Mediate Actin-Membrane Anchoring in Cells. J. Cell Biol. 2023, 222, e202203016. [Google Scholar] [CrossRef]
- Kuzmić, M.; Castro Linares, G.; Leischner Fialová, J.; Iv, F.; Salaün, D.; Llewellyn, A.; Gomes, M.; Belhabib, M.; Liu, Y.; Asano, K.; et al. Septin-Microtubule Association via a Motif Unique to Isoform 1 of Septin 9 Tunes Stress Fibers. J. Cell Sci. 2022, 135, jcs258850. [Google Scholar] [CrossRef]
- Nakos, K.; Rosenberg, M.; Spiliotis, E.T. Regulation of Microtubule plus End Dynamics by Septin 9. Cytoskeleton 2019, 76, 83–91. [Google Scholar] [CrossRef]
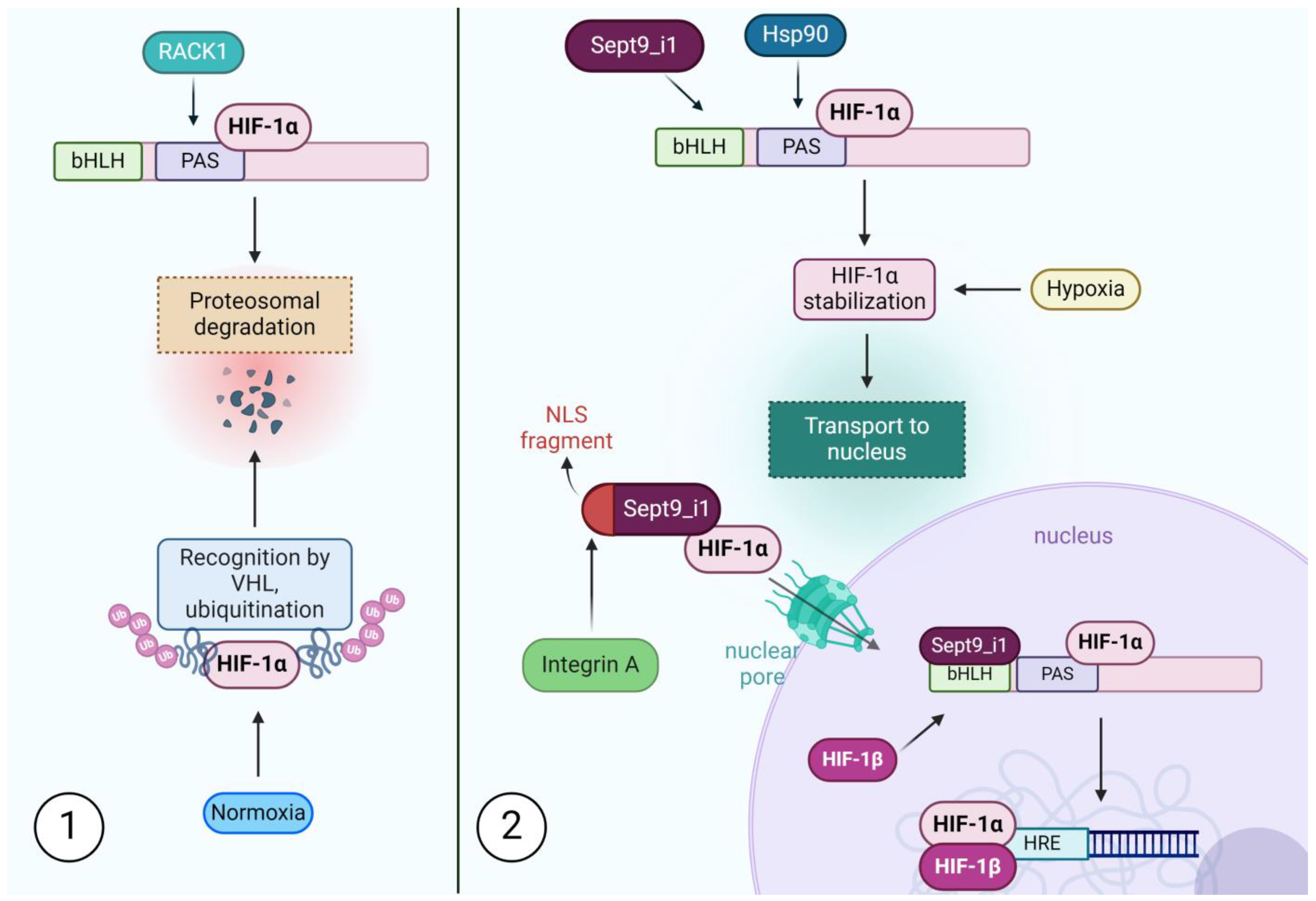
- Golan, M.; Mabjeesh, N.J. SEPT9-I1 Is Required for the Association between HIF-1α and Importin-α to Promote Efficient Nuclear Translocation. Cell Cycle 2013, 12, 2297–2308. [Google Scholar] [CrossRef]
- Lu, J.; Wu, T.; Zhang, B.; Liu, S.; Song, W.; Qiao, J.; Ruan, H. Types of Nuclear Localization Signals and Mechanisms of Protein Import into the Nucleus. Cell Commun. Signal. 2021, 19, 60. [Google Scholar] [CrossRef]
- Nölke, T.; Schwan, C.; Lehmann, F.; Østevold, K.; Pertz, O.; Aktories, K. Septins Guide Microtubule Protrusions Induced by Actin-Depolymerizing Toxins like Clostridium difficile Transferase (CDT). Proc. Natl. Acad. Sci. USA 2016, 113, 7870–7875. [Google Scholar] [CrossRef]
- Salameh, J.; Cantaloube, I.; Benoit, B.; Poüs, C.; Baillet, A. Cdc42 and Its BORG2 and BORG3 Effectors Control the Subcellular Localization of Septins between Actin Stress Fibers and Microtubules. Curr. Biol. 2021, 31, 4088–4103.e5. [Google Scholar] [CrossRef]
- Hecht, M.; Rösler, R.; Wiese, S.; Johnsson, N.; Gronemeyer, T. An Interaction Network of the Human SEPT9 Established by Quantitative Mass Spectrometry. G3 Genes|Genomes|Genet. 2019, 9, 1869–1880. [Google Scholar] [CrossRef]
- Devlin, L.; Okletey, J.; Perkins, G.; Bowen, J.R.; Nakos, K.; Montagna, C.; Spiliotis, E.T. Proteomic Profiling of the Oncogenic Septin 9 Reveals Isoform-specific Interactions in Breast Cancer Cells. Proteomics 2021, 21, 2100155. [Google Scholar] [CrossRef]
- Panagiotou, T.C.; Chen, A.; Wilde, A. An Anillin-CIN85-SEPT9 Complex Promotes Intercellular Bridge Maturation Required for Successful Cytokinesis. Cell Rep. 2022, 40, 111274. [Google Scholar] [CrossRef]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep. Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E.; et al. Modulators of HIF1α and NFkB in Cancer Treatment: Is It a Rational Approach for Controlling Malignant Progression? Front. Pharmacol. 2013, 4, 13. [Google Scholar] [CrossRef]
- Bui, B.P.; Nguyen, P.L.; Lee, K.; Cho, J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers 2022, 14, 6054. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®®) in Cancer Treatment: A Review of 15 Years of Clinical Experience and Future Outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Negri, A.L. Role of Prolyl Hydroxylase/HIF-1 Signaling in Vascular Calcification. Clin. Kidney J. 2023, 16, 205–209. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Marxsen, J.H.; Stengel, P.; Doege, K.; Heikkinen, P.; Jokilehto, T.; Wagner, T.; Jelkmann, W.; Jaakkola, P.; Metzen, E. Hypoxia-Inducible Factor-1 (HIF-1) Promotes Its Degradation by Induction of HIF-α-Prolyl-4-Hydroxylases. Biochem. J. 2004, 381, 761–767. [Google Scholar] [CrossRef]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 Competes with HSP90 for Binding to HIF-1α and Is Required for O2-Independent and HSP90 Inhibitor-Induced Degradation of HIF-1α. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef]
- Amir, S.; Wang, R.; Simons, J.W.; Mabjeesh, N.J. SEPT9_v1 Up-Regulates Hypoxia-Inducible Factor 1 by Preventing Its RACK1-Mediated Degradation. J. Biol. Chem. 2009, 284, 11142–11151. [Google Scholar] [CrossRef]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Köhler, M. Nuclear Translocation of Hypoxia-Inducible Factors (HIFs): Involvement of the Classical Importin α/β Pathway. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 394–404. [Google Scholar] [CrossRef]
- Oka, M.; Yoneda, Y. Importin α: Functions as a Nuclear Transport Factor and Beyond. Proc. Jpn. Acad. Ser. B 2018, 94, 259–274. [Google Scholar] [CrossRef]
- Tazat, K.; Schindler, S.; Depping, R.; Mabjeesh, N.J. Septin 9 Isoform 1 (SEPT9_i1) Specifically Interacts with Importin-A7 to Drive Hypoxia-Inducible Factor (HIF)-1α Nuclear Translocation. Cytoskeleton 2019, 76, 123–130. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef]
- Wood, R.A.; Barbour, M.J.; Gould, G.W.; Cunningham, M.R.; Plevin, R.J. Conflicting Evidence for the Role of JNK as a Target in Breast Cancer Cell Proliferation: Comparisons between Pharmacological Inhibition and Selective ShRNA Knockdown Approaches. Pharmacol. Res. Perspect. 2018, 6, e00376. [Google Scholar] [CrossRef]
- Liu, Z.; Xia, Y.; Zhang, X.; Liu, L.; Tu, S.; Zhu, W.; Yu, L.; Wan, H.; Yu, B.; Wan, F. Roles of the MST1-JNK Signaling Pathway in Apoptosis of Colorectal Cancer Cells Induced by Taurine. Libyan J. Med. 2018, 13, 1500346. [Google Scholar] [CrossRef]
- Zhao, J.; Jie, Q.; Li, G.; Li, Y.; Liu, B.; Li, H.; Luo, J.; Qin, X.; Li, Z.; Wei, Y. Rac1 Promotes the Survival of H9c2 Cells during Serum Deficiency Targeting JNK/c-JUN/Cyclin-D1 and AKT2/MCL1 Pathways. Int. J. Med. Sci. 2018, 15, 1062–1071. [Google Scholar] [CrossRef]
- Amir, S.; Wang, R.; Matzkin, H.; Simons, J.W.; Mabjeesh, N.J. MSF-A Interacts with Hypoxia-Lnducible Factor-1αand Augments Hypoxia-Inducible Factor Transcriptional Activation to Affect Tumorigenicity and Angiogenesis. Cancer Res. 2006, 66, 856–866. [Google Scholar] [CrossRef]
- Newell, S.; van der Watt, P.J.; Leaner, V.D. Therapeutic Targeting of Nuclear Export and Import Receptors in Cancer and Their Potential in Combination Chemotherapy. IUBMB Life 2024, 76, 4–25. [Google Scholar] [CrossRef]
- Tan, X.; Yan, Y.; Song, B.; Zhu, S.; Mei, Q.; Wu, K. Focal Adhesion Kinase: From Biological Functions to Therapeutic Strategies. Exp. Hematol. Oncol. 2023, 12, 83. [Google Scholar] [CrossRef]
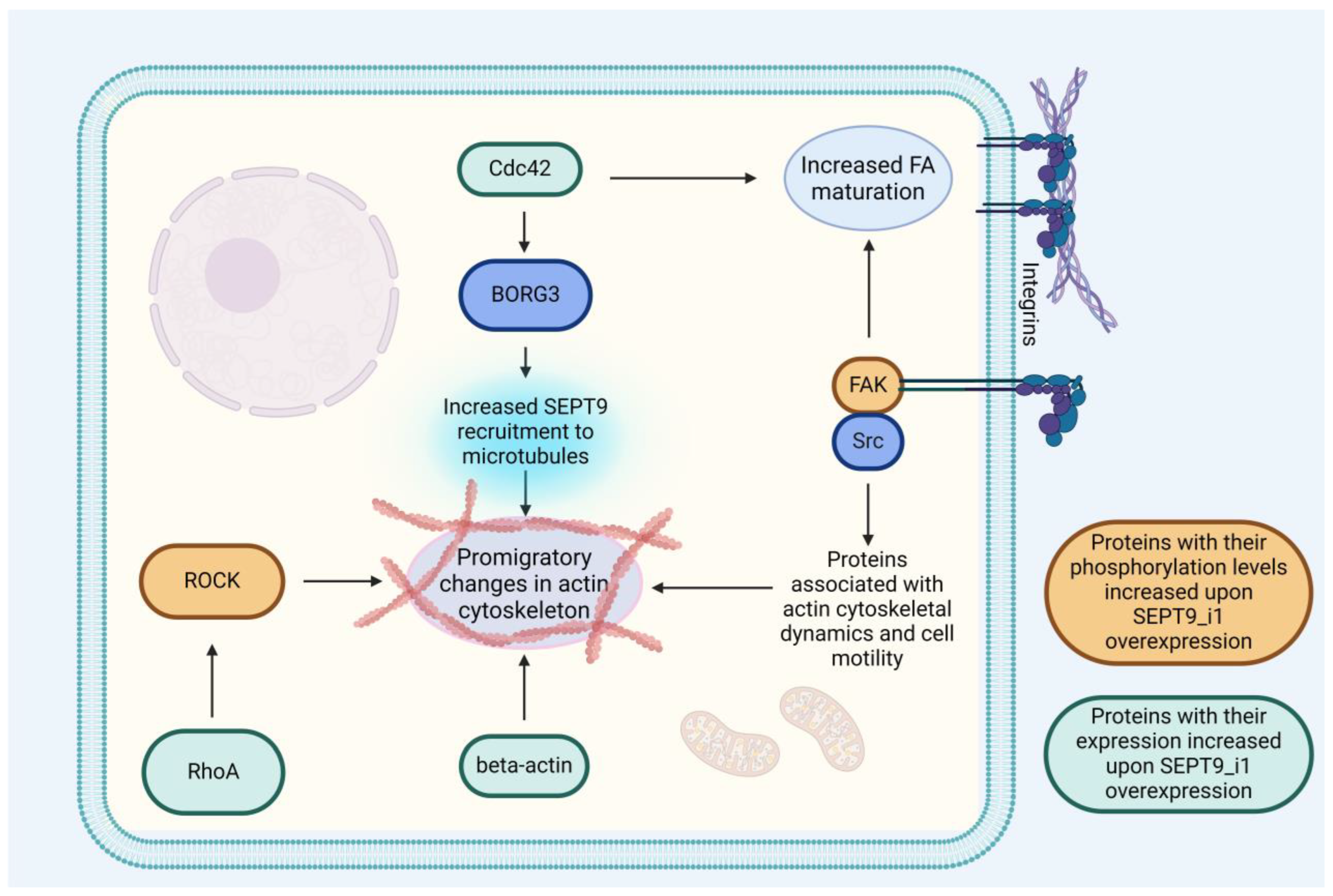
- Farrugia, A.J.; Rodríguez, J.; Orgaz, J.L.; Lucas, M.; Sanz-Moreno, V.; Calvo, F. CDC42EP5/BORG3 Modulates SEPT9 to Promote Actomyosin Function, Migration, and Invasion. J. Cell Biol. 2020, 219, e201912159. [Google Scholar] [CrossRef]
- Masi, I.; Caprara, V.; Bagnato, A.; Rosanò, L. Tumor Cellular and Microenvironmental Cues Controlling Invadopodia Formation. Front. Cell Dev. Biol. 2020, 8, 584181. [Google Scholar] [CrossRef]
- Ferrari, R.; Infante, E.; Chavrier, P. Nucleus–Invadopodia Duo During Cancer Invasion. Trends Cell Biol. 2019, 29, 93–96. [Google Scholar] [CrossRef]
- Okletey, J.; Angelis, D.; Jones, T.M.; Montagna, C.; Spiliotis, E.T. An Oncogenic Isoform of Septin 9 Promotes the Formation of Juxtanuclear Invadopodia by Reducing Nuclear Deformability. Cell Rep. 2023, 42, 112893. [Google Scholar] [CrossRef]
- Kang, N.; Matsui, T.S.; Liu, S.; Deguchi, S. ARHGAP4-SEPT2-SEPT9 Complex Enables Both up- and down-Modulation of Integrin-Mediated Focal Adhesions, Cell Migration, and Invasion. Mol. Biol. Cell 2021, 32, ar28. [Google Scholar] [CrossRef]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of Taxane Resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef]
- Amir, S.; Mabjeesh, N.J. SEPT9_V1 Protein Expression Is Associated with Human Cancer Cell Resistance to Microtubule Disrupting Agents. Cancer Biol. Ther. 2007, 6, 1926–1931. [Google Scholar] [CrossRef]
- Ghossoub, R.; Hu, Q.; Failler, M.; Rouyez, M.-C.; Spitzbarth, B.; Mostowy, S.; Wolfrum, U.; Saunier, S.; Cossart, P.; Jamesnelson, W.; et al. Septins 2, 7 and 9 and MAP4 Colocalize along the Axoneme in the Primary Cilium and Control Ciliary Length. J. Cell Sci. 2013, 126, 2583–2594. [Google Scholar] [CrossRef]
- Froidevaux-Klipfel, L.; Targa, B.; Cantaloube, I.; Ahmed-Zaïd, H.; Poüs, C.; Baillet, A. Septin Cooperation with Tubulin Polyglutamylation Contributes to Cancer Cell Adaptation to Taxanes. Oncotarget 2015, 6, 36063–36080. [Google Scholar] [CrossRef]
- Thakkar, P.V.; Kita, K.; del Castillo, U.; Galletti, G.; Madhukar, N.; Navarro, E.V.; Barasoain, I.; Goodson, H.V.; Sackett, D.; Díaz, J.F.; et al. CLIP-170S Is a Microtubule +TIP Variant That Confers Resistance to Taxanes by Impairing Drug-Target Engagement. Dev. Cell 2021, 56, 3264–3275.e7. [Google Scholar] [CrossRef]
- Ganguly, A.; Yang, H.; Pedroza, M.; Bhattacharya, R.; Cabral, F. Mitotic Centromere-Associated Kinesin (MCAK) Mediates Paclitaxel Resistance. J. Biol. Chem. 2011, 286, 36378–36384. [Google Scholar] [CrossRef]
- van der Vaart, B.; Akhmanova, A.; Straube, A. Regulation of Microtubule Dynamic Instability. Biochem. Soc. Trans. 2009, 37, 1007–1013. [Google Scholar] [CrossRef]
- Dragestein, K.A.; van Cappellen, W.A.; van Haren, J.; Tsibidis, G.D.; Akhmanova, A.; Knoch, T.A.; Grosveld, F.; Galjart, N. Dynamic Behavior of GFP–CLIP-170 Reveals Fast Protein Turnover on Microtubule plus Ends. J. Cell Biol. 2008, 180, 729–737. [Google Scholar] [CrossRef]
- Targa, B.; Klipfel, L.; Cantaloube, I.; Salameh, J.; Benoit, B.; Poüs, C.; Baillet, A. Septin Filament Coalignment with Microtubules Depends on SEPT9_i1 and Tubulin Polyglutamylation, and Is an Early Feature of Acquired Cell Resistance to Paclitaxel. Cell Death Dis. 2019, 10, 54. [Google Scholar] [CrossRef]
- Kremer, B.E.; Haystead, T.; Macara, I.G. Mammalian Septins Regulate Microtubule Stability through Interaction with the Microtubule-Binding Protein MAP4. Mol. Biol. Cell 2005, 16, 4648–4659. [Google Scholar] [CrossRef]
- Ageta-Ishihara, N.; Miyata, T.; Ohshima, C.; Watanabe, M.; Sato, Y.; Hamamura, Y.; Higashiyama, T.; Mazitschek, R.; Bito, H.; Kinoshita, M. Septins Promote Dendrite and Axon Development by Negatively Regulating Microtubule Stability via HDAC6-Mediated Deacetylation. Nat. Commun. 2013, 4, 2532. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; Peterson, E.A.; Privette, L.M.; Loffreda-Wren, J.L.; Kalikin, L.M.; Petty, E.M. High SEPT9_v1 Expression in Human Breast Cancer Cells Is Associated with Oncogenic Phenotypes. Cancer Res. 2007, 67, 8554–8564. [Google Scholar] [CrossRef]
- Verdier-Pinard, P.; Salaun, D.; Bouguenina, H.; Shimada, S.; Pophillat, M.; Audebert, S.; Agavnian, E.; Coslet, S.; Charafe-Jauffret, E.; Tachibana, T.; et al. Septin 9_i2 Is Downregulated in Tumors, Impairs Cancer Cell Migration and Alters Subnuclear Actin Filaments. Sci. Rep. 2017, 7, 44976. [Google Scholar] [CrossRef]
- Wasserkort, R.; Kalmar, A.; Valcz, G.; Spisak, S.; Krispin, M.; Toth, K.; Tulassay, Z.; Sledziewski, A.Z.; Molnar, B. Aberrant Septin 9 DNA Methylation in Colorectal Cancer Is Restricted to a Single CpG Island. BMC Cancer 2013, 13, 398. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, L.; Xiang, Q.; He, X.; Wang, Y.; Zhou, D.; Zou, C.; Chen, Q.; Peng, M.; He, J.; et al. SEPT9-v2, Frequently Silenced by Promoter Hypermethylation, Exerts Anti-Tumor Functions through Inactivation of Wnt/β-Catenin Signaling Pathway via MiR92b-3p/FZD10 in Nasopharyngeal Carcinoma Cells. Clin. Epigenetics 2020, 12, 41. [Google Scholar] [CrossRef]
- Ricci, A.; Bertoletti, C. Urea Derivatives on the Move: Cytokinin-like Activity and Adventitious Rooting Enhancement Depend on Chemical Structure. Plant Biol. 2009, 11, 262–272. [Google Scholar] [CrossRef]
- Hu, Q.; Nelson, W.J.; Spiliotis, E.T. Forchlorfenuron Alters Mammalian Septin Assembly, Organization, and Dynamics. J. Biol. Chem. 2008, 283, 29563–29571. [Google Scholar] [CrossRef]
- Angelis, D.; Karasmanis, E.P.; Bai, X.; Spiliotis, E.T. In Silico Docking of Forchlorfenuron (FCF) to Septins Suggests That FCF Interferes with GTP Binding. PLoS ONE 2014, 9, e96390. [Google Scholar] [CrossRef]
- Sun, L.; Cao, X.; Lechuga, S.; Feygin, A.; Naydenov, N.G.; Ivanov, A.I. A Septin Cytoskeleton-Targeting Small Molecule, Forchlorfenuron, Inhibits Epithelial Migration via Septin-Independent Perturbation of Cellular Signaling. Cells 2019, 9, 84. [Google Scholar] [CrossRef]
- Vardi-Oknin, D.; Golan, M.; Mabjeesh, N.J. Forchlorfenuron Disrupts SEPT9-I1 Filaments and Inhibits HIF-1. PLoS ONE 2013, 8, e73179. [Google Scholar] [CrossRef]
- Blum, W.; Henzi, T.; Pecze, L.; Diep, K.-L.; Bochet, C.G.; Schwaller, B. The Phytohormone Forchlorfenuron Decreases Viability and Proliferation of Malignant Mesothelioma Cells In Vitro and In Vivo. Oncotarget 2019, 10, 6944–6956. [Google Scholar] [CrossRef]
- Kim, K.K.; Singh, R.K.; Khazan, N.; Kodza, A.; Singh, N.A.; Jones, A.; Sivagnanalingam, U.; Towner, M.; Itamochi, H.; Turner, R.; et al. Development of Potent Forchlorfenuron Analogs and Their Cytotoxic Effect in Cancer Cell Lines. Sci. Rep. 2020, 10, 3241. [Google Scholar] [CrossRef] [PubMed]
- Henzi, T.; Diep, K.-L.; Oberson, A.; Salicio, V.; Bochet, C.; Schwaller, B. Forchlorfenuron and Novel Analogs Cause Cytotoxic Effects in Untreated and Cisplatin-Resistant Malignant Mesothelioma-Derived Cells. Int. J. Mol. Sci. 2022, 23, 3963. [Google Scholar] [CrossRef] [PubMed]
- Heasley, L.R.; Garcia, G.; McMurray, M.A. Off-Target Effects of the Septin Drug Forchlorfenuron on Nonplant Eukaryotes. Eukaryot. Cell 2014, 13, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, A.V.; Kudryavtsev, A.V.; Sokolova, O.S.; Shaitan, K.V. Procyanidin B3 as a Potential Inhibitor of Human Septin 9. Biophysics 2021, 66, 887–896. [Google Scholar] [CrossRef]
- Lee, A.Y.; St Onge, R.P.; Proctor, M.J.; Wallace, I.M.; Nile, A.H.; Spagnuolo, P.A.; Jitkova, Y.; Gronda, M.; Wu, Y.; Kim, M.K.; et al. Mapping the Cellular Response to Small Molecules Using Chemogenomic Fitness Signatures. Science 2014, 344, 208–211. [Google Scholar] [CrossRef]
- Chacko, A.D.; McDade, S.S.; Chanduloy, S.; Church, S.W.; Kennedy, R.; Price, J.; Hall, P.A.; Russell, S.E.H. Expression of the SEPT9_i4 Isoform Confers Resistance to Microtubule-Interacting Drugs. Cell Oncol. 2012, 35, 85–93. [Google Scholar] [CrossRef]
- Xu, M.; Takanashi, M.; Oikawa, K.; Nishi, H.; Isaka, K.; Yoshimoto, T.; Ohyashiki, J.; Kuroda, M. Identification of a Novel Role of S Eptin 10 in Paclitaxel-resistance in Cancers through a Functional Genomics Screen. Cancer Sci. 2012, 103, 821–827. [Google Scholar] [CrossRef]
- Song, K.; Yu, P.; Zhang, C.; Yuan, Z.; Zhang, H. The LncRNA FGD5-AS1/MiR-497-5p Axis Regulates Septin 2 (SEPT2) to Accelerate Cancer Progression and Increase Cisplatin-Resistance in Laryngeal Squamous Cell Carcinoma. Mol. Carcinog. 2021, 60, 469–480. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P.-K. HIF-1 Signaling: A Key Orchestrator of Cancer Radioresistance. Radiat. Med. Prot. 2020, 1, 7–14. [Google Scholar] [CrossRef]
- Bhushan, A.; Gonsalves, A.; Menon, J.U. Current State of Breast Cancer Diagnosis, Treatment, and Theranostics. Pharmaceutics 2021, 13, 723. [Google Scholar] [CrossRef]
- Marcus, E.A.; Tokhtaeva, E.; Turdikulova, S.; Capri, J.; Whitelegge, J.P.; Scott, D.R.; Sachs, G.; Berditchevski, F.; Vagin, O. Septin Oligomerization Regulates Persistent Expression of ErbB2/HER2 in Gastric Cancer Cells. Biochem. J. 2016, 473, 1703–1718. [Google Scholar] [CrossRef] [PubMed]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the Treatment of Breast Cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Wasim, S.; Park, J.; Nam, S.; Kim, J. Review of Current Treatment Intensification Strategies for Prostate Cancer Patients. Cancers 2023, 15, 5615. [Google Scholar] [CrossRef] [PubMed]
- Alatise, K.L.; Gardner, S.; Alexander-Bryant, A. Mechanisms of Drug Resistance in Ovarian Cancer and Associated Gene Targets. Cancers 2022, 14, 6246. [Google Scholar] [CrossRef] [PubMed]
- Saman, S.; Srivastava, N.; Yasir, M.; Chauhan, I. A Comprehensive Review on Current Treatments and Challenges Involved in the Treatment of Ovarian Cancer. Curr. Cancer Drug Targets 2024, 24, 142–166. [Google Scholar] [CrossRef]
- Scott, M.; McCluggage, W.G.; Hillan, K.J.; Hall, P.A.; Russell, S.E.H. Altered Patterns of Transcription of the Septin Gene, SEPT9, in Ovarian Tumorigenesis. Int. J. Cancer 2006, 118, 1325–1329. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; Makarova, O.; Peterson, E.A.; Privette, L.M.; Petty, E.M. Up-Regulation of SEPT9_v1 Stabilizes c-Jun-N-Terminal Kinase and Contributes to Its pro-Proliferative Activity in Mammary Epithelial Cells. Cell Signal 2009, 21, 477–487. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, J.; Wu, Q.; Cai, Y.; Zhu, L.; Lu, X.; Chen, S.; Chen, C.; Wang, Z. Acquisition of Epithelial–Mesenchymal Transition Is Associated with Skp2 Expression in Paclitaxel-Resistant Breast Cancer Cells. Br. J. Cancer 2014, 110, 1958–1967. [Google Scholar] [CrossRef]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-Metastatic and Anti-Invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Hypoxia and Hypoxia-Inducible Factors: Master Regulators of Metastasis. Clin. Cancer Res. 2010, 16, 5928–5935. [Google Scholar] [CrossRef]
- Ebelt, N.D.; Cantrell, M.A.; Van Den Berg, C.L. C-Jun N-Terminal Kinases Mediate a Wide Range of Targets in the Metastatic Cascade. Genes. Cancer 2013, 4, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Liu, L.; Fan, N.; Zhang, Q.; Wang, W.; Zheng, M.; Ma, L.; Li, Y.; Shi, L. The Requirement of SEPT2 and SEPT7 for Migration and Invasion in Human Breast Cancer via MEK/ERK Activation. Oncotarget 2016, 7, 61587–61600. [Google Scholar] [CrossRef] [PubMed]
- Arena, M.; Auteri, D.; Barmaz, S.; Bellisai, G.; Brancato, A.; Brocca, D.; Bura, L.; Byers, H.; Chiusolo, A.; Court Marques, D.; et al. Peer Review of the Pesticide Risk Assessment of the Active Substance Forchlorfenuron. EFSA J. 2017, 15, e04874. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Surma, M.; Shi, S.; Lambert-Cheatham, N.; Shi, J. Novel Insights into the Roles of Rho Kinase in Cancer. Arch. Immunol. Ther. Exp. 2016, 64, 259–278. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, Y.S.; Dong, S.M.; Kim, H.J.; Lee, D.E.; Kang, H.W.; Kim, M.J.; Park, J.S. Neural Wiskott-Aldrich Syndrome Protein (N-WASP) Promotes Distant Metastasis in Pancreatic Ductal Adenocarcinoma via Activation of LOXL2. Oncol. Res. 2024, 32, 615–624. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SEPT9_i1-Associated Protein | Mechanism of Interaction | Source |
|---|---|---|
| JNK, HIF-1 α | SEPT9_i1 increases HIF-1 α and JNK’s activity by preventing their degradation and, in the case of HIF-1 α, enhancing unclear shuttling [59,127]. | HIF-1 α and JNK have both been implicated in the development of taxane resistance [68]. |
| MAP4 | SEPT9_i1 competes for the same binding spot on the microtubules with MAP4 [99]. | MAP4 halts tubulin depolymerization—its inhibition counteracts the microtubule-stabilizing effect of taxanes [92]. |
| TTL, TTLL5, TTLL11, CCP1, CLIP-170, MCAK | SEPT9_i1 scaffolds for TTLL5, TTLL11, and CCP1, which regulate tubulin polyglutamylation, increasing the recruitment of CLIP-170 and MCAK [93,98]. | CLIP-170 and MCAK are crucial for maintaining microtubule dynamic instability—the state associated with resistance to MTAs [96,97]. |
| - | SEPT9_i1 overexpression induces EMT in tumor cells in vitro—the exact mechanism and associated proteins remain unknown [101]. | EMT is a process strongly implicated in tumor resistance to various forms of treatment, including taxane therapy [128]. |
| SEPT9_i1-Associated Protein | Mechanism of Interaction | Source |
|---|---|---|
| ARHGAP4 | SEPT9 and SEPT2 suppress ARHGAP4, which increases promigratory protein Rho activity [89]. | RhoA/ROCK pathway is crucial to various cellular processes involved in tumor cell motility and its ability to metastasize, including stress fiber stabilization and the formation of FAs [134]. |
| RhoA, ROCK | SEPT9_i1 increases activation of RhoA/ROCK pathway, leading to increased stress fiber formation and stability [27]. | |
| FAK, Src, Paxilin | FA maturation is amplified through increased recruitment of FAK, Src, and paxillin facilitated by SEPT9_i1 [27]. | Focal adhesions are involved in the formation of invadopodia, dissolution of ECM, and other processes involved in metastases [135]. |
| JNK, HIF-1 α | JNK and HIF-1 alpha, known for their promigratory properties, are protected from degradation by SEPT9_i1 [59,127]. | HIF-1 and JNK are known for their promigratory influence, with HIF-1 even being named the “master regulator of metastasis” [71,78]. |
| Cortactin, TKS5 | SEPT9_i1 recruits cortactin and TKS5 during the formation of juxtanuclear invadopodia. The exact mechanism remains unclear—stabilization of the nuclear envelope has been proposed as a contributor [88]. | Invadopodia are some of the most important structures involved in tumor cell invasion and metastases [86]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jędrzejczak, P.; Saramowicz, K.; Kuś, J.; Barczuk, J.; Rozpędek-Kamińska, W.; Siwecka, N.; Galita, G.; Wiese, W.; Majsterek, I. SEPT9_i1 and Septin Dynamics in Oncogenesis and Cancer Treatment. Biomolecules 2024, 14, 1194. https://doi.org/10.3390/biom14091194
Jędrzejczak P, Saramowicz K, Kuś J, Barczuk J, Rozpędek-Kamińska W, Siwecka N, Galita G, Wiese W, Majsterek I. SEPT9_i1 and Septin Dynamics in Oncogenesis and Cancer Treatment. Biomolecules. 2024; 14(9):1194. https://doi.org/10.3390/biom14091194
Chicago/Turabian StyleJędrzejczak, Piotr, Kamil Saramowicz, Justyna Kuś, Julia Barczuk, Wioletta Rozpędek-Kamińska, Natalia Siwecka, Grzegorz Galita, Wojciech Wiese, and Ireneusz Majsterek. 2024. "SEPT9_i1 and Septin Dynamics in Oncogenesis and Cancer Treatment" Biomolecules 14, no. 9: 1194. https://doi.org/10.3390/biom14091194