
Proteostasis Decline and Redox Imbalance in Age-Related Diseases: The Therapeutic Potential of NRF2

 ,
,  ,
,  ,
,  , , , , , , ,
, , , , , , ,  ,
,  and
and 
Abstract
1. Introduction
2. The Proteostasis Network and NRF2 Response: Molecular Regulation in Health and Disease
2.1. Proteostasis Mechanisms: The Balance Between Surveillance and Degradation
2.2. Proteostasis and Redox Regulation: Is There a Role for NRF2?
2.3. NRF2 Activation in Response to Proteotoxic-Derived Emergency Signals
3. Aging and Longevity
3.1. The Effects of NRF2 Modulation on Aging, Healthspan, and Longevity
3.2. NRF2 and Oxidative Stress from Early Developmental Stages to Senescence: A Gender Perspective
4. Cellular Dysfunctions in Age-Related Diseases and the Therapeutic Potential of NRF2
4.1. The Interplay Between Antioxidant Responses and Proteostasis in Alzheimer’s Disease, Alzheimer-like Dementia, and Parkinson’s Disease
4.2. Proteostasis and NRF2 Regulation in Cardiovascular Diseases
4.3. Metabolic Diseases (Obesity, Type 2 Diabetes Mellitus)
5. Design of Targeted Molecules and Innovative Drugs
6. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADME | Absorption, metabolism, distribution, and excretion |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| ARE | Antioxidant response element |
| ATF6 | Activating transcription factor 6 |
| Aβ | Amyloid-beta |
| BDNF | Brain-derived neurotrophic factor |
| CA | Carnosic acid |
| CNF | Central nervous system |
| CVD | Cardiovascular diseases |
| D3T | 1,2-dithiole-3-thione |
| DMF | Dimethyl fumarate |
| DS | Down syndrome |
| ER | Endoplasmic reticulum |
| HGPS | Hutchinson–Gilford progeria syndrome |
| HSE | Heat-shock factor response elements |
| HSF1 | Heat shock transcription factor 1 |
| HSP | Heat shock proteins |
| HUVEC | Human umbilical vein EC |
| I/R | Ischemia-reperfusion |
| IIS | Insulin/IGF1 signaling |
| IR | Insulin resistance |
| IRE1 | Inositol-requiring protein 1 |
| LDL | Low-density lipoprotein (LDL) |
| MetS | Metabolic syndrome |
| mHtt | Mutant huntingtin protein |
| mTOR | mammalian target of rapamycin |
| NAC | N-Acetylcysteine |
| NFTs | Neurofibrillary tangles |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| OS | Oxidative stress |
| PBMCs | Peripheral blood mononuclear cells |
| PDI | Protein disulfide isomerase |
| PEDs | Pro-electrophilic drugs |
| PERK | PKR-like ER kinase |
| PINK1 | PTEN-induced putative kinase 1 |
| PPI | Protein–protein interaction |
| ROS | Reactive oxygen species |
| RosA | Rosmarinic acid |
| SFN | Sulforaphane |
| SKN-1 | Skinhead-1 |
| T2D | Type 2 diabetes mellitus |
| UPR | Unfolded protein response |
| UPS | Ubiquitin-proteasome system |
References
- Partridge, L.; Gems, D. Mechanisms of aging: Public or private? Nat. Rev. Genet. 2002, 3, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in aging. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Hohn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Copple, I.M. Advances and challenges in therapeutic targeting of NRF2. Trends Pharmacol. Sci. 2023, 44, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Di Domenico, F. Stress Responses in Down Syndrome Neurodegeneration: State of the Art and Therapeutic Molecules. Biomolecules 2021, 11, 266. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol. 2015, 16, 322–324. [Google Scholar] [CrossRef]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. [Google Scholar] [CrossRef]
- Cecarini, V.; Ding, Q.; Keller, J.N. Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic. Res. 2007, 41, 673–680. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Di Domenico, F.; Zuliani, I.; Tramutola, A. Shining a light on defective autophagy by proteomics approaches: Implications for neurodegenerative illnesses. Expert. Rev. Proteom. 2019, 16, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.; Zhao, Y.; Ponnusamy, M.; Liu, Y. The role of ubiquitin proteasomal system and autophagy-lysosome pathway in Alzheimer’s disease. Rev. Neurosci. 2017, 28, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Gu, L.; Di Domenico, F.; Robinson, R.A. Mass spectrometry and redox proteomics: Applications in disease. Mass. Spectrom. Rev. 2014, 33, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Nagata, K. Redox-dependent protein quality control in the endoplasmic reticulum: Folding to degradation. Antioxid. Redox Signal 2012, 16, 1119–1128. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Beart, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the tightrope: Proteostasis and neurodegenerative disease. J. Neurochem. 2016, 137, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Sha, Z.; Schnell, H.M.; Ruoff, K.; Goldberg, A. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J. Cell Biol. 2018, 217, 1757–1776. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jimenez-Moreno, N.; Garcia-Yague, A.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rabano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed]
- Neef, D.W.; Jaeger, A.M.; Thiele, D.J. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 2011, 10, 930–944. [Google Scholar] [CrossRef] [PubMed]
- Oron, M.; Grochowski, M.; Jaiswar, A.; Legierska, J.; Jastrzebski, K.; Nowak-Niezgoda, M.; Kolos, M.; Kazmierczak, W.; Olesinski, T.; Lenarcik, M.; et al. The molecular network of the proteasome machinery inhibition response is orchestrated by HSP70, revealing vulnerabilities in cancer cells. Cell Rep. 2022, 40, 111428. [Google Scholar] [CrossRef]
- Chandran, A.; Oliver, H.J.; Rochet, J.C. Role of NFE2L1 in the Regulation of Proteostasis: Implications for Aging and Neurodegenerative Diseases. Biology 2023, 12, 1169. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, L.; Mesgarzadeh, J.; Xu, I.; Powers, E.T.; Wiseman, R.L.; Bollong, M.J. Defining the Functional Targets of Cap‘n’collar Transcription Factors NRF1, NRF2, and NRF3. Antioxidants 2020, 9, 1025. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Profumo, E.; Buttari, B.; Rigano, R. Oxidative stress in cardiovascular inflammation: Its involvement in autoimmune responses. Int. J. Inflam. 2011, 2011, 295705. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Glover-Cutter, K.M.; Lin, S.; Blackwell, T.K. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 2013, 9, e1003701. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernandez-Cardenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Bailey, J.T.; Worrell, S.; Glick, A.B. IRE1alpha regulates ROS and immune responses after UVB irradiation. Redox Exp. Med. 2024, 2024, e230030. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Xu, M.; Li, Y.; Xu, W.; Wu, C.; Zheng, H.; Xiao, Z.; Sun, G.; Ding, L.; Li, X.; et al. SMURF1 attenuates endoplasmic reticulum stress by promoting the degradation of KEAP1 to activate NRF2 antioxidant pathway. Cell Death Dis. 2023, 14, 361. [Google Scholar] [CrossRef]
- Binder, P.; Nguyen, B.; Collins, L.; Zi, M.; Liu, W.; Christou, F.; Luo, X.; Hille, S.S.; Frey, N.; Cartwright, E.J.; et al. Pak2 Regulation of Nrf2 Serves as a Novel Signaling Nexus Linking ER Stress Response and Oxidative Stress in the Heart. Front. Cardiovasc. Med. 2022, 9, 851419. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef]
- Zhao, G.; Zhao, J.; Lang, J.; Sun, G. Activation of NFE2L2 Alleviates Endoplasmic Reticulum Stress-Mediated Pyroptosis in Murine Hippocampus: A Bioinformatics Analysis and Experimental Validation. Mol. Neurobiol. 2024, 1–10. [Google Scholar] [CrossRef]
- Sammeta, N.; McClintock, T.S. Chemical stress induces the unfolded protein response in olfactory sensory neurons. J. Comp. Neurol. 2010, 518, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.F.; Li, X.H.; Yuan, Z.P.; Li, C.Y.; Tian, R.B.; Jia, W.; Xiao, Z.P. Allicin improves endoplasmic reticulum stress-related cognitive deficits via PERK/Nrf2 antioxidative signaling pathway. Eur. J. Pharmacol. 2015, 762, 239–246. [Google Scholar] [CrossRef]
- Mukaigasa, K.; Tsujita, T.; Nguyen, V.T.; Li, L.; Yagi, H.; Fuse, Y.; Nakajima-Takagi, Y.; Kato, K.; Yamamoto, M.; Kobayashi, M. Nrf2 activation attenuates genetic endoplasmic reticulum stress induced by a mutation in the phosphomannomutase 2 gene in zebrafish. Proc. Natl. Acad. Sci. USA 2018, 115, 2758–2763. [Google Scholar] [CrossRef]
- Lasbleiz, C.; Peyrel, A.; Tarot, P.; Sarniguet, J.; Crouzier, L.; Cubedo, N.; Delprat, B.; Rossel, M.; Maurice, T.; Lievens, J.C. Sigma-1 receptor agonist PRE-084 confers protection against TAR DNA-binding protein-43 toxicity through NRF2 signaling. Redox Biol. 2022, 58, 102542. [Google Scholar] [CrossRef]
- Park, J.S.; Oh, S.Y.; Lee, D.H.; Lee, Y.S.; Sung, S.H.; Ji, H.W.; Lee, M.J.; Lee, Y.H.; Rhee, S.G.; Bae, S.H. p62/SQSTM1 is required for the protection against endoplasmic reticulum stress-induced apoptotic cell death. Free Radic. Res. 2016, 50, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hur, E.G.; Ryoo, I.G.; Jung, K.A.; Kwak, J.; Kwak, M.K. Involvement of the Nrf2-proteasome pathway in the endoplasmic reticulum stress response in pancreatic beta-cells. Toxicol. Appl. Pharmacol. 2012, 264, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Koo, J.H.; Lee, J.M.; Joo, M.S.; Kim, T.H.; Kim, H.; Jun, D.W.; Kim, S.G. NRF2-mediated SIRT3 induction protects hepatocytes from ER stress-induced liver injury. FASEB J. 2022, 36, e22170. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Sakellari, M.; Lefaki, M.; Papaevgeniou, N.; Gonos, E.S. Proteasome activation delays aging in vitro and in vivo. Free Radic. Biol. Med. 2014, 71, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Aging as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.M.; Schafer, M.J.; LeBrasseur, N.K. Energetic interventions for healthspan and resiliency with aging. Exp. Gerontol. 2016, 86, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Volonte, D.; Liu, Z.; Musille, P.M.; Stoppani, E.; Wakabayashi, N.; Di, Y.P.; Lisanti, M.P.; Kensler, T.W.; Galbiati, F. Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by caveolin-1 promotes stress-induced premature senescence. Mol. Biol. Cell 2013, 24, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Liu, L.H.; Liu, H.; Wu, K.F.; An, J.; Wang, Q.; Liu, Y.; Bai, L.J.; Qi, B.M.; Qi, B.L.; et al. Nrf2 protects against diabetic dysfunction of endothelial progenitor cells via regulating cell senescence. Int. J. Mol. Med. 2018, 42, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Hiebert, P.; Wietecha, M.S.; Cangkrama, M.; Haertel, E.; Mavrogonatou, E.; Stumpe, M.; Steenbock, H.; Grossi, S.; Beer, H.D.; Angel, P.; et al. Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome. Dev. Cell 2018, 46, 145–161.e10. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef]
- An, J.H.; Blackwell, T.K. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003, 17, 1882–1893. [Google Scholar] [CrossRef]
- Tullet, J.M.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.P.; Baumeister, R.; Blackwell, T.K. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 2008, 132, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev. Cell 2008, 14, 76–85. [Google Scholar] [CrossRef]
- Spiers, J.G.; Breda, C.; Robinson, S.; Giorgini, F.; Steinert, J.R. Drosophila Nrf2/Keap1 Mediated Redox Signaling Supports Synaptic Function and Longevity and Impacts on Circadian Activity. Front. Mol. Neurosci. 2019, 12, 86. [Google Scholar] [CrossRef]
- Tsakiri, E.N.; Gumeni, S.; Iliaki, K.K.; Benaki, D.; Vougas, K.; Sykiotis, G.P.; Gorgoulis, V.G.; Mikros, E.; Scorrano, L.; Trougakos, I.P. Hyperactivation of Nrf2 increases stress tolerance at the cost of aging acceleration due to metabolic deregulation. Aging Cell 2019, 18, e12845. [Google Scholar] [CrossRef] [PubMed]
- Bruns, D.R.; Drake, J.C.; Biela, L.M.; Peelor, F.F., 3rd; Miller, B.F.; Hamilton, K.L. Nrf2 Signaling and the Slowed Aging Phenotype: Evidence from Long-Lived Models. Oxid. Med. Cell Longev. 2015, 2015, 732596. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Wason, E.; Edrey, Y.H.; Kristan, D.M.; Nevo, E.; Buffenstein, R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc. Natl. Acad. Sci. USA 2015, 112, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Pomatto, L.C.D.; Dill, T.; Carboneau, B.; Levan, S.; Kato, J.; Mercken, E.M.; Pearson, K.J.; Bernier, M.; de Cabo, R. Deletion of Nrf2 shortens lifespan in C57BL6/J male mice but does not alter the health and survival benefits of caloric restriction. Free Radic. Biol. Med. 2020, 152, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.J.; Lewis, K.N.; Price, N.L.; Chang, J.W.; Perez, E.; Cascajo, M.V.; Tamashiro, K.L.; Poosala, S.; Csiszar, A.; Ungvari, Z.; et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc. Natl. Acad. Sci. USA 2008, 105, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Oishi, T.; Matsumaru, D.; Ota, N.; Kitamura, H.; Zhang, T.; Honkura, Y.; Katori, Y.; Motohashi, H. Activation of the NRF2 pathway in Keap1-knockdown mice attenuates progression of age-related hearing loss. NPJ Aging Mech. Dis. 2020, 6, 14. [Google Scholar] [CrossRef]
- Wati, S.M.; Matsumaru, D.; Motohashi, H. NRF2 pathway activation by KEAP1 inhibition attenuates the manifestation of aging phenotypes in salivary glands. Redox Biol. 2020, 36, 101603. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Davinelli, S.; Willcox, D.C.; Scapagnini, G. Extending healthy aging: Nutrient sensitive pathway and centenarian population. Immun. Aging 2012, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Aging, age-related diseases and oxidative stress: What to do next? Aging Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of aging. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammaging: Chronic inflammation in aging, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Bateson, P.; Barker, D.; Clutton-Brock, T.; Deb, D.; D’Udine, B.; Foley, R.A.; Gluckman, P.; Godfrey, K.; Kirkwood, T.; Lahr, M.M.; et al. Developmental plasticity and human health. Nature 2004, 430, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, P.; Holmes, M.; Schmidt, M.V.; Cirulli, F.; Guzzardi, M.A.; Berry, A.; Balsevich, G.; Andreassi, M.G.; Wesselink, J.J.; Liistro, T.; et al. Developmental ORIgins of Healthy and Unhealthy Aging: The role of maternal obesity--introduction to DORIAN. Obes. Facts 2014, 7, 130–151. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.; Cirulli, F. The p66(Shc) gene paves the way for healthspan: Evolutionary and mechanistic perspectives. Neurosci. Biobehav. Rev. 2013, 37, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Musillo, C.; Berry, A.; Cirulli, F. Prenatal psychological or metabolic stress increases the risk for psychiatric disorders: The “funnel effect” model. Neurosci. Biobehav. Rev. 2022, 136, 104624. [Google Scholar] [CrossRef] [PubMed]
- Musillo, C.; Creutzberg, K.C.; Collacchi, B.; Ajmone-Cat, M.A.; De Simone, R.; Lepre, M.; Amrein, I.; Riva, M.A.; Berry, A.; Cirulli, F. Bdnf-Nrf-2 crosstalk and emotional behavior are disrupted in a sex-dependent fashion in adolescent mice exposed to maternal stress or maternal obesity. Transl. Psychiatry 2023, 13, 399. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Casanueva, O. Epigenetic inheritance of proteostasis and aging. Essays Biochem. 2016, 60, 191–202. [Google Scholar] [CrossRef]
- Berry, A.; Bellisario, V.; Panetta, P.; Raggi, C.; Magnifico, M.C.; Arese, M.; Cirulli, F. Administration of the Antioxidant N-Acetyl-Cysteine in Pregnant Mice Has Long-Term Positive Effects on Metabolic and Behavioral Endpoints of Male and Female Offspring Prenatally Exposed to a High-Fat Diet. Front. Behav. Neurosci. 2018, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Tower, J.; Pomatto, L.C.D.; Davies, K.J.A. Sex differences in the response to oxidative and proteolytic stress. Redox Biol. 2020, 31, 101488. [Google Scholar] [CrossRef]
- Borras, C.; Sastre, J.; Garcia-Sala, D.; Lloret, A.; Pallardo, F.V.; Vina, J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic. Biol. Med. 2003, 34, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Gambini, J.; Lopez-Grueso, R.; Abdelaziz, K.M.; Jove, M.; Borras, C. Females live longer than males: Role of oxidative stress. Curr. Pharm. Des. 2011, 17, 3959–3965. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Ramirez-Bosca, A.; Ramirez-Bosca, J.V.; Alperi, J.D. Menopause: A review on the role of oxygen stress and favorable effects of dietary antioxidants. Arch. Gerontol. Geriatr. 2006, 42, 289–306. [Google Scholar] [CrossRef]
- Martinez de Toda, I.; Gonzalez-Sanchez, M.; Diaz-Del Cerro, E.; Valera, G.; Carracedo, J.; Guerra-Perez, N. Sex differences in markers of oxidation and inflammation. Implic. Aging. Mech. Aging Dev. 2023, 211, 111797. [Google Scholar] [CrossRef]
- Tewari, D.; Stankiewicz, A.M.; Mocan, A.; Sah, A.N.; Tzvetkov, N.T.; Huminiecki, L.; Horbanczuk, J.O.; Atanasov, A.G. Ethnopharmacological Approaches for Dementia Therapy and Significance of Natural Products and Herbal Drugs. Front. Aging Neurosci. 2018, 10, 3. [Google Scholar] [CrossRef]
- Trewavas, A.; Stewart, D. Paradoxical effects of chemicals in the diet on health. Curr. Opin. Plant Biol. 2003, 6, 185–190. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, A. Neurohormetic phytochemicals: Low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci. 2006, 29, 632–639. [Google Scholar] [CrossRef]
- Gray, N.E.; Harris, C.J.; Quinn, J.F.; Soumyanath, A. Centella asiatica modulates antioxidant and mitochondrial pathways and improves cognitive function in mice. J. Ethnopharmacol. 2016, 180, 78–86. [Google Scholar] [CrossRef]
- Lee, Y.; Oh, S. Administration of red ginseng ameliorates memory decline in aged mice. J. Ginseng Res. 2015, 39, 250–256. [Google Scholar] [CrossRef]
- Musillo, C.; Borgi, M.; Saul, N.; Moller, S.; Luyten, W.; Berry, A.; Cirulli, F. Natural products improve healthspan in aged mice and rats: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2021, 121, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.; Marconi, M.; Musillo, C.; Chiarotti, F.; Bellisario, V.; Matarrese, P.; Gambardella, L.; Vona, R.; Lombardi, M.; Foglieni, C.; et al. Trehalose administration in C57BL/6N old mice affects healthspan improving motor learning and brain anti-oxidant defences in a sex-dependent fashion: A pilot study. Exp. Gerontol. 2020, 129, 110755. [Google Scholar] [CrossRef]
- Berry, A.; Amrein, I.; Notzli, S.; Lazic, S.E.; Bellisario, V.; Giorgio, M.; Pelicci, P.G.; Alleva, E.; Lipp, H.P.; Cirulli, F. Sustained hippocampal neurogenesis in females is amplified in P66(Shc-/-) mice: An animal model of healthy aging. Hippocampus 2012, 22, 2249–2259. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. Oxidative damage in neurodegeneration: Roles in the pathogenesis and progression of Alzheimer disease. Physiol. Rev. 2024, 104, 103–197. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Gureev, A.P.; Khorolskaya, V.G.; Sadovnikova, I.S.; Shaforostova, E.A.; Cherednichenko, V.R.; Burakova, I.Y.; Plotnikov, E.Y.; Popov, V.N. Age-Related Decline in Nrf2/ARE Signaling Is Associated with the Mitochondrial DNA Damage and Cognitive Impairments. Int. J. Mol. Sci. 2022, 23, 15197. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.; Malm, T.M.; Jyrkkanen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Yla-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nunez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- von Otter, M.; Landgren, S.; Nilsson, S.; Zetterberg, M.; Celojevic, D.; Bergstrom, P.; Minthon, L.; Bogdanovic, N.; Andreasen, N.; Gustafson, D.R.; et al. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech. Aging Dev. 2010, 131, 105–110. [Google Scholar] [CrossRef]
- Bresciani, G.; Manai, F.; Davinelli, S.; Tucci, P.; Saso, L.; Amadio, M. Novel potential pharmacological applications of dimethyl fumarate-an overview and update. Front. Pharmacol. 2023, 14, 1264842. [Google Scholar] [CrossRef]
- Shah, A.; Varma, M.; Bhandari, R. Exploring sulforaphane as neurotherapeutic: Targeting Nrf2-Keap & Nf-Kb pathway crosstalk in ASD. Metab. Brain Dis. 2024, 39, 373–385. [Google Scholar] [CrossRef]
- Rong, H.; Liang, Y.; Niu, Y. Rosmarinic acid attenuates beta-amyloid-induced oxidative stress via Akt/GSK-3beta/Fyn-mediated Nrf2 activation in PC12 cells. Free Radic. Biol. Med. 2018, 120, 114–123. [Google Scholar] [CrossRef]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Llewellyn, K.; Wakser, S.; Pontasch, J.; Samanich, N.; Flemer, M.; Hensley, K.; Kim, D.S.; Park, J. Mini-GAGR, an intranasally applied polysaccharide, activates the neuronal Nrf2-mediated antioxidant defense system. J. Biol. Chem. 2018, 293, 18242–18269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, Y.; Liu, H.; Yin, C.; Li, X.; Gong, Q. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: A novel mechanism mediated by the activation of Nrf2. Pharmacol. Biochem. Behav. 2016, 150–151, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, S.; Guo, H.; Jiang, H.; Liu, H.; Fu, H.; Wang, D. Forsythoside A Mitigates Alzheimer’s-like Pathology by Inhibiting Ferroptosis-mediated Neuroinflammation via Nrf2/GPX4 Axis Activation. Int. J. Biol. Sci. 2022, 18, 2075–2090. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, M.C.; Mendes, E.; Perry, M.J.; Francisco, A.P.; Marco-Contelles, J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Luchsinger, J.A.; Cirio, R.; Chen, H.; Franchino-Elder, J.; Hirsch, J.A.; Bettendorff, L.; Chen, Z.; Flowers, S.A.; Gerber, L.M.; et al. Benfotiamine and Cognitive Decline in Alzheimer’s Disease: Results of a Randomized Placebo-Controlled Phase IIa Clinical Trial. J. Alzheimers Dis. 2020, 78, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Perluigi, M.; Butterfield, D.A. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 724904. [Google Scholar] [CrossRef]
- Rueda Revilla, N.; Martinez-Cue, C. Antioxidants in Down Syndrome: From Preclinical Studies to Clinical Trials. Antioxidants 2020, 9, 692. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Tramutola, A.; Pagnotta, S.; Barone, E.; Butterfield, D.A. The BACH1/Nrf2 Axis in Brain in Down Syndrome and Transition to Alzheimer Disease-Like Neuropathology and Dementia. Antioxidants 2020, 9, 779. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Zuliani, I.; Tramutola, A.; Barone, E.; Blarzino, C.; Folgiero, V.; Caforio, M.; Valentini, D.; Villani, A.; Locatelli, F.; et al. Chronic PERK induction promotes Alzheimer-like neuropathology in Down syndrome: Insights for therapeutic intervention. Prog. Neurobiol. 2021, 196, 101892. [Google Scholar] [CrossRef]
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.A.; Head, E.; Butterfield, D.A.; et al. Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: Insights for transition to Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Greco, V.; Valentini, D.; Villani, A.; Folgiero, V.; Caforio, M.; Locatelli, F.; Pagnotta, S.; Barone, E.; Urbani, A.; et al. Proteomics Study of Peripheral Blood Mononuclear Cells in Down Syndrome Children. Antioxidants 2020, 9, 1112. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Baniowska, M.R.; Prestia, F.; Sette, C.; Nalesso, V.; Perluigi, M.; Barone, E.; Duchon, A.; Tramutola, A.; Herault, Y.; et al. Shaping down syndrome brain cognitive and molecular changes due to aging using adult animals from the Ts66Yah murine model. Neurobiol. Dis. 2024, 196, 106523. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, E.; Zamponi, N.; Coskun, P.; Quassollo, G.; Lorenzo, A.; Cannas, S.A.; Pigino, G.; Chialvo, D.R.; Gardiner, K.; Busciglio, J.; et al. Nrf2 stabilization prevents critical oxidative damage in Down syndrome cells. Aging Cell 2018, 17, e12812. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta 2013, 1832, 1249–1259. [Google Scholar] [CrossRef]
- Perluigi, M.; Pupo, G.; Tramutola, A.; Cini, C.; Coccia, R.; Barone, E.; Head, E.; Butterfield, D.A.; Di Domenico, F. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim. Biophys. Acta 2014, 1842, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Barone, E.; Arena, A.; Zuliani, I.; Mosca, L.; Blarzino, C.; Butterfield, D.A.; Perluigi, M.; Di Domenico, F. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl. Neurodegener. 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Bordi, M.; Darji, S.; Sato, Y.; Mellen, M.; Berg, M.J.; Kumar, A.; Jiang, Y.; Nixon, R.A. mTOR hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 2019, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Tramutola, A.; Barone, E.; Lanzillotta, C.; Defever, O.; Arena, A.; Zuliani, I.; Foppoli, C.; Iavarone, F.; Vincenzoni, F.; et al. Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: Focus on HNE-modified proteins in a mouse model of down syndrome. Redox Biol. 2019, 23, 101162. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Mathews, P.M.; Boiteau, A.B.; Hassinger, L.C.; Peterhoff, C.M.; Jiang, Y.; Mullaney, K.; Neve, R.L.; Gruenberg, J.; Nixon, R.A. Down syndrome fibroblast model of Alzheimer-related endosome pathology: Accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 2008, 173, 370–384. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Pensalfini, A.; Jiang, Y.; Nixon, R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic. Biol. Med. 2018, 114, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Hasle, H.; Friedman, J.M.; Olsen, J.H.; Rasmussen, S.A. Low risk of solid tumors in persons with Down syndrome. Genet. Med. 2016, 18, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Rabin, K.R.; Whitlock, J.A. Malignancy in children with trisomy 21. Oncologist 2009, 14, 164–173. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Caligiore, D.; Giocondo, F.; Silvetti, M. The Neurodegenerative Elderly Syndrome (NES) hypothesis: Alzheimer and Parkinson are two faces of the same disease. IBRO Neurosci. Rep. 2022, 13, 330–343. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rabano, A.; Kirik, D.; Cuadrado, A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Hwang, V.; Ando, D.M.; Daub, A.; Lee, A.K.; Ravisankar, A.; Modan, S.; Finucane, M.M.; Shaby, B.A.; Finkbeiner, S. Nrf2 mitigates LRRK2- and alpha-synuclein-induced neurodegeneration by modulating proteostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 1165–1170. [Google Scholar] [CrossRef]
- Pickering, A.M.; Linder, R.A.; Zhang, H.; Forman, H.J.; Davies, K.J.A. Nrf2-dependent induction of proteasome and Pa28alphabeta regulator are required for adaptation to oxidative stress. J. Biol. Chem. 2012, 287, 10021–10031. [Google Scholar] [CrossRef]
- Moscovitz, O.; Ben-Nissan, G.; Fainer, I.; Pollack, D.; Mizrachi, L.; Sharon, M. The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nat. Commun. 2015, 6, 6609. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Kuruvilla, J.; Tan, E.K. Mitophagy and reactive oxygen species interplay in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Gumeni, S.; Papanagnou, E.D.; Manola, M.S.; Trougakos, I.P. Nrf2 activation induces mitophagy and reverses Parkin/Pink1 knock down-mediated neuronal and muscle degeneration phenotypes. Cell Death Dis. 2021, 12, 671. [Google Scholar] [CrossRef] [PubMed]
- Pinjala, P.; Tryphena, K.P.; Kulkarni, A.; Goswami, P.G.; Khatri, D.K. Dimethyl Fumarate Exerts a Neuroprotective Effect by Enhancing Mitophagy via the NRF2/BNIP3/PINK1 Axis in the MPP(+) Iodide-Induced Parkinson’s Disease Mice Model. J. Alzheimers Dis. Rep. 2024, 8, 329–344. [Google Scholar] [CrossRef]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The Neuroprotective Effect of Dimethyl Fumarate in an MPTP-Mouse Model of Parkinson’s Disease: Involvement of Reactive Oxygen Species/Nuclear Factor-kappaB/Nuclear Transcription Factor Related to NF-E2. Antioxid. Redox Signal 2017, 27, 453–471. [Google Scholar] [CrossRef]
- Teng, Y.; Zhao, J.; Ding, L.; Ding, Y.; Zhou, P. Complex of EGCG with Cu(II) Suppresses Amyloid Aggregation and Cu(II)-Induced Cytotoxicity of alpha-Synuclein. Molecules 2019, 24, 2940. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nehru, B. Curcumin affords neuroprotection and inhibits alpha-synuclein aggregation in lipopolysaccharide-induced Parkinson’s disease model. Inflammopharmacology 2018, 26, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Lapidus, L.J. Curcumin prevents aggregation in alpha-synuclein by increasing reconfiguration rate. J. Biol. Chem. 2012, 287, 9193–9199. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhang, J.; Dong, M. Nrf2 as a potential target for Parkinson’s disease therapy. J. Mol. Med. 2021, 99, 917–931. [Google Scholar] [CrossRef]
- McFarthing, K.; Rafaloff, G.; Baptista, M.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2022 Update. J. Park. Dis. 2022, 12, 1073–1082. [Google Scholar] [CrossRef]
- Nakaso, K.; Horikoshi, Y.; Takahashi, T.; Hanaki, T.; Nakasone, M.; Kitagawa, Y.; Koike, T.; Matsura, T. Estrogen receptor-mediated effect of delta-tocotrienol prevents neurotoxicity and motor deficit in the MPTP mouse model of Parkinson’s disease. Neurosci. Lett. 2016, 610, 117–122. [Google Scholar] [CrossRef]
- Atia, A.; Alrawaiq, N.S.; Abdullah, A. Tocotrienols Activate Nrf2 Nuclear Translocation and Increase the Antioxidant- Related Hepatoprotective Mechanism in Mice Liver. Curr. Pharm. Biotechnol. 2021, 22, 1085–1098. [Google Scholar] [CrossRef] [PubMed]
- Kaji, H.; Matsui-Yuasa, I.; Matsumoto, K.; Omura, A.; Kiyomoto, K.; Kojima-Yuasa, A. Sesaminol prevents Parkinson’s disease by activating the Nrf2-ARE signaling pathway. Heliyon 2020, 6, e05342. [Google Scholar] [CrossRef]
- Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Aging Hallmarks and the Role of Oxidative Stress. Antioxidants 2023, 12, 651. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kim, K.J.; Choi, J. Smoking, alcohol intake, and frailty in older Korean adult men: Cross-sectional study with nationwide data. Eur. Geriatr. Med. 2020, 11, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Ng Fat, L.; Bell, S.; Britton, A. A life-time of hazardous drinking and harm to health among older adults: Findings from the Whitehall II prospective cohort study. Addiction 2020, 115, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Liu, C.Y.; Peng, L.N.; Lin, C.H.; Lin, H.P.; Chen, L.K. PM air pollution contributes to the burden of frailty. Sci. Rep. 2020, 10, 14478. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintron, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W., 2nd; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Korantzopoulos, P.; Letsas, K.; Fragakis, N.; Tse, G.; Liu, T. Oxidative stress and atrial fibrillation: An update. Free Radic. Res. 2018, 52, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, F.; Agnetti, G. Protein post-translational modifications and misfolding: New concepts in heart failure. Proteom. Clin. Appl. 2014, 8, 534–542. [Google Scholar] [CrossRef]
- Prinsen, J.K.; Kannankeril, P.J.; Sidorova, T.N.; Yermalitskaya, L.V.; Boutaud, O.; Zagol-Ikapitte, I.; Barnett, J.V.; Murphy, M.B.; Subati, T.; Stark, J.M.; et al. Highly Reactive Isolevuglandins Promote Atrial Fibrillation Caused by Hypertension. JACC Basic. Transl. Sci. 2020, 5, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef] [PubMed]
- Sergin, I.; Bhattacharya, S.; Emanuel, R.; Esen, E.; Stokes, C.J.; Evans, T.D.; Arif, B.; Curci, J.A.; Razani, B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci. Signal 2016, 9, ra2. [Google Scholar] [CrossRef]
- Howlett, G.J.; Moore, K.J. Untangling the role of amyloid in atherosclerosis. Curr. Opin. Lipidol. 2006, 17, 541–547. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Torezani, G.S.; Braga, C.A.; Palhano, F.L.; Kelly, J.W.; Saraiva, E.M.; Foguel, D. Amyloid fibrils trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation by NET-associated elastase. J. Biol. Chem. 2012, 287, 37206–37218. [Google Scholar] [CrossRef]
- Friedrich, R.P.; Tepper, K.; Ronicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fandrich, M. Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Kloska, D.; Targosz-Korecka, M.; Zapotoczny, B.; Cysewski, D.; Personnic, N.; Werner, E.; Hajduk, K.; Jozkowicz, A.; Grochot-Przeczek, A. Keap1 governs aging-induced protein aggregation in endothelial cells. Redox Biol. 2020, 34, 101572. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, Y.; Huang, T. Human age-related cataracts: Epigenetic suppression of the nuclear factor erythroid 2-related factor 2-mediated antioxidant system. Mol. Med. Rep. 2015, 11, 1442–1447. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Munzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Ide, T.; Kinugawa, S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid. Redox Signal 2006, 8, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, M.; Xia, K.; Colon, W.; Vieira, S.I.; Ribeiro, F. Protein aggregation, cardiovascular diseases, and exercise training: Where do we stand? Aging Res. Rev. 2017, 40, 1–10. [Google Scholar] [CrossRef]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280, C53–C60. [Google Scholar] [CrossRef]
- Sirish, P.; Diloretto, D.A.; Thai, P.N.; Chiamvimonvat, N. The Critical Roles of Proteostasis and Endoplasmic Reticulum Stress in Atrial Fibrillation. Front. Physiol. 2021, 12, 793171. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.G.; Kirk, J.A. Under construction: The dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J. Mol. Cell Cardiol. 2020, 148, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Immunoglobulin light chain amyloidosis: 2024 update on diagnosis, prognosis, and treatment. Am. J. Hematol. 2024, 99, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Liz, M.A.; Coelho, T.; Bellotti, V.; Fernandez-Arias, M.I.; Mallaina, P.; Obici, L. A Narrative Review of the Role of Transthyretin in Health and Disease. Neurol. Ther. 2020, 9, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Vergaro, G.; Aimo, A.; Rapezzi, C.; Castiglione, V.; Fabiani, I.; Pucci, A.; Buda, G.; Passino, C.; Lupon, J.; Bayes-Genis, A.; et al. Atrial amyloidosis: Mechanisms and clinical manifestations. Eur. J. Heart Fail. 2022, 24, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Mercanti, F.; Wang, X.; Mackintosh, S.G.; Tackett, A.J.; Prayaga, S.V.; Romeo, F.; Shmookler Reis, R.J.; Mehta, J.L. Age- and Hypertension-Associated Protein Aggregates in Mouse Heart Have Similar Proteomic Profiles. Hypertension 2016, 67, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Buttari, B.; Profumo, E.; Capozzi, A.; Facchiano, F.; Saso, L.; Sorice, M.; Rigano, R. Advanced glycation end products of human beta(2) glycoprotein I modulate the maturation and function of DCs. Blood 2011, 117, 6152–6161. [Google Scholar] [CrossRef] [PubMed]
- Tahara, K.; Kim, H.D.; Jin, J.J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of toll-like receptor signaling in Abeta uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Schisler, J.C.; Portbury, A.L.; Patterson, C. Build it up-Tear it down: Protein quality control in the cardiac sarcomere. Cardiovasc. Res. 2009, 81, 439–448. [Google Scholar] [CrossRef]
- Willis, M.S.; Patterson, C. Proteotoxicity and cardiac dysfunction—Alzheimer’s disease of the heart? N. Engl. J. Med. 2013, 368, 455–464. [Google Scholar] [CrossRef]
- Fan, G.C.; Ren, X.; Qian, J.; Yuan, Q.; Nicolaou, P.; Wang, Y.; Jones, W.K.; Chu, G.; Kranias, E.G. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation 2005, 111, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Tanonaka, K.; Furuhama, K.I.; Yoshida, H.; Kakuta, K.; Miyamoto, Y.; Toga, W.; Takeo, S. Protective effect of heat shock protein 72 on contractile function of perfused failing heart. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H215–H222. [Google Scholar] [CrossRef] [PubMed]
- Ke, L.; Meijering, R.A.; Hoogstra-Berends, F.; Mackovicova, K.; Vos, M.J.; Van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS ONE 2011, 6, e20395. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.J.; Shiroshita-Takeshita, A.; Qi, X.; Yeh, Y.H.; Chartier, D.; van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Nattel, S. Induction of heat shock response protects the heart against atrial fibrillation. Circ. Res. 2006, 99, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, S.W.; Ramos, K.S.; Brundel, B. Cardioprotective Role of Heat Shock Proteins in Atrial Fibrillation: From Mechanism of Action to Therapeutic and Diagnostic Target. Int. J. Mol. Sci. 2021, 22, 442. [Google Scholar] [CrossRef]
- Morimoto, R.I. The heat shock response: Systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Stalder, R.; McKercher, S.R.; Williamson, R.E.; Roth, G.P.; Lipton, S.A. Nrf2 and HSF-1 Pathway Activation via Hydroquinone-Based Proelectrophilic Small Molecules is Regulated by Electrochemical Oxidation Potential. ASN Neuro 2015, 7, 1759091415593294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ahn, Y.H.; Benjamin, I.J.; Honda, T.; Hicks, R.J.; Calabrese, V.; Cole, P.A.; Dinkova-Kostova, A.T. HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem. Biol. 2011, 18, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Rezaie, T.; Seki, M.; Sunico, C.R.; Tabuchi, T.; Kitagawa, T.; Yanagitai, M.; Senzaki, M.; Kosegawa, C.; Taira, H.; et al. Dual neuroprotective pathways of a pro-electrophilic compound via HSF-1-activated heat-shock proteins and Nrf2-activated phase 2 antioxidant response enzymes. J. Neurochem. 2011, 119, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, I.; Oguiza, A.; Recio, C.; Mallavia, B.; Madrigal-Matute, J.; Blanco, J.; Egido, J.; Martin-Ventura, J.L.; Gomez-Guerrero, C. Targeting HSP90 Ameliorates Nephropathy and Atherosclerosis Through Suppression of NF-kappaB and STAT Signaling Pathways in Diabetic Mice. Diabetes 2015, 64, 3600–3613. [Google Scholar] [CrossRef] [PubMed]
- Businaro, R.; Profumo, E.; Tagliani, A.; Buttari, B.; Leone, S.; D’Amati, G.; Ippoliti, F.; Leopizzi, M.; D’Arcangelo, D.; Capoano, R.; et al. Heat-shock protein 90: A novel autoantigen in human carotid atherosclerosis. Atherosclerosis 2009, 207, 74–83. [Google Scholar] [CrossRef]
- Rigano, R.; Profumo, E.; Buttari, B.; Tagliani, A.; Petrone, L.; D’Amati, G.; Ippoliti, F.; Capoano, R.; Fumagalli, L.; Salvati, B.; et al. Heat shock proteins and autoimmunity in patients with carotid atherosclerosis. Ann. N. Y Acad. Sci. 2007, 1107, 1–10. [Google Scholar] [CrossRef]
- Profumo, E.; Buttari, B.; Tinaburri, L.; D’Arcangelo, D.; Sorice, M.; Capozzi, A.; Garofalo, T.; Facchiano, A.; Businaro, R.; Kumar, P.; et al. Oxidative Stress Induces HSP90 Upregulation on the Surface of Primary Human Endothelial Cells: Role of the Antioxidant 7,8-Dihydroxy-4-methylcoumarin in Preventing HSP90 Exposure to the Immune System. Oxid. Med. Cell Longev. 2018, 2018, 2373167. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Kleindienst, R.; Schett, G.; Amberger, A.; Xu, Q. Role of heat shock protein 65/60 in the pathogenesis of atherosclerosis. Int. Arch. Allergy Immunol. 1995, 107, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Wick, C. Tolerization against atherosclerosis using heat shock protein 60. Cell Stress. Chaperones 2016, 21, 201–211. [Google Scholar] [CrossRef]
- Grundtman, C.; Kreutmayer, S.B.; Almanzar, G.; Wick, M.C.; Wick, G. Heat shock protein 60 and immune inflammatory responses in atherosclerosis. Arter. Thromb. Vasc. Biol. 2011, 31, 960–968. [Google Scholar] [CrossRef]
- Lazaro, I.; Oguiza, A.; Recio, C.; Lopez-Sanz, L.; Bernal, S.; Egido, J.; Gomez-Guerrero, C. Interplay between HSP90 and Nrf2 pathways in diabetes-associated atherosclerosis. Clin. Investig. Arter. 2017, 29, 51–59. [Google Scholar] [CrossRef]
- Dayalan Naidu, S.; Kostov, R.V.; Dinkova-Kostova, A.T. Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol. Sci. 2015, 36, 6–14. [Google Scholar] [CrossRef]
- Paul, S.; Ghosh, S.; Mandal, S.; Sau, S.; Pal, M. NRF2 transcriptionally activates the heat shock factor 1 promoter under oxidative stress and affects survival and migration potential of MCF7 cells. J. Biol. Chem. 2018, 293, 19303–19316. [Google Scholar] [CrossRef] [PubMed]
- Russ, D.W.; Boyd, I.M.; McCoy, K.M.; McCorkle, K.W. Muscle-specificity of age-related changes in markers of autophagy and sphingolipid metabolism. Biogerontology 2015, 16, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of aging and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef]
- Dai, D.F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef]
- Sazonova, M.; Budnikov, E.; Khasanova, Z.; Sobenin, I.; Postnov, A.; Orekhov, A. Studies of the human aortic intima by a direct quantitative assay of mutant alleles in the mitochondrial genome. Atherosclerosis 2009, 204, 184–190. [Google Scholar] [CrossRef]
- Zhu, L.; Wu, G.; Yang, X.; Jia, X.; Li, J.; Bai, X.; Li, W.; Zhao, Y.; Li, Y.; Cheng, W.; et al. Low density lipoprotein mimics insulin action on autophagy and glucose uptake in endothelial cells. Sci. Rep. 2019, 9, 3020. [Google Scholar] [CrossRef] [PubMed]
- Fetterman, J.L.; Holbrook, M.; Flint, N.; Feng, B.; Breton-Romero, R.; Linder, E.A.; Berk, B.D.; Duess, M.A.; Farb, M.G.; Gokce, N.; et al. Restoration of autophagy in endothelial cells from patients with diabetes mellitus improves nitric oxide signaling. Atherosclerosis 2016, 247, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kitada, M.; Ogura, Y.; Koya, D. Relationship Between Autophagy and Metabolic Syndrome Characteristics in the Pathogenesis of Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 641852. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; De Meyer, G.R. Autophagy in atherosclerosis: A cell survival and death phenomenon with therapeutic potential. Circ. Res. 2009, 104, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wang, X.; Schnackenberg, L.; Khaidakov, M.; Liu, S.; Singla, S.; Dai, Y.; Mehta, J.L. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7 g. Int. J. Cardiol. 2013, 168, 1378–1385. [Google Scholar] [CrossRef]
- Zhai, C.; Cheng, J.; Mujahid, H.; Wang, H.; Kong, J.; Yin, Y.; Li, J.; Zhang, Y.; Ji, X.; Chen, W. Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PLoS ONE 2014, 9, e90563. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Song, L.; Yan, H.; Liu, M.; Zhang, L.; Ma, Y.; Yuan, J.; Hu, J.; Ji, Z.; Zhang, R.; et al. Low dose tunicamycin enhances atherosclerotic plaque stability by inducing autophagy. Biochem. Pharmacol. 2016, 100, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Le Guezennec, X.; Brichkina, A.; Huang, Y.F.; Kostromina, E.; Han, W.; Bulavin, D.V. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab. 2012, 16, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Stypmann, J.; Janssen, P.M.; Prestle, J.; Engelen, M.A.; Kogler, H.; Lullmann-Rauch, R.; Eckardt, L.; von Figura, K.; Landgrebe, J.; Mleczko, A.; et al. LAMP-2 deficient mice show depressed cardiac contractile function without significant changes in calcium handling. Basic. Res. Cardiol. 2006, 101, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy Cell Death: Innocent Convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; D’Amore, A.; Curl, C.L.; Allen, A.M.; Harrap, S.B.; Thomas, W.G.; Delbridge, L.M. Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension 2009, 53, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, M.; Henning, R.H.; Brundel, B.J. Derailed Proteostasis as a Determinant of Cardiac Aging. Can. J. Cardiol. 2016, 32, 1166.e11–1166.e20. [Google Scholar] [CrossRef]
- Cao, X.; Chen, A.; Yang, P.; Song, X.; Liu, Y.; Li, Z.; Wang, X.; Wang, L.; Li, Y. Alpha-lipoic acid protects cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy. Biochem. Biophys. Res. Commun. 2013, 441, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Hang, S.; Zhang, W.; Mao, C.; Li, D. An update on the bridging factors connecting autophagy and Nrf2 antioxidant pathway. Front. Cell Dev. Biol. 2023, 11, 1232241. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ding, Z.; Du, K.; Ye, X.; Cheng, S. Reactive Oxygen Species as a Link between Antioxidant Pathways and Autophagy. Oxid. Med. Cell Longev. 2021, 2021, 5583215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Feng, C.; Jiang, H. Novel target for treating Alzheimer’s Diseases: Crosstalk between the Nrf2 pathway and autophagy. Aging Res. Rev. 2021, 65, 101207. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, S.; Zhao, Y.; Ma, X.; Zhang, K.; He, X.; Wang, Z. Interaction domains of p62: A bridge between p62 and selective autophagy. DNA Cell Biol. 2013, 32, 220–227. [Google Scholar] [CrossRef]
- Zakkar, M.; Van der Heiden, K.; Luong, L.A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arter. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef]
- Juan, S.H.; Lee, T.S.; Tseng, K.W.; Liou, J.Y.; Shyue, S.K.; Wu, K.K.; Chau, L.Y. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation 2001, 104, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Ito, K.; Hosoya, T.; Mimura, J.; Maruyama, A.; Noguchi, N.; Yagami, K.; Morito, N.; Takahashi, S.; Maher, J.M.; et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic. Biol. Med. 2012, 53, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef]
- Bories, G.; Colin, S.; Vanhoutte, J.; Derudas, B.; Copin, C.; Fanchon, M.; Daoudi, M.; Belloy, L.; Haulon, S.; Zawadzki, C.; et al. Liver X receptor activation stimulates iron export in human alternative macrophages. Circ. Res. 2013, 113, 1196–1205. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Cheng, L.; Jin, Z.; Zhao, R.; Ren, K.; Deng, C.; Yu, S. Resveratrol attenuates inflammation and oxidative stress induced by myocardial ischemia-reperfusion injury: Role of Nrf2/ARE pathway. Int. J. Clin. Exp. Med. 2015, 8, 10420–10428. [Google Scholar]
- Ashrafian, H.; Czibik, G.; Bellahcene, M.; Aksentijevic, D.; Smith, A.C.; Mitchell, S.J.; Dodd, M.S.; Kirwan, J.; Byrne, J.J.; Ludwig, C.; et al. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 2012, 15, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Piao, C.S.; Gao, S.; Lee, G.H.; Kim, D.S.; Park, B.H.; Chae, S.W.; Chae, H.J.; Kim, S.H. Sulforaphane protects ischemic injury of hearts through antioxidant pathway and mitochondrial K(ATP) channels. Pharmacol. Res. 2010, 61, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, A.A.; Beka, V.; Prado, C.M. The effect of caloric restriction on blood pressure and cardiovascular function: A systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. 2021, 40, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Parrado-Fernandez, C.; Csiszar, A.; de Cabo, R. Mechanisms underlying caloric restriction and lifespan regulation: Implications for vascular aging. Circ. Res. 2008, 102, 519–528. [Google Scholar] [CrossRef]
- Maloyan, A.; Gulick, J.; Glabe, C.G.; Kayed, R.; Robbins, J. Exercise reverses preamyloid oligomer and prolongs survival in alphaB-crystallin-based desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 5995–6000. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Jiang, Y.N.; Guan, X.; Ren, F.F.; Wu, S.J.; Chu, M.P.; Wu, L.P.; Lai, T.F.; Li, L. Aerobic Exercise Attenuates Pressure Overload-Induced Myocardial Remodeling and Myocardial Inflammation via Upregulating miR-574-3p in Mice. Circ. Heart Fail. 2024, 17, e010569. [Google Scholar] [CrossRef] [PubMed]
- Mohammadkhani, R.; Ranjbar, K.; Salehi, I.; Komaki, A.; Zarrinkalam, E.; Amiri, P. Comparison of the preconditioning effect of different exercise training modalities on myocardial ischemia-reperfusion injury. PLoS ONE 2023, 18, e0295169. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhu, Y.; Zheng, C.; Hu, D.; Ma, S.; Chen, L.; Wang, Q.; Chen, Z.; Xie, J.; Yan, Y.; et al. Antihypertrophic Memory After Regression of Exercise-Induced Physiological Myocardial Hypertrophy Is Mediated by the Long Noncoding RNA Mhrt779. Circulation 2021, 143, 2277–2292. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Mao, Y.; Chen, Z.; Kang, L.; Xu, B.; Wang, K. Exercise-induced myocardial hypertrophy preconditioning promotes fibroblast senescence and improves myocardial fibrosis through Nrf2 signaling pathway. Cell Cycle 2023, 22, 1529–1543. [Google Scholar] [CrossRef]
- Shanmugam, G.; Challa, A.K.; Litovsky, S.H.; Devarajan, A.; Wang, D.; Jones, D.P.; Darley-Usmar, V.M.; Rajasekaran, N.S. Enhanced Keap1-Nrf2 signaling protects the myocardium from isoproterenol-induced pathological remodeling in mice. Redox Biol. 2019, 27, 101212. [Google Scholar] [CrossRef]
- Queisser, M.A.; Yao, D.; Geisler, S.; Hammes, H.P.; Lochnit, G.; Schleicher, E.D.; Brownlee, M.; Preissner, K.T. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 2010, 59, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Catalgol, B.; Grune, T. The proteasomal system. Mol. Asp. Med. 2009, 30, 191–296. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Giannelli, S.; Sottero, B.; Staurenghi, E.; Giaccone, G.; Caroppo, P.; Gamba, P.; Leonarduzzi, G. 24-Hydroxycholesterol Induces Tau Proteasome-Dependent Degradation via the SIRT1/PGC1alpha/Nrf2 Pathway: A Potential Mechanism to Counteract Alzheimer’s Disease. Antioxidants 2023, 12, 631. [Google Scholar] [CrossRef]
- Chapple, S.J.; Siow, R.C.; Mann, G.E. Crosstalk between Nrf2 and the proteasome: Therapeutic potential of Nrf2 inducers in vascular disease and aging. Int. J. Biochem. Cell Biol. 2012, 44, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Pomatto, L.C.D.; Cline, M.; Woodward, N.; Pakbin, P.; Sioutas, C.; Morgan, T.E.; Finch, C.E.; Forman, H.J.; Davies, K.J.A. Aging attenuates redox adaptive homeostasis and proteostasis in female mice exposed to traffic-derived nanoparticles (‘vehicular smog’). Free Radic. Biol. Med. 2018, 121, 86–97. [Google Scholar] [CrossRef]
- Kuntic, M.; Kuntic, I.; Krishnankutty, R.; Gericke, A.; Oelze, M.; Junglas, T.; Bayo Jimenez, M.T.; Stamm, P.; Nandudu, M.; Hahad, O.; et al. Co-exposure to urban particulate matter and aircraft noise adversely impacts the cerebro-pulmonary-cardiovascular axis in mice. Redox Biol. 2023, 59, 102580. [Google Scholar] [CrossRef] [PubMed]
- Manola, M.S.; Gumeni, S.; Trougakos, I.P. Differential Dose- and Tissue-Dependent Effects of foxo on Aging, Metabolic and Proteostatic Pathways. Cells 2021, 10, 3577. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef]
- Zhang, D.D. Bardoxolone brings Nrf2-based therapies to light. Antioxid. Redox Signal 2013, 19, 517–518. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Townshend, T.; Lake, A. Obesogenic environments: Current evidence of the built and food environments. Perspect. Public. Health 2017, 137, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Gimenez, L.; Becerril, S.; Camoes, S.P.; da Silva, I.V.; Rodrigues, C.; Moncada, R.; Valenti, V.; Catalan, V.; Gomez-Ambrosi, J.; Miranda, J.P.; et al. Role of aquaporin-7 in ghrelin- and GLP-1-induced improvement of pancreatic beta-cell function after sleeve gastrectomy in obese rats. Int. J. Obes. 2017, 41, 1394–1402. [Google Scholar] [CrossRef]
- Garus-Pakowska, A. Metabolic Diseases-A Challenge for Public Health in the 21st Century. Int. J. Env. Res. Public. Health 2023, 20, 6789. [Google Scholar] [CrossRef] [PubMed]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef]
- Lasker, S.; Rahman, M.M.; Parvez, F.; Zamila, M.; Miah, P.; Nahar, K.; Kabir, F.; Sharmin, S.B.; Subhan, N.; Ahsan, G.U.; et al. High-fat diet-induced metabolic syndrome and oxidative stress in obese rats are ameliorated by yogurt supplementation. Sci. Rep. 2019, 9, 20026. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Shah, J.; Orosz, T.; Singh, A.; Laxma, S.P.; Gross, R.E.; Smith, N.; Vroegop, S.; Sudler, S.; Porter, J.T.; Colon, M.; et al. Influence of Exercise and Genistein to Mitigate the Deleterious Effects of High-Fat High-Sugar Diet on Alzheimer’s Disease-Related Markers in Male Mice. Int. J. Mol. Sci. 2024, 25, 9019. [Google Scholar] [CrossRef]
- Lefebvre, C.; Tiffay, A.; Breemeersch, C.E.; Dreux, V.; Bole-Feysot, C.; Guerin, C.; Breton, J.; Maximin, E.; Monnoye, M.; Dechelotte, P.; et al. Sex-dependent effects of a high fat diet on metabolic disorders, intestinal barrier function and gut microbiota in mouse. Sci. Rep. 2024, 14, 19835. [Google Scholar] [CrossRef] [PubMed]
- Janoutova, J.; Machaczka, O.; Zatloukalova, A.; Janout, V. Is Alzheimer’s disease a type 3 diabetes? A review. Cent. Eur. J. Public. Health 2022, 30, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Jaisson, S.; Gillery, P. Impaired proteostasis: Role in the pathogenesis of diabetes mellitus. Diabetologia 2014, 57, 1517–1527. [Google Scholar] [CrossRef]
- Dodson, M.; Shakya, A.; Anandhan, A.; Chen, J.; Garcia, J.G.N.; Zhang, D.D. NRF2 and Diabetes: The Good, the Bad, and the Complex. Diabetes 2022, 71, 2463–2476. [Google Scholar] [CrossRef]
- Bentanachs, R.; Blanco, L.; Montesinos, M.; Sala-Vila, A.; Lazaro, I.; Rodriguez-Morato, J.; Sanchez, R.M.; Laguna, J.C.; Roglans, N.; Alegret, M. Adipose Tissue Protects against Hepatic Steatosis in Male Rats Fed a High-Fat Diet plus Liquid Fructose: Sex-Related Differences. Nutrients 2023, 15, 3909. [Google Scholar] [CrossRef]
- Velazquez, A.M.; Bentanachs, R.; Sala-Vila, A.; Lazaro, I.; Rodriguez-Morato, J.; Sanchez, R.M.; Alegret, M.; Roglans, N.; Laguna, J.C. ChREBP-driven DNL and PNPLA3 Expression Induced by Liquid Fructose are Essential in the Production of Fatty Liver and Hypertriglyceridemia in a High-Fat Diet-Fed Rat Model. Mol. Nutr. Food Res. 2022, 66, e2101115. [Google Scholar] [CrossRef]
- Di Veroli, B.; Bentanachs, R.; Roglans, N.; Alegret, M.; Giona, L.; Profumo, E.; Berry, A.; Saso, L.; Laguna, J.C.; Buttari, B. Sex Differences Affect the NRF2 Signaling Pathway in the Early Phase of Liver Steatosis: A High-Fat-Diet-Fed Rat Model Supplemented with Liquid Fructose. Cells 2024, 13, 1247. [Google Scholar] [CrossRef]
- Giona, L.; Musillo, C.; De Cristofaro, G.; Ristow, M.; Zarse, K.; Siems, K.; Tait, S.; Cirulli, F.; Berry, A. Western diet-induced cognitive and metabolic dysfunctions in aged mice are prevented by rosmarinic acid in a sex-dependent fashion. Clin. Nutr. 2024, 43, 2236–2248. [Google Scholar] [CrossRef]
- Boesch, S.; Indelicato, E. Approval of omaveloxolone for Friedreich ataxia. Nat. Rev. Neurol. 2024, 20, 313–314. [Google Scholar] [CrossRef]
- Wang, Q.; Chuikov, S.; Taitano, S.; Wu, Q.; Rastogi, A.; Tuck, S.J.; Corey, J.M.; Lundy, S.K.; Mao-Draayer, Y. Dimethyl Fumarate Protects Neural Stem/Progenitor Cells and Neurons from Oxidative Damage through Nrf2-ERK1/2 MAPK Pathway. Int. J. Mol. Sci. 2015, 16, 13885–13907. [Google Scholar] [CrossRef]
- Brennan, M.S.; Matos, M.F.; Richter, K.E.; Li, B.; Scannevin, R.H. The NRF2 transcriptional target, OSGIN1, contributes to monomethyl fumarate-mediated cytoprotection in human astrocytes. Sci. Rep. 2017, 7, 42054. [Google Scholar] [CrossRef]
- Chaves, C.; Ganguly, R.; Ceresia, C.; Camac, A. Lymphocyte subtypes in relapsing-remitting multiple sclerosis patients treated with dimethyl fumarate. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317702933. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Lipton, S. Recent advances in understanding NRF2 as a druggable target: Development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate. F1000Research 2017, 6, 2138. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A. Sulforaphane: Its “Coming of Age” as a Clinically Relevant Nutraceutical in the Prevention and Treatment of Chronic Disease. Oxid. Med. Cell Longev. 2019, 2019, 2716870. [Google Scholar] [CrossRef]
- Hu, Y.; Luo, Y.; Zheng, Y. Nrf2 Pathway and Autophagy Crosstalk: New Insights into Therapeutic Strategies for Ischemic Cerebral Vascular Diseases. Antioxidants 2022, 11, 1747. [Google Scholar] [CrossRef]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rezaie, T.; Nutter, A.; Lopez, K.M.; Parker, J.; Kosaka, K.; Satoh, T.; McKercher, S.R.; Masliah, E.; Nakanishi, N. Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 2016, 7, e2499. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Trudler, D.; Oh, C.K.; Lipton, S.A. Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome. Antioxidants 2022, 11, 124. [Google Scholar] [CrossRef]
- Cores, A.; Carmona-Zafra, N.; Clerigue, J.; Villacampa, M.; Menendez, J.C. Quinones as Neuroprotective Agents. Antioxidants 2023, 12, 1464. [Google Scholar] [CrossRef]
- Iegre, J.; Krajcovicova, S.; Gunnarsson, A.; Wissler, L.; Kack, H.; Luchniak, A.; Tangefjord, S.; Narjes, F.; Spring, D.R. A cell-active cyclic peptide targeting the Nrf2/Keap1 protein-protein interaction. Chem. Sci. 2023, 14, 10800–10805. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Colarusso, S.; De Simone, D.; Frattarelli, T.; Andreini, M.; Cerretani, M.; Missineo, A.; Moretti, D.; Tambone, S.; Kempf, G.; Augustin, M.; et al. Optimization of linear and cyclic peptide inhibitors of KEAP1-NRF2 protein-protein interaction. Bioorg. Med. Chem. 2020, 28, 115738. [Google Scholar] [CrossRef] [PubMed]
- Carrow, K.P.; Hamilton, H.L.; Hopps, M.P.; Li, Y.; Qiao, B.; Payne, N.C.; Thompson, M.P.; Zhang, X.; Magassa, A.; Fattah, M.; et al. Inhibiting the Keap1/Nrf2 Protein-Protein Interaction with Protein-Like Polymers. Adv. Mater. 2024, 36, e2311467. [Google Scholar] [CrossRef]
- Crisman, E.; Duarte, P.; Dauden, E.; Cuadrado, A.; Rodriguez-Franco, M.I.; Lopez, M.G.; Leon, R. KEAP1-NRF2 protein-protein interaction inhibitors: Design, pharmacological properties and therapeutic potential. Med. Res. Rev. 2023, 43, 237–287. [Google Scholar] [CrossRef] [PubMed]
- Shahcheraghi, S.H.; Salemi, F.; Peirovi, N.; Ayatollahi, J.; Alam, W.; Khan, H.; Saso, L. Nrf2 Regulation by Curcumin: Molecular Aspects for Therapeutic Prospects. Molecules 2021, 27, 167. [Google Scholar] [CrossRef]
- Kim, A.N.; Jeon, W.K.; Lee, J.J.; Kim, B.C. Up-regulation of heme oxygenase-1 expression through CaMKII-ERK1/2-Nrf2 signaling mediates the anti-inflammatory effect of bisdemethoxycurcumin in LPS-stimulated macrophages. Free Radic. Biol. Med. 2010, 49, 323–331. [Google Scholar] [CrossRef]
- Xiao, Y.; Xia, J.; Wu, S.; Lv, Z.; Huang, S.; Huang, H.; Su, X.; Cheng, J.; Ke, Y. Curcumin Inhibits Acute Vascular Inflammation through the Activation of Heme Oxygenase-1. Oxid. Med. Cell Longev. 2018, 2018, 3295807. [Google Scholar] [CrossRef]
- Moratilla-Rivera, I.; Sanchez, M.; Valdes-Gonzalez, J.A.; Gomez-Serranillos, M.P. Natural Products as Modulators of Nrf2 Signaling Pathway in Neuroprotection. Int. J. Mol. Sci. 2023, 24, 3748. [Google Scholar] [CrossRef] [PubMed]
- Calixto-Campos, C.; Correa, M.P.; Carvalho, T.T.; Zarpelon, A.C.; Hohmann, M.S.; Rossaneis, A.C.; Coelho-Silva, L.; Pavanelli, W.R.; Pinge-Filho, P.; Crespigio, J.; et al. Quercetin reduces Ehrlich tumor-induced cancer pain in mice. Anal. Cell Pathol. 2015, 2015, 285708. [Google Scholar] [CrossRef]
- Kim, K.H.; Sadikot, R.T.; Joo, M. Therapeutic effect of ent-kaur-16-en-19-oic acid on neutrophilic lung inflammation and sepsis is mediated by Nrf2. Biochem. Biophys. Res. Commun. 2016, 474, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Zinovkin, R.A.; Kondratenko, N.D.; Zinovkina, L.A. Does Nrf2 Play a Role of a Master Regulator of Mammalian Aging? Biochemistry 2022, 87, 1465–1476. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cui, J.Y.; Lu, Y.F.; Corton, J.C.; Klaassen, C.D. Sex-, Age-, and Race/Ethnicity-Dependent Variations in Drug-Processing and NRF2-Regulated Genes in Human Livers. Drug Metab. Dispos. 2021, 49, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Cucci, M.A.; Grattarola, M.; Dianzani, C.; Muzio, G.; Pizzimenti, S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants 2021, 10, 510. [Google Scholar] [CrossRef] [PubMed]
- Camina, N.; Penning, T.M. Genetic and epigenetic regulation of the NRF2-KEAP1 pathway in human lung cancer. Br. J. Cancer 2022, 126, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Mendez, I.; Diaz-Munoz, M. Circadian and Metabolic Perspectives in the Role Played by NADPH in Cancer. Front. Endocrinol 2018, 9, 93. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes. Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Lv, B.; Xing, S.; Wang, Z.; Zhang, A.; Wang, Q.; Bian, Y.; Pei, Y.; Sun, H.; Chen, Y. NRF2 inhibitors: Recent progress, future design and therapeutic potential. Eur. J. Med. Chem. 2024, 279, 116822. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, H.; Fan, L.; Wu, X.; Xin, A.; Ren, H.; Wang, X.J. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic. Biol. Med. 2011, 50, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Jung, B.J.; Lee, S.H.; Yoo, H.S.; Shin, E.A.; Ko, H.J.; Chang, S.; Kim, S.Y.; Jeon, S.M. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017, 36, 5285–5295. [Google Scholar] [CrossRef]
- Yu, C.; Jiao, Y.; Xue, J.; Zhang, Q.; Yang, H.; Xing, L.; Chen, G.; Wu, J.; Zhang, S.; Zhu, W.; et al. Metformin Sensitizes Non-small Cell Lung Cancer Cells to an Epigallocatechin-3-Gallate (EGCG) Treatment by Suppressing the Nrf2/HO-1 Signaling Pathway. Int. J. Biol. Sci. 2017, 13, 1560–1569. [Google Scholar] [CrossRef]
- Simov, V.; Altman, M.D.; Bianchi, E.; DelRizzo, S.; DiNunzio, E.N.; Feng, G.; Goldenblatt, P.; Ingenito, R.; Johnson, S.A.; Mansueto, M.S.; et al. Discovery and characterization of novel peptide inhibitors of the NRF2/MAFG/DNA ternary complex for the treatment of cancer. Eur. J. Med. Chem. 2021, 224, 113686. [Google Scholar] [CrossRef] [PubMed]
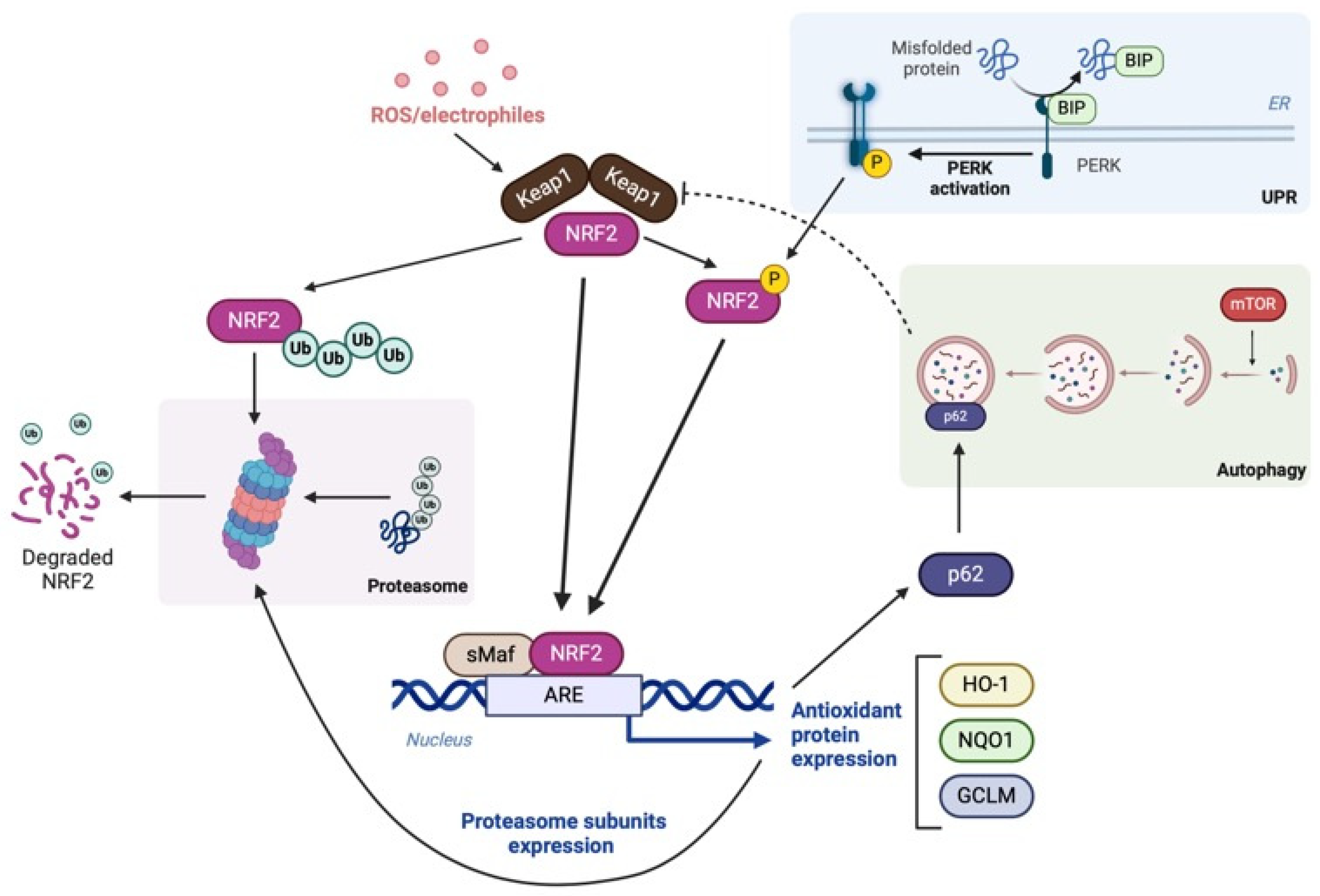
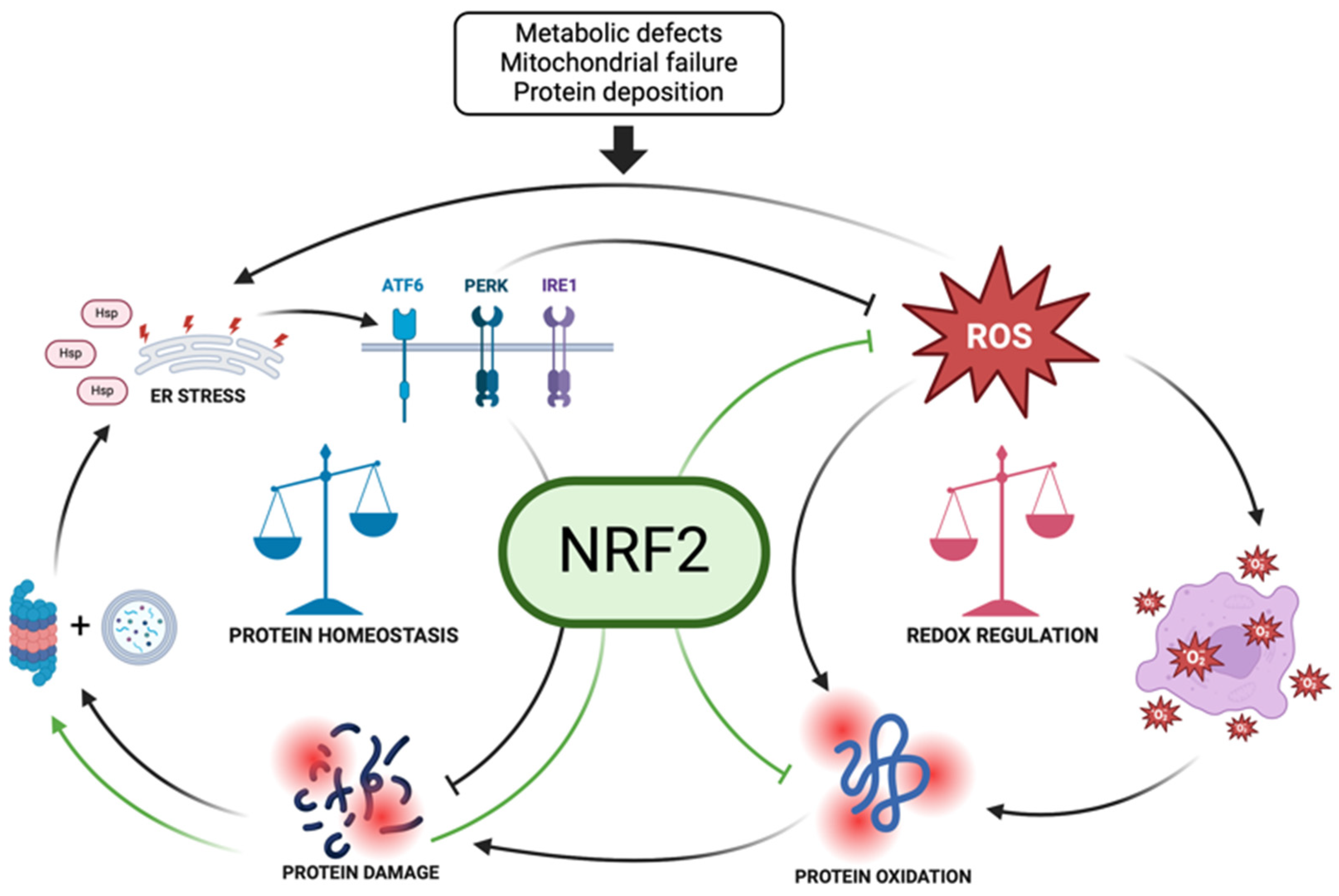

{kind=link}
{kind=link}
| NRF2 Activators | Drug Name | Mechanism of Action | Clinical Use | References | |
| Tecfidera | Dimethyl fumarate (DMF) | Electrophilic modification of KEAP1-Cys-151 | Multiple sclerosis | [284] | |
| Diroximel fumarate | Monomethyl fumarate (MMF) | Electrophilic modification of KEAP1-Cys-151 | Multiple sclerosis (phase III clinical trials) | NCT03093324 | |
| Tepilamide fumarate | Monomethyl fumarate (MMF) | Electrophilic modification of KEAP1-Cys-151 | Plaque psoriasis (phase II clinical trials) | NCT02173301 | |
| Sulforaphane (SFN) | 1-isothiocyanato-4-(methylsulfonyl)-butane | Electrophilic modification of KEAP1-Cys-151 | Cancer, autism, chronic kidney disease, and type 2 diabetes | [288] | |
| Benfotiamine | Synthetic thiamine precursor | Direct actions on multiple metabolic enzymes, inflammation and oxidative stress | Alzheimer’s disease | NCT02292238 | |
| Carnosic Acid (CA) | Active ingredient in the herb rosemary (Rosmarinus officinalis) | Pro-electrophilic drug with antioxidative and ant inflammatory effects | Alzheimer’s disease | [118,291,292,293] | |
| Zonarol and Isozonarol | Found in seaweed (Dictyopteris undulata) | Pro-electrophilic drug with antioxidative and ant inflammatory effects | Alzheimer’s disease | [287] | |
| Omaveloxolone | KEAP1-NRF2 protein–protein interaction inhibitors | DLG and ETGE motifs on NRF2, which bind to the Kelch domain of KEAP1, are primary targets for non-covalent inhibitors of the KEAP1-NRF2 PPI | Friedreich’s ataxia | [300] | |
| KEAP1-NRF2 PPI inhibitors | DLG and ETGE motifs on NRF2, which bind to the Kelch domain of KEAP1, are primary targets for the KEAP1-NRF2 PPI | Neurodegenerative disease | [295] | ||
| NRF2 Activators | Natural Antioxidant Compounds | Mechanism of Action | Experimental Models | References | |
| Curcumin | Polyphenol-derived compound | Anti-inflammatory effects | Renal epithelial cells, lung mesenchymal stem cells, macrophages, Leydig cells | [301] | |
| Bisdemethoxycurcumin | Curcumin analogue | Anti-inflammatory effects | Macrophages, β-cells | [302,303] | |
| Tiliroside | A glycoside containing kaempferol | Anti-inflammatory and antioxidant effects | Neurons (HT-22 cell line) and microglia (BV22 cell line) | [304] | |
| Engeletin | Dihydrokaempferol 3-rhamnoside | Anti-inflammatory and antioxidant effects | Microglia (BV22 cell line) | [304] | |
| Quercetin | Anti-inflammatory and antioxidant effects | Microglia (BV22 cell line) and mouse model for chronic pain | [117,305] | ||
| Kaurenoic acid | Ent-kaur-16-en-19-oic acid (diterpene) | Anti-inflammatory effects | Mouse model for sepsis and with a chronic lung inflammation | [306] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buttari, B.; Tramutola, A.; Rojo, A.I.; Chondrogianni, N.; Saha, S.; Berry, A.; Giona, L.; Miranda, J.P.; Profumo, E.; Davinelli, S.; et al. Proteostasis Decline and Redox Imbalance in Age-Related Diseases: The Therapeutic Potential of NRF2. Biomolecules 2025, 15, 113. https://doi.org/10.3390/biom15010113
Buttari B, Tramutola A, Rojo AI, Chondrogianni N, Saha S, Berry A, Giona L, Miranda JP, Profumo E, Davinelli S, et al. Proteostasis Decline and Redox Imbalance in Age-Related Diseases: The Therapeutic Potential of NRF2. Biomolecules. 2025; 15(1):113. https://doi.org/10.3390/biom15010113
Chicago/Turabian StyleButtari, Brigitta, Antonella Tramutola, Ana I. Rojo, Niki Chondrogianni, Sarmistha Saha, Alessandra Berry, Letizia Giona, Joana P. Miranda, Elisabetta Profumo, Sergio Davinelli, and et al. 2025. "Proteostasis Decline and Redox Imbalance in Age-Related Diseases: The Therapeutic Potential of NRF2" Biomolecules 15, no. 1: 113. https://doi.org/10.3390/biom15010113
APA StyleButtari, B., Tramutola, A., Rojo, A. I., Chondrogianni, N., Saha, S., Berry, A., Giona, L., Miranda, J. P., Profumo, E., Davinelli, S., Daiber, A., Cuadrado, A., & Di Domenico, F. (2025). Proteostasis Decline and Redox Imbalance in Age-Related Diseases: The Therapeutic Potential of NRF2. Biomolecules, 15(1), 113. https://doi.org/10.3390/biom15010113