

Developmental and Epileptic Encephalopathy: Pathogenesis of Intellectual Disability Beyond Channelopathies

 , , , and
, , , and
Abstract
1. Introduction
2. Pathogenesis of Developmental Delay and Intellectual Disability in Developmental and Epileptic Encephalopathy
3. Molecular Mechanisms Underlying Developmental and Epileptic Encephalopathy
3.1. Malformations of Cortical Development as a Cause of DEE
3.1.1. Neuronal Progenitor Proliferation Disruption
3.1.2. Neuronal Differentiation Disruption
3.1.3. Neuronal Migration Disorders
3.1.4. Dendrito- and Axonogenesis Disorders
3.2. Synaptopathies—Synaptic Transmission Disorders
3.3. Metabolic Disorders
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Engel, J. A Proposed Diagnostic Scheme for People with Epileptic Seizures and with Epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001, 42, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Elliott, K.S.; Shetty, J.; Armstrong, M.; Brunklaus, A.; Cutcutache, I.; A Diver, L.; Dorris, L.; Gardiner, S.; Jollands, A.; et al. Early childhood epilepsies: Epidemiology, classification, aetiology, and socio-economic determinants. Brain 2021, 144, 2879–2891. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, S.M.; Wirrell, E.; Yozawitz, E.; Wilmshurst, J.M.; Specchio, N.; Riney, K.; Pressler, R.; Auvin, S.; Samia, P.; Hirsch, E.; et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1349–1397. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Raga, S.; Specchio, N.; Rheims, S.; Wilmshurst, J.M. Developmental and epileptic encephalopathies: Recognition and approaches to care. Epileptic Disord. 2021, 23, 40–52. [Google Scholar] [CrossRef]
- Guerrini, R.; Conti, V.; Mantegazza, M.; Balestrini, S.; Galanopoulou, A.S.; Benfenati, F. Developmental and epileptic encephalopathies: From genetic heterogeneity to phenotypic continuum. Physiol. Rev. 2023, 103, 433–513. [Google Scholar] [CrossRef]
- Surdi, P.; Trivisano, M.; De Dominicis, A.; Mercier, M.; Piscitello, L.M.; Pavia, G.C.; Calabrese, C.; Cappelletti, S.; Correale, C.; Mazzone, L.; et al. Unveiling the Disease Progression in Developmental and Epileptic Encephalopathies: Insights from EEG and Neuropsychology. Epilepsia 2024, 65, 3279–3292. [Google Scholar] [CrossRef]
- Keezer, M.R.; Sisodiya, S.M.; Sander, J.W. Comorbidities of Epilepsy: Current Concepts and Future Perspectives. Lancet Neurol. 2016, 15, 106–115. [Google Scholar] [CrossRef]
- Nariai, H.; Duberstein, S.; Shinnar, S. Treatment of Epileptic Encephalopathies: Current State of the Art. J. Child Neurol. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Jeffrey, J.S.; Leathem, J.; King, C.; Mefford, H.C.; Ross, K.; Sadleir, L.G. Developmental and epileptic encephalopathy: Personal utility of a genetic diagnosis for families. Epilepsia Open 2020, 6, 149–159. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Zhao, Y.; Bao, X.; Wei, L.; Wang, J. Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy. Clin. Genet. 2017, 91, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.B.; Eggers, S.; Dalziel, K.; Riseley, J.; Mandelstam, S.; Myers, C.T.; McMahon, J.M.; Schneider, A.; Carvill, G.L.; Mefford, H.C.; et al. A population-based cost-effectiveness study of early genetic testing in severe epilepsies of infancy. Epilepsia 2018, 59, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Happ, H.C.; Carvill, G.L. A 2020 View on the Genetics of Developmental and Epileptic Encephalopathies. Epilepsy Curr. 2020, 20, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Borowicz-Reutt, K.; Czernia, J.; Krawczyk, M. Genetic Background of Epilepsy and Antiepileptic Treatments. Int. J. Mol. Sci. 2023, 24, 16280. [Google Scholar] [CrossRef] [PubMed]
- Syrbe, S. Developmental and epileptic encephalopathies–therapeutic consequences of genetic testing. Med. Genet. 2022, 34, 215–224. [Google Scholar] [CrossRef]
- Nieh, S.E.; Sherr, E.H. Epileptic Encephalopathies: New Genes and New Pathways. Neurotherapeutics 2014, 11, 796–806. [Google Scholar] [CrossRef]
- Symonds, J.D.; McTague, A. Epilepsy and developmental disorders: Next generation sequencing in the clinic. Eur. J. Paediatr. Neurol. 2019, 24, 15–23. [Google Scholar] [CrossRef]
- Nickels, K.C.; Wirrell, E.C. Cognitive and Social Outcomes of Epileptic Encephalopathies. Semin. Pediatr. Neurol. 2017, 24, 264–275. [Google Scholar] [CrossRef]
- Raspall-Chaure, M.; Chin, R.F.M.; Neville, B.G.; Bedford, H.; Scott, R.C. The Epidemiology of Convulsive Status Epilepticus in Children: A Critical Review. Epilepsia 2007, 48, 1652–1663. [Google Scholar] [CrossRef]
- Holmes, G.L. Cognitive impairment in epilepsy: The role of network abnormalities. Epileptic Disord. 2015, 17, 101–116. [Google Scholar] [CrossRef]
- Lenck-Santini, P.-P.; Scott, R.C. Mechanisms Responsible for Cognitive Impairment in Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022772. [Google Scholar] [CrossRef] [PubMed]
- Lado, F.A.; Moshé, S.L. How Do Seizures Stop? Epilepsia 2008, 49, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Mathalon, D.H.; Sohal, V.S. Neural Oscillations and Synchrony in Brain Dysfunction and Neuropsychiatric Disorders It’s about Time. JAMA Psychiatry 2015, 72, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Ademuwagun, I.A.; Rotimi, S.O.; Syrbe, S.; Ajamma, Y.U.; Adebiyi, E. Voltage Gated Sodium Channel Genes in Epilepsy: Mutations, Functional Studies, and Treatment Dimensions. Front. Neurol. 2021, 12, 600050. [Google Scholar] [CrossRef]
- Ohba, C.; Kato, M.; Takahashi, S.; Lerman-Sagie, T.; Lev, D.; Terashima, H.; Kubota, M.; Kawawaki, H.; Matsufuji, M.; Kojima, Y.; et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 2014, 55, 994–1000. [Google Scholar] [CrossRef]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef]
- Johannessen, K.; Marini, C.; Pfeffer, S.; Møller, R.S.; Dorn, T.; Niturad, C.; Gardella, E.; Weber, Y.; Søndergård, M.; Hjalgrim, H.; et al. Phenotypic Spectrum of GABRA1: From Generalized Epilepsies to Severe Epileptic Encephalopathies. Neurology 2016, 87, 1140–1151. [Google Scholar] [CrossRef]
- Menezes, L.F.S.; Sabiá Júnior, E.F.; Tibery, D.V.; dos Carneiro, L.A.; Schwartz, E.F. Epilepsy-Related Volt-age-Gated Sodium Channelopathies: A review. Front. Pharmacol. 2020, 11, 1276. [Google Scholar] [CrossRef]
- Bonsi, P.; De Jaco, A.; Fasano, L.; Gubellini, P. Postsynaptic autism spectrum disorder genes and synaptic dysfunction. Neurobiol. Dis. 2021, 162, 105564. [Google Scholar] [CrossRef]
- Goto, A. Synaptic plasticity during systems memory consolidation. Neurosci. Res. 2022, 183, 1–6. [Google Scholar] [CrossRef]
- Spoto, G.; Valentini, G.; Saia, M.C.; Butera, A.; Amore, G.; Salpietro, V.; Nicotera, A.G.; Di Rosa, G. Synapto-pathies in Developmental and Epileptic Encephalopathies: A Focus on Pre-Synaptic Dysfunction. Front. Neurol. 2022, 13, 826211. [Google Scholar] [CrossRef] [PubMed]
- Berecki, G.; Bryson, A.; Terhag, J.; Maljevic, S.; Gazina, E.V.; Hill, S.L.; Petrou, S. SCN1A gain of function in early infantile encephalopathy. Ann. Neurol. 2019, 85, 514–525. [Google Scholar] [CrossRef]
- Pearson-Smith, J.N.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef] [PubMed]
- Hurni, N.; Kolodziejczak, M.; Tomasello, U.; Badia, J.; Jacobshagen, M.; Prados, J.; Dayer, A. Transient Cell-intrinsic Activity Regulates the Migration and Laminar Positioning of Cortical Projection Neurons. Cereb. Cortex 2017, 27, 3052–3063. [Google Scholar] [CrossRef] [PubMed]
- Vitaliti, G.; Pavone, P.; Marino, S.; Saporito, M.A.N.; Corsello, G.; Falsaperla, R. Molecular Mechanism In-volved in the Pathogenesis of Early-Onset Epileptic Encephalopathy. Front. Mol. Neurosci. 2019, 12, 118. [Google Scholar] [CrossRef]
- Lynch, M.A. Long-Term Potentiation and Memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef]
- Langille, J.J.; Brown, R.E. The Synaptic Theory of Memory: A Historical Survey and Reconciliation of Recent Opposition. Front. Syst. Neurosci. 2018, 12, 52. [Google Scholar] [CrossRef]
- Han, T.; Qin, Y.; Mou, C.; Wang, M.; Jiang, M.; Liu, B. Seizure Induced Synaptic Plasticity Alteration in Hip-pocampus Is Mediated by IL-1β Receptor through PI3K/Akt Pathway. Am. J. Transl. Res. 2016, 8, 4499. [Google Scholar]
- Lin, H.; Hangya, B.; Fox, S.E.; Muller, R.U. Repetitive Convulsant-Induced Seizures Reduce the Number But Not Precision of Hippocampal Place Cells. J. Neurosci. 2012, 32, 4163–4178. [Google Scholar] [CrossRef]
- Mitsui, Y.; Sato, H.; Togi, S.; Ura, H.; Niida, Y. A case of SCN8A-related developmental epileptic encephalopathy diagnosed by clinical speculation driven targeted DNA sequencing and remission of epilepsy by sodium channel blockers combination therapy. Brain Dev. Case Rep. 2024, 2, 100015. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Liao, J. When Monogenic Isn’t Monogenic—Unravelling the Oligogenic Architecture of the Developmental and Epileptic Encephalopathies. Epilepsy Curr. 2019, 19, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Hong, S.-Y.; Lin, W.-D.; Lin, C.-H.; Lin, S.-S.; Tsai, F.-J.; Chou, I.-C. Genetic Testing in Children with Developmental and Epileptic Encephalopathies: A Review of Advances in Epilepsy Genomics. Children 2023, 10, 556. [Google Scholar] [CrossRef] [PubMed]
- Papuc, S.M.; Abela, L.; Steindl, K.; Begemann, A.; Simmons, T.L.; Schmitt, B.; Zweier, M.; Oneda, B.; Socher, E.; Crowther, L.M.; et al. The role of recessive inheritance in early-onset epileptic encephalopathies: A combined whole-exome sequencing and copy number study. Eur. J. Hum. Genet. 2018, 27, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.S.; Loddenkemper, T.; Gaínza-Lein, M.; Sheidley, B.R.; Poduri, A. Diagnostic yield of genetic tests in epilepsy. Neurology 2019, 92, E418–E428. [Google Scholar] [CrossRef]
- Bartolini, E. Inherited Developmental and Epileptic Encephalopathies. Neurol. Int. 2021, 13, 555–568. [Google Scholar] [CrossRef]
- Han, X.; Deng, J.; Chen, C.; Wang, X.; Fang, F.; Li, H.; Luo, J.; Wu, J. Developmental and Epileptic Encephalopathy 76: Case Report and Review of Literature. Children 2022, 9, 1967. [Google Scholar] [CrossRef]
- Peñagarikano, O.; Geschwind, D.H. What Does CNTNAP2 Reveal about Autism Spectrum Disorder? Trends Mol. Med. 2012, 18, 156–163. [Google Scholar] [CrossRef]
- Chatron, N.; Møller, R.S.; Champaigne, N.L.; Schneider, A.L.; Kuechler, A.; Labalme, A.; Simonet, T.; Baggett, L.; Bardel, C.; Kamsteeg, E.J.; et al. The epilepsy phenotypic spectrum associated with a recurrent CUX2 variant. Ann. Neurol. 2018, 83, 926–934. [Google Scholar] [CrossRef]
- Begemann, A.; Sticht, H.; Begtrup, A.; Vitobello, A.; Faivre, L.; Banka, S.; Alhaddad, B.; Asadollahi, R.; Becker, J.; Bierhals, T.; et al. New insights into the clinical and molecular spectrum of the novel CYFIP2-related neurodevelopmental disorder and impairment of the WRC-mediated actin dynamics. Anesth. Analg. 2021, 23, 543–554. [Google Scholar] [CrossRef]
- Reiner, O.; Coquelle, F.M.; Peter, B.; Levy, T.; Kaplan, A.; Sapir, T.; Orr, I.; Barkai, N.; Eichele, G.; Bergmann, S. The evolving doublecortin (DCX) superfamily. BMC Genom. 2006, 7, 188. [Google Scholar] [CrossRef]
- Scoto, M.; Rossor, A.M.; Harms, M.B.; Cirak, S.; Calissano, M.; Robb, S.; Manzur, A.Y.; Arroyo, A.M.; Sanz, A.R.; Mansour, S.; et al. Novel mutations expand the clinical spectrum of DYNC1H1 -associated spinal muscular atrophy. Neurology 2015, 84, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.W.; Millichap, J.J.; Soares, D.C.; Chin, R.; McLellan, A.; FitzPatrick, D.R.; Elmslie, F.; Lees, M.M.; Schaefer, G.B.; DDD Study; et al. Novel de novo EEF1A2 missense mutations causing epilepsy and intellectual disability. Mol. Genet. Genom. Med. 2016, 4, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.H.; Pandey, U.B. Function and Dysfunction of GEMIN5: Understanding a Novel Neurodevelopmental Disorder. Neural Regen. Res. 2024, 19, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Al Masseri, Z.; AlSayed, M. Gonadal mosaicism in GNAO1 causing neurodevelopmental disorder with involuntary movements; two additional variants. Mol. Genet. Metab. Rep. 2022, 31, 100864. [Google Scholar] [CrossRef]
- Taylor, J.; Spiller, M.; Ranguin, K.; Vitobello, A.; Philippe, C.; Bruel, A.; Cappuccio, G.; Brunetti-Pierri, N.; Willems, M.; Isidor, B.; et al. Expanding the phenotype of HNRNPU-related neurodevelopmental disorder with emphasis on seizure phenotype and review of literature. Am. J. Med. Genet. Part A 2022, 188, 1497–1514. [Google Scholar] [CrossRef]
- Hecher, L.; Harms, F.L.; Lisfeld, J.; Alawi, M.; Denecke, J.; Kutsche, K. INPP4A-related genetic and phenotypic spectrum and functional relevance of subcellular targeting of INPP4A isoforms. Neurogenetics 2023, 24, 79–93. [Google Scholar] [CrossRef]
- Langhammer, F.; Maroofian, R.; Badar, R.; Gregor, A.; Rochman, M.; Ratliff, J.B.; Koopmans, M.; Herget, T.; Hempel, M.; Kortüm, F.; et al. Genotype-phenotype correlations in RHOBTB2-associated neurodevelopmental disorders. Anesth. Analg. 2023, 25, 100885. [Google Scholar] [CrossRef]
- Liu, J.; Feldman, R.; Zhang, Z.; Deardorff, M.A.; Haverfield, E.V.; Kaur, M.; Li, J.R.; Clark, D.; Kline, A.D.; Waggoner, D.J.; et al. SMC1A expression and mechanism of pathogenicity in probands with X-Linked Cornelia de Lange syndrome. Hum. Mutat. 2009, 30, 1535–1542. [Google Scholar] [CrossRef]
- Tessarech, M.; Friocourt, G.; Marguet, F.; Lecointre, M.; Le Mao, M.; Díaz, R.M.; Mignot, C.; Keren, B.; Héron, B.; De Bie, C.; et al. De novo variants in SP9 cause a novel form of interneuronopathy characterized by intellectual disability, autism spectrum disorder, and epilepsy with variable expressivity. Anesthesia Analg. 2024, 26, 101087. [Google Scholar] [CrossRef]
- Van de Vondel, L.; De Winter, J.; Beijer, D.; Coarelli, G.; Wayand, M.; Palvadeau, R.; Pauly, M.G.; Klein, K.; Rautenberg, M.; Guillot-Noël, L.; et al. De Novo and Dominantly Inherited SPTAN1 Mutations Cause Spastic Paraplegia and Cerebellar Ataxia. Mov. Disord. 2022, 37, 1175–1186. [Google Scholar] [CrossRef]
- Hebebrand, M.; Hüffmeier, U.; Trollmann, R.; Hehr, U.; Uebe, S.; Ekici, A.B.; Kraus, C.; Krumbiegel, M.; Reis, A.; Thiel, C.T.; et al. The mutational and phenotypic spectrum of TUBA1A-associated tubulinopathy. Orphanet J. Rare Dis. 2019, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Zara, F. ARHGEF9 mutations cause a specific recognizable X-linked intellectual disability syndrome. Neurol. Genet. 2017, 3, e159. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, B.K.; Veenma, D.C.M.; Chang, K.; Schulman, H.; Van Woerden, G.M. Case Report: Developmental Delay and Acute Neuropsychiatric Episodes Associated With a de novo Mutation in the CAMK2B Gene (c.328G>A p.Glu110Lys). Front. Pharmacol. 2022, 13, 794008. [Google Scholar] [CrossRef] [PubMed]
- Wonkam-Tingang, E.; Schrauwen, I.; Esoh, K.K.; Bharadwaj, T.; Nouel-Saied, L.M.; Acharya, A.; Nasir, A.; Leal, S.M.; Wonkam, A. A novel variant in DMXL2 gene is associated with autosomal dominant non-syndromic hearing impairment (DFNA71) in a Cameroonian family. Exp. Biol. Med. 2021, 246, 1524–1532. [Google Scholar] [CrossRef]
- Strehlow, V.; O Heyne, H.; Vlaskamp, D.R.M.; Marwick, K.F.M.; Rudolf, G.; de Bellescize, J.; Biskup, S.; Brilstra, E.H.; Brouwer, O.F.; Callenbach, P.M.C.; et al. GRIN2A-related disorders: Genotype and functional consequence predict phenotype. Brain 2018, 142, 80–92. [Google Scholar] [CrossRef]
- Conroy, J.; Allen, N.M.; Gorman, K.; Shahwan, A.; Ennis, S.; Lynch, S.A.; King, M.D.; King, M.D. NAPB–a novel SNARE-associated protein for early-onset epileptic encephalopathy. Clin. Genet. 2015, 89, E1–E3. [Google Scholar] [CrossRef]
- Schubert, J.; Siekierska, A.; Langlois, M.; May, P.; Huneau, C.; Becker, F.; Muhle, H.; Suls, A.; Lemke, J.R.; de Kovel, C.G.F.; et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet. 2014, 46, 1327–1332. [Google Scholar] [CrossRef]
- Vlaskamp, D.R.; Shaw, B.J.; Burgess, R.; Mei, D.; Montomoli, M.; Xie, H.; Myers, C.T.; Bennett, M.F.; XiangWei, W.; Williams, D.; et al. SYNGAP1 encephalopathy. Neurology 2019, 92, e96–e107. [Google Scholar] [CrossRef]
- Balestrini, S.; Milh, M.; Castiglioni, C.; Lüthy, K.; Finelli, M.J.; Verstreken, P.; Cardon, A.; Stražišar, B.G.; Holder, J.L.; Lesca, G.; et al. TBC1D24 genotype–phenotype correlation: Epilepsies and other neurologic features. Neurology 2016, 87, 77–85. [Google Scholar] [CrossRef]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N.; et al. AGC1 Deficiency Causes Infantile Epilepsy, Abnormal Myelination, and Reduced N-Acetylaspartate. In JIMD Reports; SSIEM and Springer: Berlin/Heidelberg, Germany, 2014; Volume 14. [Google Scholar]
- Alsharhan, H.; He, M.; Edmondson, A.C.; Daniel, E.J.P.; Chen, J.; Donald, T.; Bakhtiari, S.; Amor, D.J.; Jones, E.A.; Vassallo, G.; et al. ALG13 X-linked intellectual disability: New variants, glycosylation analysis, and expanded phenotypes. J. Inherit. Metab. Dis. 2021, 44, 1001–1012. [Google Scholar] [CrossRef]
- Okur, V.; Cho, M.T.; van Wijk, R.; van Oirschot, B.; Picker, J.; Coury, S.A.; Grange, D.; Manwaring, L.; Krantz, I.; Muraresku, C.C.; et al. De novo variants in HK1 associated with neurodevelopmental abnormalities and visual impairment. Eur. J. Hum. Genet. 2019, 27, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucińska-Więckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosińska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, J.; Guo, C.; Duan, Y.; Liu, C.; Tan, Y.; Pan, Y. Clinical report and genetic analysis of a Chinese patient with developmental and epileptic encephalopathy associated with novel biallelic variants in the ST3GAL3 gene. Mol. Genet. Genom. Med. 2023, 12, e2322. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.T.; Wilkinson, K.D.; Reines, D.; Warren, S.T. FMR1 Protein: Conserved RNP Family Domains and Selective RNA Binding. Science 1993, 262, 563–566. [Google Scholar] [CrossRef]
- Zweier, M.; Gregor, A.; Zweier, C.; Engels, H.; Sticht, H.; Wohlleber, E.; Bijlsma, E.K.; Holder, S.E.; Zenker, M.; Rossier, E.; et al. Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum. Mutat. 2010, 31, 722–733. [Google Scholar] [CrossRef]
- Carvill, G.L.; Heavin, S.B.; Yendle, S.C.; McMahon, J.M.; O’Roak, B.J.; Cook, J.; Khan, A.; Dorschner, M.O.; Weaver, M.; Calvert, S.; et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP. Nat. Genet. 2013, 45, 825–830. [Google Scholar] [CrossRef]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 Mutations Define a Subtype of Autism Early in Development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef]
- Cassina, M.; Cappellari, A.; Toldo, I.; Nosadini, M.; Rigon, C.; Suppiej, A.; Sartori, S.; Bertossi, C. Forkhead Box G1 Gene Haploinsufficiency: An Emerging Cause of Dyskinetic Encephalopathy of Infancy. Neuropediatrics 2015, 46, 056–064. [Google Scholar] [CrossRef]
- Sapir, T.; Kshirsagar, A.; Gorelik, A.; Olender, T.; Porat, Z.; Scheffer, I.E.; Goldstein, D.B.; Devinsky, O.; Reiner, O. Heterogeneous nuclear ribonucleoprotein U (HNRNPU) safeguards the developing mouse cortex. Nat. Commun. 2022, 13, 4209. [Google Scholar] [CrossRef]
- Dugger, S.A.; Dhindsa, R.S.; Sampaio, G.D.A.; Ressler, A.K.; Rafikian, E.E.; Petri, S.; Letts, V.A.; Teoh, J.; Ye, J.; Colombo, S.; et al. Neurodevelopmental deficits and cell-type-specific transcriptomic perturbations in a mouse model of HNRNPU haploinsufficiency. PLoS Genet. 2023, 19, e1010952. [Google Scholar] [CrossRef]
- Wu, J.I.; Lessard, J.; Olave, I.A.; Qiu, Z.; Ghosh, A.; Graef, I.A.; Crabtree, G.R. Regulation of Dendritic Development by Neuron-Specific Chromatin Remodeling Complexes. Neuron 2007, 56, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Staahl, B.T.; Tang, J.; Wu, W.; Sun, A.; Gitler, A.D.; Yoo, A.S.; Crabtree, G.R. Kinetic Analysis of npBAF to nBAF Switching Reveals Exchange of SS18 with CREST and Integration with Neural Developmental Pathways. J. Neurosci. 2013, 33, 10348–10361. [Google Scholar] [CrossRef] [PubMed]
- Vogel-Ciernia, A.; Matheos, D.P.; Barrett, R.M.; A Kramár, E.; Azzawi, S.; Chen, Y.; Magnan, C.N.; Zeller, M.; Sylvain, A.; Haettig, J.; et al. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat. Neurosci. 2013, 16, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Vogel-Ciernia, A.; Wood, M.A. Neuron-specific chromatin remodeling: A missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology 2013, 80, 18–27. [Google Scholar] [CrossRef]
- Bell, S.; Rousseau, J.; Peng, H.; Aouabed, Z.; Priam, P.; Theroux, J.-F.; Jefri, M.; Tanti, A.; Wu, H.; Kolobova, I.; et al. Mutations in ACTL6B Cause Neurodevelopmental Deficits and Epilepsy and Lead to Loss of Dendrites in Human Neurons. Am. J. Hum. Genet. 2019, 104, 815–834. [Google Scholar] [CrossRef]
- Karaca, E.; Harel, T.; Pehlivan, D.; Jhangiani, S.N.; Gambin, T.; Akdemir, Z.C.; Gonzaga-Jauregui, C.; Erdin, S.; Bayram, Y.; Campbell, I.M.; et al. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron 2015, 88, 499–513. [Google Scholar] [CrossRef]
- Yüksel, Z.; Yazol, M.; Gümüş, E. Pathogenic homozygous variations in ACTL6B cause DECAM syndrome: Developmental delay, Epileptic encephalopathy, Cerebral Atrophy, and abnormal Myelination. Am. J. Med. Genet. Part A 2019, 179, 1603–1608. [Google Scholar] [CrossRef]
- Wenderski, W.; Wang, L.; Krokhotin, A.; Walsh, J.J.; Li, H.; Shoji, H.; Ghosh, S.; George, R.D.; Miller, E.L.; Elias, L.; et al. Loss of the neural-specific BAF subunit ACTL6B relieves repression of early response genes and causes recessive autism. Proc. Natl. Acad. Sci. USA 2020, 117, 10055–10066. [Google Scholar] [CrossRef]
- Ahn, L.Y.; Coatti, G.C.; Liu, J.; Gumus, E.; Schaffer, A.E.; Miranda, H.C. An epilepsy-associated ACTL6B variant captures neuronal hyperexcitability in a human induced pluripotent stem cell model. J. Neurosci. Res. 2021, 99, 110–123. [Google Scholar] [CrossRef]
- Olave, I.; Wang, W.; Xue, Y.; Kuo, A.; Crabtree, G.R. Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev. 2002, 16, 2509–2517. [Google Scholar] [CrossRef]
- Lessard, J.; Wu, J.I.; Ranish, J.A.; Wan, M.; Winslow, M.M.; Staahl, B.T.; Wu, H.; Aebersold, R.; Graef, I.A.; Crabtree, G.R. An Essential Switch in Subunit Composition of a Chromatin Remodeling Complex during Neural Development. Neuron 2007, 55, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.C.; Popp, D.; Gebhard, W.; Kabsch, W. Atomic model of the actin filament. Nature 1990, 347, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Fichera, M.; Failla, P.; Saccuzzo, L.; Miceli, M.; Salvo, E.; Castiglia, L.; Galesi, O.; Grillo, L.; Calì, F.; Greco, D.; et al. Mutations in ACTL6B, coding for a subunit of the neuron-specific chromatin remodeling complex nBAF, cause early onset severe developmental and epileptic encephalopathy with brain hypomyelination and cerebellar atrophy. Hum. Genet. 2019, 138, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Maddirevula, S.; Alzahrani, F.; Al-Owain, M.; Al Muhaizea, M.A.; Kayyali, H.R.; AlHashem, A.; Rahbeeni, Z.; Al-Otaibi, M.; Alzaidan, H.I.; Balobaid, A.; et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 2018, 21, 736–742. [Google Scholar] [CrossRef]
- Al-Ubaidi, M.R.; Hollyfield, J.G.; A Overbeek, P.; Baehr, W. Photoreceptor degeneration induced by the expression of simian virus 40 large tumor antigen in the retina of transgenic mice. Proc. Natl. Acad. Sci. USA 1992, 89, 1194–1198. [Google Scholar] [CrossRef]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of Neuronal Survival by the Serine-Threonine Protein Kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef]
- Feddersen, R.M.; Yunis, W.S.; O’Donnell, M.A.; Ebner, T.J.; Shenb, L.; Iadecolac, C.; Orr, H.T.; Clarka, H.B. Susceptibility to Cell Death Induced by Mutant SV40 T-Antigen Correlates with Purkinje Neuron Functional Development. Mol. Cell Neurosci. 1997, 9, 42–62. [Google Scholar] [CrossRef]
- Nystuen, A.; Legare, M.E.; Shultz, L.D.; Frankel, W.N. A Null Mutation in Inositol Polyphosphate 4-Phosphatase Type I Causes Selective Neuronal Loss in Weeble Mutant Mice. Neuron 2001, 32, 203–212. [Google Scholar] [CrossRef]
- Sasaki, J.; Kofuji, S.; Itoh, R.; Momiyama, T.; Takayama, K.; Murakami, H.; Chida, S.; Tsuya, Y.; Takasuga, S.; Eguchi, S.; et al. The PtdIns(3,4)P2 phosphatase INPP4A is a suppressor of excitotoxic neuronal death. Nature 2010, 465, 497–501. [Google Scholar] [CrossRef]
- Hiebert, S.W.; Lutterbach, B.; Amann, J. Role of co-repressors in transcriptional repression mediated by the t(8;21), t(16;21), t(12;21), and inv(16) fusion proteins. Curr. Opin. Hematol. 2001, 8, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Batra, J.; Mabalirajan, U.; Sharma, S.; Nagarkatti, R.; Aich, J.; Sharma, S.K.; Niphadkar, P.V.; Ghosh, B. A Genetic Variation in Inositol Polyphosphate 4 Phosphatase A Enhances Susceptibility to Asthma. Am. J. Respir. Crit. Care Med. 2008, 177, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Duan, C.; Yang, Q. Inositol Phosphatase INPP4A Inhibits the Apoptosis of in Vitro Neu-rons with Characteristic of Intractable Epilepsy by Reducing Intracellular Ca 2+ Concentration. Int. J. Clin. Exp. Pathol. 2018, 11, 1999. [Google Scholar] [PubMed]
- Verrotti, A.; Agostinelli, S.; Prezioso, G.; Coppola, G.; Capovilla, G.; Romeo, A.; Striano, P.; Parisi, P.; Grosso, S.; Spalice, A.; et al. Epilepsy in patients with Cornelia de Lange syndrome: A clinical series. Seizre 2013, 22, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.-M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. Part A 2017, 173, 2108–2125. [Google Scholar] [CrossRef]
- Musio, A. The Multiple Facets of the SMC1A Gene. Gene 2020, 743, 144612. [Google Scholar] [CrossRef]
- Elwan, M.; Fowkes, R.; Lewis-Smith, D.; Winder, A.; Baker, M.R.; Thomas, R.H. Late-onset cluster seizures and intellectual disability associated with a novel truncation variant in SMC1A. Epilepsy Behav. Rep. 2022, 19, 100556. [Google Scholar] [CrossRef]
- Bozarth, X.L.; Lopez, J.; Fang, H.; Lee-Eng, J.; Duan, Z.; Deng, X. Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy. Genes 2023, 14, 852. [Google Scholar] [CrossRef]
- Parmeggiani, L.; Stanzial, F.; Menna, E.; Boni, E.; Manzoni, F.; Benedicenti, F.; Pellegrin, S. Early onset developmental and epileptic encephalopathy and Rett-like phenotype in a 15-year-old girl affected by Cornelia de Lange syndrome type 2 due to a SMC1A gene mutation. Epilepsy Behav. Rep. 2023, 24, 100634. [Google Scholar] [CrossRef]
- Carico, Z.M.; Stefan, H.C.; Justice, M.; Yimit, A.; Dowen, J.M. A cohesin cancer mutation reveals a role for the hinge domain in genome organization and gene expression. PLoS Genet. 2021, 17, e1009435. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, M.H.; Wu, X.; Kodani, A.; Fan, J.; Doan, R.; Ozawa, M.; Ma, J.; Yoshida, N.; Reiter, J.F.; et al. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 2016, 166, 1147–1162.e15. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; Van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Mastropasqua, F.; Oksanen, M.; Soldini, C.; Alatar, S.; Arora, A.; Ballarino, R.; Molinari, M.; Agostini, F.; Poulet, A.; Watts, M.; et al. Deficiency of the Heterogeneous Nuclear Ribonucleoprotein U locus leads to delayed hindbrain neurogenesis. Biol. Open 2023, 12, bio060113. [Google Scholar] [CrossRef]
- Pacheco, A.; Lopez de Quinto, S.; Ramajo, J.; Fernández, N.; Martínez-Salas, E. A novel role for Gemin5 in mRNA translation. Nucleic Acids Res. 2009, 37, 582–590. [Google Scholar] [CrossRef]
- Yong, J.; Kasim, M.; Bachorik, J.L.; Wan, L.; Dreyfuss, G. Gemin5 Delivers snRNA Precursors to the SMN Complex for snRNP Biogenesis. Mol. Cell 2010, 38, 551–562. [Google Scholar] [CrossRef]
- Workman, E.; Kalda, C.; Patel, A.; Battle, D.J. Gemin5 Binds to the Survival Motor Neuron mRNA to Regulate SMN Expression. J. Biol. Chem. 2015, 290, 15662–15669. [Google Scholar] [CrossRef]
- Jin, W.; Wang, Y.; Liu, C.-P.; Yang, N.; Jin, M.; Cong, Y.; Wang, M.; Xu, R.-M. Structural basis for snRNA recognition by the double-WD40 repeat domain of Gemin. Genes Dev. 2016, 30, 2391–2403. [Google Scholar] [CrossRef]
- Xu, C.; Ishikawa, H.; Izumikawa, K.; Li, L.; He, H.; Nobe, Y.; Yamauchi, Y.; Shahjee, H.M.; Wu, X.-H.; Yu, Y.-T.; et al. Structural insights into Gemin5-guided selection of pre-snRNAs for snRNP assembly. Genes Dev. 2016, 30, 2376–2390. [Google Scholar] [CrossRef]
- Francisco-Velilla, R.; Fernandez-Chamorro, J.; Dotu, I.; Martinez-Salas, E. The landscape of the non-canonical RNA-binding site of Gemin5 unveils a feedback loop counteracting the negative effect on translation. Nucleic Acids Res. 2018, 46, 7339–7353. [Google Scholar] [CrossRef]
- Moreno-Morcillo, M.; Francisco-Velilla, R.; Embarc-Buh, A.; Fernández-Chamorro, J.; Ramón-Maiques, S.; Martinez-Salas, E. Structural basis for the dimerization of Gemin5 and its role in protein recruitment and translation control. Nucleic Acids Res. 2019, 48, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zhao, S.; Francisco-Velilla, R.; Zhang, J.; Embarc-Buh, A.; Abellan, S.; Lv, M.; Tang, P.; Gong, Q.; Shen, H.; et al. Structural basis for Gemin5 decamer-mediated mRNA binding. Nat. Commun. 2022, 13, 5166. [Google Scholar] [CrossRef] [PubMed]
- Francisco-Velilla, R.; Abellan, S.; Oliveros, J.C.; Martinez-Salas, E. Alternative Splicing Events Driven by Altered Levels of GEMIN5 Undergo Translation. RNA Biol. 2024, 21, 23–24. [Google Scholar] [CrossRef]
- Ibrahim, N.; Naz, S.; Mattioli, F.; Guex, N.; Sharif, S.; Iqbal, A.; Ansar, M.; Reymond, A. A Biallelic Truncating Variant in the TPR Domain of GEMIN5 Associated with Intellectual Disability and Cerebral Atrophy. Genes 2023, 14, 707. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Zhu, G.; Wan, L.; Liang, Y.; Huang, M.; Yang, G. Expand-ing the Clinical Phenotype and Genetic Spectrum of GEMIN5 Disorders: Early-Infantile Developmental and Epileptic Encephalopathies. Brain Behav. 2024, 14, e3535. [Google Scholar] [CrossRef]
- Gubitz, A.K.; Mourelatos, Z.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Gemin5, a Novel WD Repeat Protein Component of the SMN Complex That Binds Sm Proteins. J. Biol. Chem. 2002, 277, 5631–5636. [Google Scholar] [CrossRef]
- Fernandez-Chamorro, J.; Piñeiro, D.; Gordon, J.M.B.; Ramajo, J.; Francisco-Velilla, R.; Macias, M.J.; Martinez-Salas, E. Identification of novel non-canonical RNA-binding sites in Gemin5 involved in internal initiation of translation. Nucleic Acids Res. 2014, 42, 5742–5754. [Google Scholar] [CrossRef]
- Kour, S.; Rajan, D.S.; Fortuna, T.R.; Anderson, E.N.; Ward, C.; Lee, Y.; Lee, S.; Shin, Y.B.; Chae, J.-H.; Choi, M.; et al. Loss of function mutations in GEMIN5 cause a neurodevelopmental disorder. Nat. Commun. 2021, 12, 2558. [Google Scholar] [CrossRef]
- Gates, J.; Lam, G.; Ortiz, J.A.; Losson, R.; Thummel, C.S. Rigor Mortis Encodes a Novel Nuclear Receptor In-teracting Protein Required for Ecdysone Signaling during Drosophila Larval Development. Development 2004, 131, 25–36. [Google Scholar] [CrossRef]
- Rajan, D.S.; Kour, S.; Fortuna, T.R.; Cousin, M.A.; Barnett, S.S.; Niu, Z.; Babovic-Vuksanovic, D.; Klee, E.W.; Kirmse, B.; Innes, M.; et al. Autosomal Recessive Cerebellar Atrophy and Spastic Ataxia in Patients With Pathogenic Biallelic Variants in GEMIN. Front. Cell Dev. Biol. 2022, 10, 783762. [Google Scholar] [CrossRef]
- Kamma, H.; Portman, D.S.; Dreyfuss, G. Cell Type-Specific Expression of hnRNP Proteins. Exp. Cell Res. 1995, 221, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Kukalev, A.; Nord, Y.; Palmberg, C.; Bergman, T.; Percipalle, P. Actin and hnRNP U cooperate for productive transcription by RNA polymerase II. Nat. Struct. Mol. Biol. 2005, 12, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Obrdlik, A.; Kukalev, A.; Louvet, E.; Farrants, A.-K.; Caputo, L.; Percipalle, P. The Histone Acetyltransferase PCAF Associates with Actin and hnRNP U for RNA Polymerase II Transcription. Mol. Cell Biol. 2008, 28, 6342–6357. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Miyaji, M.; Ichiyasu, S.; Tsutsui, K.M.; Tsutsui, K. Regulation of DNA Topoisomerase IIβ through RNA-dependent Association with Heterogeneous Nuclear Ribonucleoprotein U (hnRNP U). J. Biol. Chem. 2010, 285, 26451–26460. [Google Scholar] [CrossRef]
- Huelga, S.C.; Vu, A.Q.; Arnold, J.D.; Liang, T.Y.; Liu, P.P.; Yan, B.Y.; Donohue, J.P.; Shiue, L.; Hoon, S.; Brenner, S.; et al. Integrative Genome-wide Analysis Reveals Cooperative Regulation of Alternative Splicing by hnRNP Proteins. Cell Rep. 2012, 1, 167–178. [Google Scholar] [CrossRef]
- Ye, J.; Beetz, N.; O’keeffe, S.; Tapia, J.C.; Macpherson, L.; Chen, W.V.; Bassel-Duby, R.; Olson, E.N.; Maniatis, T. hnRNP U protein is required for normal pre-mRNA splicing and postnatal heart development and function. Proc. Natl. Acad. Sci. USA 2015, 112, E3020–E3029. [Google Scholar] [CrossRef]
- Fan, H.; Lv, P.; Huo, X.; Wu, J.; Wang, Q.; Cheng, L.; Liu, Y.; Tang, Q.-Q.; Zhang, L.; Zhang, F.; et al. The nuclear matrix protein HNRNPU maintains 3D genome architecture globally in mouse hepatocytes. Genome Res. 2017, 28, 192–202. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Depienne, C.; DDD Study; Nava, C.; Keren, B.; Heide, S.; Rastetter, A.; Passemard, S.; Chantot-Bastaraud, S.; Moutard, M.-L.; Agrawal, P.B.; et al. Genetic and phenotypic dissection of 1q43q44 microdeletion syndrome and neurodevelopmental phenotypes associated with mutations in ZBTB18 and HNRNPU. Hum. Genet. 2017, 136, 463–479. [Google Scholar] [CrossRef]
- I, D.V.; Aysina, V.A. Early infantile epileptic encephalopathy type 54: Clinical and neurophysiological aspects. Epilepsy Paroxysmal Cond. 2021, 13, 132–139. [Google Scholar] [CrossRef]
- Bramswig, N.C.; Lüdecke, H.-J.; Hamdan, F.F.; Altmüller, J.; Beleggia, F.; Elcioglu, N.H.; Freyer, C.; Gerkes, E.H.; Demirkol, Y.K.; Knupp, K.G.; et al. Heterozygous HNRNPU variants cause early onset epilepsy and severe intellectual disability. Hum. Genet. 2017, 136, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Yates, T.M.; Vasudevan, P.C.; Chandler, K.E.; E Donnelly, D.; Stark, Z.; Sadedin, S.; Willoughby, J.; Genomics, B.C.F.M.; DDD Study; Balasubramanian, M. De novo mutations in HNRNPU result in a neurodevelopmental syndrome. Am. J. Med. Genet. Part A 2017, 173, 3003–3012. [Google Scholar] [CrossRef] [PubMed]
- Leduc, M.S.; Chao, H.; Qu, C.; Walkiewicz, M.; Xiao, R.; Magoulas, P.; Pan, S.; Beuten, J.; He, W.; Bernstein, J.A.; et al. Clinical and molecular characterization of de novo loss of function variants in HNRNPU. Am. J. Med. Genet. Part A 2017, 173, 2680–2689. [Google Scholar] [CrossRef] [PubMed]
- Roshon, M.J.; Ruley, H.E. Hypomorphic mutation in hnRNP U results in post-implantation lethality. Transgenic Res. 2005, 14, 179–192. [Google Scholar] [CrossRef]
- Lalli, M.A.; Avey, D.; Dougherty, J.D.; Milbrandt, J.; Mitra, R.D. High-throughput single-cell functional elucidation of neurodevelopmental disease–associated genes reveals convergent mechanisms altering neuronal differentiation. Genome Res. 2020, 30, 1317–1331. [Google Scholar] [CrossRef]
- Pilaz, L.-J.; McMahon, J.J.; Miller, E.E.; Lennox, A.L.; Suzuki, A.; Salmon, E.; Silver, D.L. Prolonged Mitosis of Neural Progenitors Alters Cell Fate in the Developing Brain. Neuron 2016, 89, 83–99. [Google Scholar] [CrossRef]
- Zhang, F.; Li, F.; Chen, F.; Huang, J.; Luo, Q.; Du, X.; Zhou, J.; Gu, W.; Xu, K. Novel Variant Expands the Clinical Spectrum of CUX2-Associated Developmental and Epileptic Encephalopathies. Front. Genet. 2022, 13, 808181. [Google Scholar] [CrossRef]
- Cubelos, B.; Sebastián-Serrano, A.; Kim, S.; Moreno-Ortiz, C.; Redondo, J.M.; Walsh, C.A.; Nieto, M. Cux-2 Controls the Proliferation of Neuronal Intermediate Precursors of the Cortical Subventricular Zone. Cereb. Cortex 2007, 18, 1758–1770. [Google Scholar] [CrossRef]
- Cubelos, B.; Sebastián-Serrano, A.; Beccari, L.; Calcagnotto, M.E.; Cisneros, E.; Kim, S.; Dopazo, A.; Alvarez-Dolado, M.; Redondo, J.M.; Bovolenta, P.; et al. Cux1 and Cux2 Regulate Dendritic Branching, Spine Morphology, and Synapses of the Upper Layer Neurons of the Cortex. Neuron 2010, 66, 523–535. [Google Scholar] [CrossRef]
- Magno, L.; Asgarian, Z.; Pendolino, V.; Velona, T.; Mackintosh, A.; Lee, F.; Stryjewska, A.; Zimmer, C.; Guillemot, F.; Farrant, M.; et al. Transient developmental imbalance of cortical interneuron subtypes presages long-term changes in behavior. Cell Rep. 2021, 35, 109249. [Google Scholar] [CrossRef]
- Nieto, M.; Monuki, E.S.; Tang, H.; Imitola, J.; Haubst, N.; Khoury, S.J.; Cunningham, J.; Gotz, M.; Walsh, C.A. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II–IV of the cerebral cortex. J. Comp. Neurol. 2004, 479, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, C.; Tiveron, M.-C.; Bodmer, R.; Cremer, H. Dynamics of Cux2 Expression Suggests that an Early Pool of SVZ Precursors is Fated to Become Upper Cortical Layer Neurons. Cereb. Cortex 2004, 14, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Iulianella, A.; Sharma, M.; Durnin, M.; Heuvel, G.B.V.; Trainor, P.A. Cux2(Cutl2) integrates neural progenitor development with cell-cycle progression during spinal cord neurogenesis. Development 2008, 135, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kodera, H.; Akita, T.; Shiina, M.; Kato, M.; Hoshino, H.; Terashima, H.; Osaka, H.; Nakamura, S.; Tohyama, J.; et al. De Novo Mutations in GNAO1, Encoding a Gαo Subunit of Heterotrimeric G Proteins, Cause Epileptic Encephalopathy. Am. J. Hum. Genet. 2013, 93, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Fukai, R.; Ben-Zeev, B.; Sakai, Y.; Mimaki, M.; Okamoto, N.; Suzuki, Y.; Monden, Y.; Saito, H.; Tziperman, B.; et al. Phenotypic spectrum of GNAO1 variants: Epileptic encephalopathy to involuntary movements with severe developmental delay. Eur. J. Hum. Genet. 2015, 24, 129–134. [Google Scholar] [CrossRef]
- Schirinzi, T.; Garone, G.; Travaglini, L.; Vasco, G.; Galosi, S.; Rios, L.; Castiglioni, C.; Barassi, C.; Battaglia, D.; Gambardella, M.L.; et al. Phenomenology and clinical course of movement disorder in GNAO1 variants: Results from an analytical review. Park. Relat. Disord. 2019, 61, 19–25. [Google Scholar] [CrossRef]
- Morrison-Levy, N.; Borlot, F.; Jain, P.; Whitney, R. Early-Onset Developmental and Epileptic Encephalopathies of Infancy: An Overview of the Genetic Basis and Clinical Features. Pediatric Neurol. 2021, 116, 85–94. [Google Scholar] [CrossRef]
- Solis, G.P.; Kozhanova, T.V.; Koval, A.; Zhilina, S.S.; Mescheryakova, T.I.; Abramov, A.A.; Ishmuratov, E.V.; Bolshakova, E.S.; Osipova, K.V.; Ayvazyan, S.O.; et al. Pediatric Encephalopathy: Clinical, Biochemical and Cellular Insights into the Role of Gln52 of GNAO1 and GNAI1 for the Dominant Disease. Cells 2021, 10, 2749. [Google Scholar] [CrossRef]
- Pérez-Dueñas, B.; Gorman, K.; Marcé-Grau, A.; Ortigoza-Escobar, J.D.; Macaya, A.; Danti, F.R.; Barwick, K.; Papandreou, A.; Ng, J.; Meyer, E.; et al. The Genetic Landscape of Complex Childhood-Onset Hyperkinetic Movement Disorders. Mov. Disord. 2022, 37, 2197–2209. [Google Scholar] [CrossRef]
- Thiel, M.; Bamborschke, D.; Janzarik, W.G.; Assmann, B.; Zittel, S.; Patzer, S.; Auhuber, A.; Opp, J.; Matzker, E.; Bevot, A.; et al. Genotype–phenotype correlation and treatment effects in young patients with GNAO1-associated disorders. J. Neurol. Neurosurg. Psychiatry 2023, 94, 806–815. [Google Scholar] [CrossRef]
- Taira, R.; Akamine, S.; Okuzono, S.; Fujii, F.; Hatai, E.; Yonemoto, K.; Takemoto, R.; Kato, H.; Masuda, K.; Kato, T.A.; et al. Gnao1 Is a Molecular Switch That Regulates the Rho Signaling Pathway in Differentiating Neurons. Sci. Rep. 2024, 14, 17097. [Google Scholar] [CrossRef] [PubMed]
- Ghil, S.; Kim, B.; Lee, Y.; Suh-Kim, H. Neurite Outgrowth Induced by Cyclic AMP Can Be Modulated by the α Subunit of Go. J. Neurochem. 2000, 74, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Nakata, H.; Kozasa, T. Functional Characterization of Gαo Signaling through G Protein-Regulated Inducer of Neurite Outgrowth. Mol. Pharmacol. 2004, 67, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Hwangpo, T.A.; Jordan, J.D.; Premsrirut, P.K.; Jayamaran, G.; Licht, J.D.; Iyengar, R.; Neves, S.R. G Protein-regulated Inducer of Neurite Outgrowth (GRIN) Modulates Sprouty Protein Repression of Mitogen-activated Protein Kinase (MAPK) Activation by Growth Factor Stimulation. J. Biol. Chem. 2012, 287, 13674–13685. [Google Scholar] [CrossRef]
- Wettschureck, N.; Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef]
- Huang, Y.; Thathiah, A. Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 2015, 589, 1607–1619. [Google Scholar] [CrossRef]
- Larasati, Y.A.; Savitsky, M.; Koval, A.; Solis, G.P.; Valnohova, J.; Katanaev, V.L. Restoration of the GTPase activity and cellular interactions of Gαo mutants by Zn2+ in GNAO1 encephalopathy models. Sci. Adv. 2022, 8, eabn9350. [Google Scholar] [CrossRef]
- Akamine, S.; Okuzono, S.; Yamamoto, H.; Setoyama, D.; Sagata, N.; Ohgidani, M.; Kato, T.A.; Ishitani, T.; Kato, H.; Masuda, K.; et al. GNAO1 organizes the cytoskeletal remodeling and firing of developing neurons. FASEB J. 2020, 34, 16601–16621. [Google Scholar] [CrossRef]
- Domínguez-Carral, J.; Ludlam, W.G.; Segarra, M.J.; Marti, M.F.; Balsells, S.; Muchart, J.; Petrović, D.; Espinoza, I.; Ortigoza-Escobar, J.D.; Martemyanov, K.A.; et al. Severity of GNAO1-Related Disorder Correlates with Changes in G-Protein Function. Ann. Neurol. 2023, 94, 987–1004. [Google Scholar] [CrossRef]
- Silachev, D.; Koval, A.; Savitsky, M.; Padmasola, G.; Quairiaux, C.; Thorel, F.; Katanaev, V.L. Mouse models characterize GNAO1 encephalopathy as a neurodevelopmental disorder leading to motor anomalies: From a severe G203R to a milder C215Y mutation. Acta Neuropathol. Commun. 2022, 10, 9. [Google Scholar] [CrossRef]
- Benedetti, M.C.; D’Andrea, T.; Colantoni, A.; Silachev, D.; de Turris, V.; Boussadia, Z.; Babenko, V.A.; Volovikov, E.A.; Belikova, L.; Bogomazova, A.N.; et al. Cortical neurons obtained from patient-derived iPSCs with GNAO1 p.G203R variant show altered differentiation and functional properties. Heliyon 2024, 10, e26656. [Google Scholar] [CrossRef] [PubMed]
- Suske, G.; Bruford, E.; Philipsen, S. Mammalian SP/KLF transcription factors: Bring in the family. Genomics 2005, 85, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Friocourt, G.; Parnavelas, J.G. Mutations in ARX result in several defects involving GABAergic neurons. Front. Cell Neurosci. 2010, 4, 1437. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Z.; Lindtner, S.; Li, Z.; Xu, Z.; Wei, S.; Liang, Q.; Wen, Y.; Tao, G.; You, Y.; et al. Sp9 Regulates Medial Ganglionic Eminence-Derived Cortical Interneuron Development. Cereb. Cortex 2018, 29, 2653–2667. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Dobyns, W.B. Malformations of cortical development: Clinical features and genetic causes. Lancet Neurol. 2014, 13, 710–726. [Google Scholar] [CrossRef]
- Lasser, M.; Tiber, J.; Lowery, L.A. The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disor-ders. Front. Cell Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the De-veloping Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef]
- Romaniello, R.; Arrigoni, F.; Fry, A.E.; Bassi, M.T.; Rees, M.I.; Borgatti, R.; Pilz, D.T.; Cushion, T.D. Tubulin genes and malformations of cortical development. Eur. J. Med. Genet. 2018, 61, 744–754. [Google Scholar] [CrossRef]
- Hoff, K.J.; E Aiken, J.; A Gutierrez, M.; Franco, S.J.; Moore, J.K.; Biology, D.; States, U. TUBA1A tubulinopathy mutants disrupt neuron morphogenesis and override XMAP215/Stu2 regulation of microtubule dynamics. eLife 2022, 11, e76189. [Google Scholar] [CrossRef]
- Gloster, A.; El-Bizri, H.; Bamji, S.X.; Rogers, D.; Miller, F.D. Early Induction of Tα1 α-Tubulin Transcription in Neurons of the Developing Nervous System. J. Comp. Neurol. 1999, 405, 45–60. [Google Scholar] [CrossRef]
- Schröter, J.; Döring, J.H.; Garbade, S.F.; Hoffmann, G.F.; Kölker, S.; Ries, M.; Syrbe, S. Cross-sectional quantitative analysis of the natural history of TUBA1A and TUBB2B tubulinopathies. Anesth. Analg. 2021, 23, 516–523. [Google Scholar] [CrossRef]
- Aiken, J.; Buscaglia, G.; Bates, E.A.; Moore, J.K. The α-Tubulin gene TUBA1A in Brain Development: A Key Ingredient in the Neuronal Isotype Blend. J. Dev. Biol. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Poirier, K.; Boddaert, N.; Saillour, Y.; Castelnau, L.; Philip, N.; Buyse, G.; Villard, L.; Joriot, S.; Marret, S.; et al. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J. Med. Genet. 2008, 45, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Keays, D.A.; Tian, G.; Poirier, K.; Huang, G.-J.; Siebold, C.; Cleak, J.; Oliver, P.L.; Fray, M.; Harvey, R.J.; Molnár, Z.; et al. Mutations in α-Tubulin Cause Abnormal Neuronal Migration in Mice and Lissencephaly in Humans. Cell 2007, 128, 45–57. [Google Scholar] [CrossRef]
- Bodakuntla, S.; Jijumon, A.S.; Villablanca, C.; Gonzalez-Billault, C.; Janke, C. Microtubule-Associated Pro-teins: Structuring the Cytoskeleton. Trends Cell Biol. 2019, 29, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.A.; Pilz, D.T.; Babatz, T.D.; Cushion, T.D.; Harvey, K.; Topf, M.; Yates, L.; Robb, S.; Uyanik, G.; Mancini, G.M.; et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 2010, 19, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Fallet-Bianco, C.; Laquerrière, A.; Poirier, K.; Razavi, F.; Guimiot, F.; Dias, P.; Loeuillet, L.; Lascelles, K.; Beldjord, C.; Carion, N.; et al. Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly. Acta Neuropathol. Commun. 2014, 2, 69. [Google Scholar] [CrossRef]
- Hung, K.-L.; Lu, J.-F.; Su, D.-J.; Hsu, S.-J.; Wang, L.-C. Tubulinopathy Presenting as Developmental and Epileptic Encephalopathy. Children 2022, 9, 1105. [Google Scholar] [CrossRef]
- Belvindrah, R.; Natarajan, K.; Shabajee, P.; Bruel-Jungerman, E.; Bernard, J.; Goutierre, M.; Moutkine, I.; Jaglin, X.H.; Savariradjane, M.; Irinopoulou, T.; et al. Mutation of the α-tubulin Tuba1a leads to straighter microtubules and perturbs neuronal migration. J. Cell Biol. 2017, 216, 2443–2461. [Google Scholar] [CrossRef]
- Aiken, J.; Buscaglia, G.; Aiken, A.S.; Moore, J.K.; Bates, E.A. Tubulin mutations in brain development disorders: Why haploinsufficiency does not explain TUBA1A tubulinopathies. Cytoskeleton 2020, 77, 40–54. [Google Scholar] [CrossRef]
- Bittermann, E.; Abdelhamed, Z.; Liegel, R.P.; Menke, C.; Timms, A.; Beier, D.R.; Stottmann, R.W. Differential requirements of tubulin genes in mammalian forebrain development. PLoS Genet. 2019, 15, e1008243. [Google Scholar] [CrossRef] [PubMed]
- Leca, I.; Phillips, A.W.; Ushakova, L.; Cushion, T.D.; Keays, D.A. Codon modification of Tuba1a alters mRNA levels and causes a severe neurodevelopmental phenotype in mice. Sci. Rep. 2023, 13, 1215. [Google Scholar] [CrossRef] [PubMed]
- Francis, F.; Koulakoff, A.; Boucher, D.; Chafey, P.; Schaar, B.; Vinet, M.-C.; Friocourt, G.; McDonnell, N.; Reiner, O.; Kahn, A.; et al. Doublecortin Is a Developmentally Regulated, Microtubule-Associated Protein Expressed in Migrating and Differentiating Neurons. Neuron 1999, 23, 247–256. [Google Scholar] [CrossRef]
- Gleeson, J.G.; Lin, P.T.; A Flanagan, L.; A Walsh, C. Doublecortin Is a Microtubule-Associated Protein and Is Expressed Widely by Migrating Neurons. Neuron 1999, 23, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Ramos, R.L.; Ackman, J.B.; Thomas, A.M.; Lee, R.V.; LoTurco, J.J. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 2003, 6, 1277–1283. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Souville, I.; Fourniol, F.J.; Toussaint, A.; Moores, C.A.; Houdusse, A.; Lemaitre, J.Y.; Poirier, K.; Khalaf-Nazzal, R.; Hully, M.; et al. New insights into genotype–phenotype correlations for the doublecortin-related lissencephaly spectrum. Brain 2013, 136, 223–244. [Google Scholar] [CrossRef]
- Katsarou, A.; Moshé, S.L.; Galanopoulou, A.S. Interneuronopathies and their role in early life epilepsies and neurodevelopmental disorders. Epilepsia Open 2017, 2, 284–306. [Google Scholar] [CrossRef]
- Procopio, R.; Fortunato, F.; Gagliardi, M.; Talarico, M.; Sammarra, I.; Sarubbi, M.C.; Malanga, D.; Annesi, G.; Gambardell, A. Phenotypic Variability in Novel Doublecortin Gene Variants Associated with Subcortical Band Heterotopia. Int. J. Mol. Sci. 2024, 25, 5505. [Google Scholar] [CrossRef]
- Mahmud, R. Subcortical Band Heterotopia Presented With Refractory Epilepsy and Reversible Aphasia. Cureus 2021, 13, e16990. [Google Scholar] [CrossRef]
- Lin, J.; Cheng, J.; Liu, Y.; Hsu, T.; Lin, K.; Chen, C.; Lin, C.; Tsai, M.; Tsai, J. Novel lissencephaly-associated DCX variants in the C-terminal DCX domain affect microtubule binding and dynamics. Epilepsia 2022, 63, 1253–1265. [Google Scholar] [CrossRef]
- Stouffer, M.; Khalaf-Nazzal, R.; Cifuentes-Diaz, C.; Albertini, G.; Bandet, E.; Grannec, G.; Lavilla, V.; Deleuze, J.-F.; Olaso, R.; Nosten-Bertrand, M.; et al. Doublecortin mutation leads to persistent defects in the Golgi apparatus and mitochondria in adult hippocampal pyramidal cells. Neurobiol. Dis. 2022, 168, 105702. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Leventer, R.; Kuc, J.; Mewborn, S.; Dudlicek, L.L.; Ramocki, M.B.; Pilz, D.T.; Mills, P.L.; Das, S.; Ross, M.E.; et al. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur. J. Hum. Genet. 2001, 9, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Leventer, R.J. Genotype-Phenotype Correlation in Lissencephaly and Subcortical Band Heterotopia: The Key Questions Answered. J. Child Neurol. 2005, 20, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Cuccurullo, C.; Rubino, M.; De Vita, G.; Terrone, G.; Bilo, L.; Coppola, A.-N. X-Linked Epilepsies: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 4110. [Google Scholar] [CrossRef]
- Cioni, J.-M.; Wong, H.H.-W.; Bressan, D.; Kodama, L.; Harris, W.A.; Holt, C.E. Axon-Axon Interactions Regulate Topographic Optic Tract Sorting via CYFIP2-Dependent WAVE Complex Function. Neuron 2018, 97, 1078–1093.e6. [Google Scholar] [CrossRef]
- Chen, B.; Chou, H.-T.; Brautigam, C.A.; Xing, W.; Yang, S.; Henry, L.; Doolittle, L.K.; Walz, T.; Rosen, M.K. Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. eLife 2017, 6, e29795. [Google Scholar] [CrossRef]
- Schaks, M.; Reinke, M.; Witke, W.; Rottner, K. Molecular Dissection of Neurodevelopmental Disorder-Causing Mutations in CYFIP. Cells 2020, 9, 1355. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, D.; Ryu, J.R.; Zhang, Y.; Kim, S.; Kim, Y.; Lee, B.; Sun, W.; Han, K. Phosphorylation of CYFIP2, a component of the WAVE-regulatory complex, regulates dendritic spine density and neurite outgrowth in cultured hippocampal neurons potentially by affecting the complex assembly. NeuroReport 2017, 28, 749–754. [Google Scholar] [CrossRef]
- Schaks, M.; Singh, S.P.; Kage, F.; Thomason, P.; Klünemann, T.; Steffen, A.; Blankenfeldt, W.; Stradal, T.E.; Insall, R.H.; Rottner, K. Distinct Interaction Sites of Rac GTPase with WAVE Regulatory Complex Have Non-redundant Functions in Vivo. Curr. Biol. 2018, 28, 3674–3684.e6. [Google Scholar] [CrossRef]
- Abekhoukh, S.; Bardoni, B. CYFIP family proteins between autism and intellectual disability: Links with Fragile X syndrome. Front. Cell Neurosci. 2014, 8, 81. [Google Scholar] [CrossRef]
- Nakashima, M.; Kato, M.; Aoto, K.; Shiina, M.; Belal, H.; Mukaida, S.; Kumada, S.; Sato, A.; Zerem, A.; Lerman-Sagie, T.; et al. De novo hotspot variants in CYFIP2 cause early-onset epileptic encephalopathy. Ann. Neurol. 2018, 83, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Xiao, R.; Bekheirnia, M.R.; Kanani, F.; Parker, M.J.; Koenig, M.K.; van Haeringen, A.; Ruivenkamp, C.; Rosmaninho-Salgado, J.; Almeida, P.M.; et al. Heterozygous variants in SPTBN1 cause intellectual disability and autism. Am. J. Med. Genet. Part A 2021, 185, 2037–2045. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.-M.; Zhang, C.; Zollinger, D.R.; Leterrier, C.; Rasband, M.N. An αII Spectrin-Based Cytoskeleton Protects Large-Diameter Myelinated Axons from Degeneration. J. Neurosci. 2017, 37, 11323–11334. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, J.; Nakashima, M.; Nabatame, S.; Gaik-Siew, C.; Miyata, R.; Rener-Primec, Z.; Kato, M.; Matsumoto, N.; Saitsu, H. SPTAN1 encephalopathy: Distinct phenotypes and genotypes. J. Hum. Genet. 2015, 60, 167–173. [Google Scholar] [CrossRef]
- Leveille, E.; Estiar, M.A.; Krohn, L.; Spiegelman, D.; Dionne-Laporte, A.; Dupré, N.; Trempe, J.F.; Rouleau, G.A.; Gan-Or, Z. SPTAN1 variants as a potential cause for autosomal recessive hereditary spastic paraplegia. J. Hum. Genet. 2019, 64, 1145–1151. [Google Scholar] [CrossRef]
- Siripurapu, V.; Meth, J.; Kobayashi, N.; Hamaguchi, M. DBC2 Significantly Influences Cell-cycle, Apoptosis, Cytoskeleton and Membrane-trafficking Pathways. J. Mol. Biol. 2005, 346, 83–89. [Google Scholar] [CrossRef]
- Freeman, S.N.; Ma, Y.; Cress, W.D. RhoBTB2 (DBC2) Is a Mitotic E2F1 Target Gene with a Novel Role in Apoptosis. J. Biol. Chem. 2008, 283, 2353–2362. [Google Scholar] [CrossRef]
- Santos, P.K.F.; Araujo, N.d.S.; Françoso, E.; Zuntini, A.R.; Arias, M.C. Diapause in a tropical oil-collecting bee: Molecular basis unveiled by RNA-Seq. BMC Genom. 2018, 19, 305. [Google Scholar] [CrossRef]
- Straub, J.; Konrad, E.D.; Grüner, J.; Toutain, A.; Bok, L.A.; Cho, M.T.; Crawford, H.P.; Dubbs, H.; Douglas, G.; Jobling, R.; et al. Missense Variants in RHOBTB2 Cause a Developmental and Epileptic Encephalopathy in Humans, and Altered Levels Cause Neurological Defects in Drosophila. Am. J. Hum. Genet. 2017, 102, 44–57. [Google Scholar] [CrossRef]
- Chung, C.-T.; Lee, N.-C.; Fan, S.-P.; Hung, M.-Z.; Lin, Y.-H.; Chen, C.-H.; Jao, T. DYNC1H1 variant associated with epilepsy: Expanding the phenotypic spectrum. Epilepsy Behav. Rep. 2022, 21, 100580. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed]
- Mentis, A.-F.; Vlachakis, D.; Papakonstantinou, E.; Zaganas, I.; Patrinos, G.P.; Chrousos, G.P.; Dardiotis, E. A novel variant in DYNC1H1 could contribute to human amyotrophic lateral sclerosis-frontotemporal dementia spectrum. Mol. Case Stud. 2021, 8, a006096. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Pierre, B.S.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef]
- Gandhi, T.; Canepa, C.R.; Adeyelu, T.T.; Adeniyi, P.A.; Lee, C.C. Neuroanatomical Alterations in the CNTNAP2 Mouse Model of Autism Spectrum Disorder. Brain Sci. 2023, 13, 891. [Google Scholar] [CrossRef] [PubMed]
- Macrì, S.; Onori, M.P.; Roessner, V.; Laviola, G. Animal models recapitulating the multifactorial origin of Tourette syndrome. Int. Rev. Neurobiol. 2013, 112, 211–237. [Google Scholar]
- Ramanathan, S.; Al-Diwani, A.; Waters , P.; Irania, S.R. The autoantibody-mediated encephalitides: From clinical observations to molecular pathogenesis. J. Neurol. 2021, 268, 1689–1707. [Google Scholar]
- Poot, M. Intragenic CNTNAP2 Deletions: A Bridge Too Far? Mol. Syndromol. 2017, 8, 118–130. [Google Scholar]
- Mann, R.S.; Allman, B.L.; Schmid, S. Developmental changes in electrophysiological properties of auditory cortical neurons in the Cntnap2 knockout rat. J. Neurophysiol. 2023, 129, 937–947. [Google Scholar] [CrossRef]
- Gao, R.; Zaccard, C.R.; Shapiro, L.P.; Dionisio, L.E.; Martin-De-Saavedra, M.D.; Piguel, N.H.; Pratt, C.P.; Horan, K.E.; Penzes, P. The CNTNAP2-CASK complex modulates GluA1 subcellular distribution in interneurons. Neurosci. Lett. 2019, 701, 92–99. [Google Scholar] [CrossRef]
- Martín-De-Saavedra, M.D.; Dos Santos, M.; Culotta, L.; Varea, O.; Spielman, B.P.; Parnell, E.; Forrest, M.P.; Gao, R.; Yoon, S.; McCoig, E.; et al. Shed CNTNAP2 ectodomain is detectable in CSF and regulates Ca2+ homeostasis and network synchrony via PMCA2/ATP2B. Neuron 2021, 110, 627–643.e9. [Google Scholar] [CrossRef]
- Vogt, L.M.; Lorenzo, M.; Prendergast, D.B.; Jobling, R.; Gill, P.J. EEF1A2 pathogenic variant presenting in an infant with failure to thrive and frequent apneas requiring respiratory support. Am. J. Med. Genet. Part A 2022, 188, 3106–3109. [Google Scholar] [CrossRef]
- Mohamed, M.S.; Klann, E. Autism- and epilepsy-associated EEF1A2 mutations lead to translational dysfunction and altered actin bundling. Proc. Natl. Acad. Sci. USA 2023, 120, e2307704120. [Google Scholar] [CrossRef]
- Rumpansuwon, K.; Prommahom, A.; Dharmasaroja, P. eEF1A2 knockdown impairs neuronal proliferation and inhibits neurite outgrowth of differentiating neurons. NeuroReport 2022, 33, 336–344. [Google Scholar] [CrossRef]
- Cali, E.; Rocca, C.; Salpietro, V.; Houlden, H. Epileptic Phenotypes Associated with SNAREs and Related Synaptic Vesicle Exocytosis Machinery. Front. Neurol. 2022, 12, 806506. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. Neurotransmitter Release: The Last Millisecond in the Life of a Synaptic Vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Khvotchev, M.; Dulubova, I.; Sun, J.; Dai, H.; Rizo, J.; Südhof, T.C. Dual Modes of Munc18-1/SNARE Interactions Are Coupled by Functionally Critical Binding to Syntaxin-1 N Terminus. J. Neurosci. 2007, 27, 12147–12155. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.H.; Rah, J.-C.; Min, S.-W.; Liu, X.; de Wit, H.; Dulubova, I.; Meyer, A.C.; Rizo, J.; Arancillo, M.; Hammer, R.E.; et al. Conformational Switch of Syntaxin-1 Controls Synaptic Vesicle Fusion. Science 2008, 321, 1507–1510. [Google Scholar] [CrossRef]
- Wolking, S.; May, P.; Mei, D.; Møller, R.S.; Balestrini, S.; Helbig, K.L.; Altuzarra, C.D.; Chatron, N.; Kaiwar, C.; Stöhr, K.; et al. Clinical spectrum of STX1B -related epileptic disorders. Neurology 2019, 92, e1238–e1249. [Google Scholar] [CrossRef]
- Mishima, T.; Fujiwara, T.; Sanada, M.; Kofuji, T.; Kanai-Azuma, M.; Akagawa, K. Syntaxin 1B, but Not Syntaxin 1A, Is Necessary for the Regulation of Synaptic Vesicle Exocytosis and of the Readily Releasable Pool at Central Synapses. PLoS ONE 2014, 9, e90004. [Google Scholar] [CrossRef]
- Constable, J.R.L.; Graham, M.E.; Morgan, A.; Burgoyne, R.D. Amisyn Regulates Exocytosis and Fusion Pore Stability by Both Syntaxin-dependent and Syntaxin-independent Mechanisms. J. Biol. Chem. 2005, 280, 31615–31623. [Google Scholar] [CrossRef]
- Vinci, M.; Costanza, C.; Rando, R.G.; Treccarichi, S.; Saccone, S.; Carotenuto, M.; Roccella, M.; Calì, F.; Elia, M.; Vetri, L. STXBP6 Gene Mutation: A New Form of SNAREopathy Leads to Developmental Epileptic Encephalopathy. Int. J. Mol. Sci. 2023, 24, 16436. [Google Scholar] [CrossRef]
- Liu, C.; Hu, Q.; Chen, Y.; Wu, L.; Liu, X.; Liang, D. Behavioral and Gene Expression Analysis of Stxbp6-Knockout Mice. Brain Sci. 2021, 11, 436. [Google Scholar] [CrossRef]
- Jahn, R.; Scheller, R.H. SNAREs-Engines for Membrane Fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. [Google Scholar] [CrossRef]
- Burgalossi, A.; Jung, S.; Meyer, G.; Jockusch, W.J.; Jahn, O.; Taschenberger, H.; O’Connor, V.M.; Nishiki, T.-I.; Takahashi, M.; Brose, N.; et al. SNARE Protein Recycling by αSNAP and βSNAP Supports Synaptic Vesicle Priming. Neuron 2010, 68, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Mignon-Ravix, C.; Riccardi, F.; Daquin, G.; Cacciagli, P.; Lamoureux-Toth, S.; Villard, L.; Villeneuve, N.; Molinari, F. NAPB and developmental and epileptic encephalopathy: Description of the electroclinical profile associated with a novel pathogenic variant. Epilepsia 2023, 64, E127–E134. [Google Scholar] [CrossRef]
- AbdelAleem, A.; Haddad, N.; Al-Ettribi, G.; Crunk, A.; Elsotouhy, A. Cohen syndrome and early-onset epileptic encephalopathy in male triplets: Two disease-causing mutations in VPS13B and NAPB. Neurogenetics 2023, 24, 103–112. [Google Scholar] [CrossRef]
- Chen, H.-J.; Rojas-Soto, M.; Oguni, A.; Kennedy, M.B. A Synaptic Ras-GTPase Activating Protein (p135 SynGAP) Inhibited by CaM Kinase II. Neuron 1998, 20, 895–904. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, H.-K.; Takamiya, K.; Huganir, R.L. The Role of Synaptic GTPase-Activating Protein in Neuronal Development and Synaptic Plasticity. J. Neurosci. 2003, 23, 1119–1124. [Google Scholar] [CrossRef]
- Carlisle, H.J.; Manzerra, P.; Marcora, E.; Kennedy, M.B. SynGAP Regulates Steady-State and Activity-Dependent Phosphorylation of Cofilin. J. Neurosci. 2008, 28, 13673–13683. [Google Scholar] [CrossRef]
- Araki, Y.; Zeng, M.; Zhang, M.; Huganir, R.L. Rapid Dispersion of SynGAP from Synaptic Spines Triggers AMPA Receptor Insertion and Spine Enlargement during LTP. Neuron 2015, 85, 173–189. [Google Scholar] [CrossRef]
- Clement, J.P.; Aceti, M.; Creson, T.K.; Ozkan, E.D.; Shi, Y.; Reish, N.J.; Almonte, A.G.; Miller, B.H.; Wiltgen, B.J.; Miller, C.A.; et al. Pathogenic SYNGAP1 Mutations Impair Cognitive Development by Disrupting Maturation of Dendritic Spine Synapses. Cell 2012, 151, 709–723. [Google Scholar] [CrossRef]
- McMahon, A.; Barnett, M.; O’Leary, T.; Stoney, P.; Collins, M.; Papadia, S.; Choudhary, J.; Komiyama, N.; Grant, S.; Hardingham, G.; et al. SynGAP isoforms exert opposing effects on synaptic strength. Nat. Commun. 2012, 3, 900. [Google Scholar] [CrossRef]
- Araki, Y.; Hong, I.; Gamache, T.R.; Ju, S.; Collado-Torres, L.; Shin, J.H.; Huganir, R.L.; States, U. SynGAP isoforms differentially regulate synaptic plasticity and dendritic development. eLife 2020, 9, e56273. [Google Scholar] [CrossRef]
- Morimune, T.; Tano, A.; Tanaka, Y.; Yukiue, H.; Yamamoto, T.; Tooyama, I.; Maruo, Y.; Nishimura, M.; Mori, M. Gm14230 controls Tbc1d24 cytoophidia and neuronal cellular juvenescence. PLoS ONE 2021, 16, e0248517. [Google Scholar] [CrossRef] [PubMed]
- Aprile, D.; Fruscione, F.; Baldassari, S.; Fadda, M.; Ferrante, D.; Falace, A.; Buhler, E.; Sartorelli, J.; Represa, A.; Baldelli, P.; et al. TBC1D24 regulates axonal outgrowth and membrane trafficking at the growth cone in rodent and human neurons. Cell Death Differ. 2019, 26, 2464–2478. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Denef, N.; Schüpbach, T. The Vacuolar Proton Pump, V-ATPase, Is Required for Notch Signaling and Endosomal Trafficking in Drosophila. Dev. Cell 2009, 17, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Faronato, M.; Nguyen, V.T.; Patten, D.K.; Lombardo, Y.; Steel, J.H.; Patel, N.; Woodley, L.; Shousha, S.; Pruneri, G.; Coombes, R.C.; et al. DMXL2 drives epithelial to mesenchymal transition in hormonal therapy resistant breast cancer through notch hyper-activation. Oncotarget 2015, 6, 22467–22479. [Google Scholar] [CrossRef]
- Shah, A.A.; Amjad, M.; Hassan, J.-U.; Ullah, A.; Mahmood, A.; Deng, H.; Ali, Y.; Gul, F.; Xia, K. Molecular Insights into the Role of Pathogenic nsSNPs in GRIN2B Gene Provoking Neurodevelopmental Disorders. Genes 2022, 13, 1332. [Google Scholar] [CrossRef]
- Zong, P.; Feng, J.; Yue, Z.; Li, Y.; Wu, G.; Sun, B.; He, Y.; Miller, B.; Yu, A.S.; Su, Z.; et al. Functional coupling of TRPM2 and extrasynaptic NMDARs exacerbates excitotoxicity in ischemic brain injury. Neuron 2022, 110, 1944–1958.e8. [Google Scholar] [CrossRef]
- Myers, S.J.; Yuan, H.; Kang, J.-Q.; Tan, F.C.K.; Traynelis, S.F.; Low, C.-M. Distinct roles of GRIN2A and GRIN2B variants in neurological conditions. F1000Research 2019, 8, 1940. [Google Scholar] [CrossRef]
- Korinek, M.; Serra, M.C.; Abdelrahman, F.E.S.; Dobrovolski, M.; Kuchtiak, V.; Abramova, V.; Fili, K.; Tomovic; Krausova, B.H.; Krusek, J.; et al. Dis-ease-Associated Variants in GRIN1, GRIN2A and GRIN2B Genes: Insights into NMDA Receptor Structure, Function, and Pathophysiology. Physiol. Res. 2024, 73, 413–434. [Google Scholar] [CrossRef]
- Hines, D.J.; Contreras, A.; Garcia, B.; Barker, J.S.; Boren, A.J.; El Achkar, C.M.; Moss, S.J.; Hines, R.M. Human ARHGEF9 intellectual disability syndrome is phenocopied by a mutation that disrupts collybistin binding to the GABAA receptor α2 subunit. Mol. Psychiatry 2022, 27, 1729–1741. [Google Scholar] [CrossRef]
- Alber, M.; Kalscheuer, V.M.; Marco, E.; Sherr, E.; Lesca, G.; Till, M.; Gradek, G.; Wiesener, A.; Korenke, C.; Mercier, S.; et al. ARHGEF9 disease. Neurol. Genet. 2017, 3, e148. [Google Scholar] [CrossRef]
- Hines, R.M.; Maric, H.M.; Hines, D.J.; Modgil, A.; Panzanelli, P.; Nakamura, Y.; Nathanson, A.J.; Cross, A.; Deeb, T.; Brandon, N.J.; et al. Developmental seizures and mortality result from reducing GABAA receptor α2-subunit interaction with collybistin. Nat. Commun. 2018, 9, 3130. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.J.; Onori, M.P.; Borgesius, N.Z.; van de Bree, J.E.; Elgersma-Hooisma, M.; Nio, E.; Bezstarosti, K.; Buitendijk, G.H.; Jolfaei, M.A.; Demmers, J.A.; et al. CAMK2-Dependent Signaling in Neurons Is Essential for Survival. J. Neurosci. 2019, 39, 5424–5439. [Google Scholar] [CrossRef] [PubMed]
- Küry, S.; van Woerden, G.M.; Besnard, T.; Onori, M.P.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef] [PubMed]
- Akita, T.; Aoto, K.; Kato, M.; Shiina, M.; Mutoh, H.; Nakashima, M.; Kuki, I.; Okazaki, S.; Magara, S.; Shiihara, T.; et al. De novo variants in CAMK2A and CAMK2B cause neurodevelopmental disorders. Ann. Clin. Transl. Neurol. 2018, 5, 280–296. [Google Scholar] [CrossRef]
- Rumian, N.L.; Freund, R.K.; Dell’acqua, M.L.; Coultrap, S.J.; Bayer, K.U. Decreased nitrosylation of CaMKII causes aging-associated impairments in memory and synaptic plasticity in mice. Sci. Signal. 2023, 16, eade5892. [Google Scholar] [CrossRef]
- Stephenson, J.R.; Wang, X.; Perfitt, T.L.; Parrish, W.P.; Shonesy, B.C.; Marks, C.R.; Mortlock, D.P.; Nakagawa, T.; Sutcliffe, J.S.; Colbran, R.J. A Novel Human CAMK2A Mutation Disrupts Dendritic Morphology and Synaptic Transmission, and Causes ASD-Related Behaviors. J. Neurosci. 2017, 37, 2216–2233. [Google Scholar] [CrossRef]
- Lee, L.-C.; Su, M.-T.; Huang, H.-Y.; Cho, Y.-C.; Yeh, T.-K.; Chang, C.-Y. Association of CaMK2A and MeCP2 signaling pathways with cognitive ability in adolescents. Mol. Brain 2021, 14. [Google Scholar] [CrossRef]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, 243–256. [Google Scholar] [CrossRef]
- Murakami, K.; Kanno, H.; Tancabelic, J.; Fujii, H. Gene Expression and Biological Significance of Hexokinase in Erythroid Cells. Acta Haematol. 2002, 108, 204–209. [Google Scholar] [CrossRef]
- E Aleshin, A.; Zeng, C.; Bourenkov, G.P.; Bartunik, H.D.; Fromm, H.J.; Honzatko, R.B. The mechanism of regulation of hexokinase: New insights from the crystal structure of recombinant human brain hexokinase complexed with glucose and glucose-6-phosphate. Structure 1998, 6, 39–50. [Google Scholar] [CrossRef]
- Rosano, C.; Sabini, E.; Rizzi, M.; Deriu, D.; Murshudov, G.; Bianchi, M.; Serafini, G.; Magnani, M.; Bolognesi, M. Binding of non-catalytic ATP to human hexokinase I highlights the structural components for enzyme–membrane association control. Structure 1999, 7, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.-Y.; Alechina, O.; Aleshin, A.E.; Fromm, H.J.; Honzatko, R.B. Identification of a Phosphate Regulatory Site and a Low Affinity Binding Site for Glucose 6-Phosphate in the N-terminal Half of Human Brain Hexokinase. J. Biol. Chem. 1998, 273, 19548–19553. [Google Scholar] [CrossRef] [PubMed]
- Sui, D.; E Wilson, J. Functional interactions between the noncovalently associated N- and C-terminal halves of mammalian Type I hexokinase. Arch. Biochem. Biophys. 2002, 401, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.L.; Badonyi, M.; Cozens, A.; Foulds, N.; Marsh, J.A.; Rahman, S.; Ross, A.; Schooley, J.; Straub, V.; Quigley, A.J.; et al. Expanding the neurodevelopmental phenotype associated with HK1 de novo heterozygous missense variants. Eur. J. Med. Genet. 2023, 66, 104696. [Google Scholar] [CrossRef]
- Kane, M.S.; Alban, J.; Desquiret-Dumas, V.; Gueguen, N.; Ishak, L.; Ferre, M.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Lenaers, G.; et al. Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J. Cell Mol. Med. 2017, 21, 2284–2297. [Google Scholar] [CrossRef]
- Indiveri, C.; Krämer, R.; Palmieri, F. Reconstitution of the malate/aspartate shuttle from mitochondria. J. Biol. Chem. 1987, 262, 15979–15983. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.; Walker, J.; Saheki, T.; Satrústegui, J.; et al. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; del Arco, A.; Saheki, T.; Pardo, B.; Satrústegui, J. Calcium-Regulation of Mitochondrial Respiration Maintains ATP Homeostasis and Requires ARALAR/AGC1-Malate Aspartate Shuttle in Intact Cortical Neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef]
- Poeta, E.; Petralla, S.; Babini, G.; Renzi, B.; Celauro, L.; Magnifico, M.C.; Barile, S.N.; Masotti, M.; De Chirico, F.; Massenzio, F.; et al. Histone Acetylation Defects in Brain Precursor Cells: A Potential Pathogenic Mechanism Causing Proliferation and Differentiation Dysfunctions in Mitochondrial Aspartate-Glutamate Carrier Isoform 1 Deficiency. Front. Cell Neurosci. 2022, 15, 773709. [Google Scholar] [CrossRef]
- Prasun, P.; Young, S.; Salomons, G.; Werneke, A.; Jiang, Y.; Struys, E.; Paige, M.; Avantaggiati, M.L.; McDonald, M. Expanding the Clinical Spectrum of Mitochondrial Citrate Carrier (SLC25A1) Deficiency: Facial Dysmorphism in Siblings with Epileptic Encephalopathy and Combined D,L-2-Hydroxyglutaric Aciduria. JIMD Rep. 2015, 19, 111–115. [Google Scholar]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Thangaratnarajah, C.; Ruprecht, J.J.; Kunji, E.R. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat. Commun. 2014, 5, 5491. [Google Scholar] [CrossRef] [PubMed]
- Pierri, C.L.; Palmieri, F.; De Grassi, A. Single-nucleotide evolution quantifies the importance of each site along the structure of mitochondrial carriers. Cell Mol. Life Sci. 2013, 71, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Amoedo, N.D.; Punzi, G.; Obre, E.; Lacombe, D.; De Grassi, A.; Pierri, C.L.; Rossignol, R. AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim. Biophys. Acta 2016, 1863, 2394–2412. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.; Basu, S.; Bogenhagen, D.F. A Quantitative Proteomic Analysis of Mitochondrial Participation in P19 Cell Neuronal Differentiation. J. Proteome Res. 2007, 7, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Mekala, P.; Yahya, D.; Wu, G.; Ledeen, R.W. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: Evidence for myelin-associated aspartoacylase. J. Neurochem. 2001, 78, 736–745. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wang, J.; Wu, G.; Lu, Z.H.; Chakraborty, G.; Meyenhofer, M.; Tyring, S.K.; Matalon, R. Physiological role of N-acetylaspartate: Contribution to myelinogenesis. Adv. Exp. Med. Biol. 2006, 576, 131–143, discussion 361–363. [Google Scholar]
- Wibom, R.; Lasorsa, F.M.; Töhönen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 Deficiency Associated with Global Cerebral Hypomyelination. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef]
- Ramos, M.; Pardo, B.; Llorente-Folch, I.; Saheki, T.; del Arco, A.; Satrústegui, J. Deficiency of the mitochondrial transporter of aspartate/glutamate aralar/AGC1 causes hypomyelination and neuronal defects unrelated to myelin deficits in mouse brain. J. Neurosci. Res. 2011, 89, 2008–2017. [Google Scholar] [CrossRef]
- Gómez-Galán, M.; Makarova, J.; Llorente-Folch, I.; Saheki, T.; Pardo, B.; Satrústegui, J.; Herreras, O. Altered Postnatal Development of Cortico—Hippocampal Neuronal Electric Activity in Mice Deficient for the Mitochondrial Aspartate—Glutamate Transporter. J. Cereb. Blood Flow Metab. 2011, 32, 306–317. [Google Scholar] [CrossRef]
- Contreras, L.; Ramirez, L.; Du, J.; Hurley, J.B.; Satrústegui, J.; de la Villa, P. Deficient Glucose and Glutamine Metabolism in Aralar/AGC1/Slc25a12 Knockout Mice Contributes to Altered Visual Function. Mol. Vis. 2016, 22, 1198. [Google Scholar]
- Petralla, S.; Peña-Altamira, L.E.; Poeta, E.; Massenzio, F.; Virgili, M.; Barile, S.N.; Sbano, L.; Profilo, E.; Corricelli, M.; Danese, A.; et al. Deficiency of Mitochondrial Aspartate-Glutamate Carrier 1 Leads to Oligodendrocyte Precursor Cell Proliferation Defects Both In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 4486. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010, 11, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Vautrin, J. The Synaptomatrix: A Solid Though Dynamic Contact Disconnecting Transmissions from Exocy-totic Events. Neurochem. Int. 2010, 57, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Dani, N.; Broadie, K. Glycosylated synaptomatrix regulation of trans-synaptic signaling. Dev. Neurobiol. 2011, 72, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Wasser, C.R.; Masiulis, I.; Durakoglugil, M.S.; Lane-Donovan, C.; Xian, X.; Beffert, U.; Agarwala, A.; Hammer, R.E.; Herz, J. Differential splicing and glycosylation of Apoer2 alters synaptic plasticity and fear learning. Sci. Signal. 2014, 7, ra113. [Google Scholar] [CrossRef]
- Gao, P.; Wang, F.; Huo, J.; Wan, D.; Zhang, J.; Niu, J.; Wu, J.; Yu, B.; Sun, T. ALG13 Deficiency Associated with Increased Seizure Susceptibility and Severity. Neuroscience 2019, 409, 204–221. [Google Scholar] [CrossRef]
- De Ligt, J.; Willemsen, M.H.; Van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; De Vries, P.; Gilissen, C.; et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.M.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Datta, A.N.; Bahi-Buisson, N.; Bienvenu, T.; Buerki, S.E.; Gardiner, F.; Cross, J.H.; Heron, B.; Kaminska, A.; Korff, C.M.; Lepine, A.; et al. The phenotypic spectrum of X-linked, infantile onset ALG13-related developmental and epileptic encephalopathy. Epilepsia 2021, 62, 325–334. [Google Scholar] [CrossRef]
- Gao, X.-D.; Tachikawa, H.; Sato, T.; Jigami, Y.; Dean, N. Alg14 Recruits Alg13 to the Cytoplasmic Face of the Endoplasmic Reticulum to Form a Novel Bipartite UDP-N-acetylglucosamine Transferase Required for the Second Step of N-Linked Glycosylation. J. Biol. Chem. 2005, 280, 36254–36262. [Google Scholar] [CrossRef]
- Averbeck, N.; Gao, X.-D.; Nishimura, S.-I.; Dean, N. Alg13p, the Catalytic Subunit of the Endoplasmic Reticulum UDP-GlcNAc Glycosyltransferase, Is a Target for Proteasomal Degradation. Mol. Biol. Cell 2008, 19, 2169–2178. [Google Scholar] [CrossRef]
- Gao, X.-D.; Moriyama, S.; Miura, N.; Dean, N.; Nishimura, S.-I. Interaction between the C Termini of Alg13 and Alg14 Mediates Formation of the Active UDP-N-acetylglucosamine Transferase Complex. J. Biol. Chem. 2008, 283, 32534–32541. [Google Scholar] [CrossRef] [PubMed]
- Timal, S.; Hoischen, A.; Lehle, L.; Adamowicz, M.; Huijben, K.; Sykut-Cegielska, J.; Paprocka, J.; Jamroz, E.; van Spronsen, F.J.; Körner, C.; et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet. 2012, 21, 4151–4161. [Google Scholar] [CrossRef] [PubMed]
- Mitusińska, K.; Góra, A.; Bogdańska, A.; Rożdżyńska-Świątkowska, A.; Tylki-Szymańska, A.; Jezela-Stanek, A. Structural Analysis of the Effect of Asn107Ser Mutation on Alg13 Activity and Alg13-Alg14 Complex Formation and Expanding the Phenotypic Variability of ALG13-CDG. Biomolecules 2022, 12, 398. [Google Scholar] [CrossRef]
- Kukuruzinska, M.; Lennon, K. Protein N-Glycosylation: Molecular Genetics and Functional Significance. Crit. Rev. Oral Biol. Med. 1998, 9, 415–448. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Intracellular Functions of N-Linked Glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef]
- Dennis, J.W.; Nabi, I.R.; Demetriou, M. Metabolism, Cell Surface Organization, and Disease. Cell 2009, 139, 1229–1241. [Google Scholar] [CrossRef]
- Epi4K Consortium; Epilepsy Phenome/Genome Project. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef]
- Bissar-Tadmouri, N.; Donahue, W.L.; Al-Gazali, L.; Nelson, S.F.; Bayrak-Toydemir, P.; Kantarci, S. X chromosome exome sequencing reveals a novel ALG13 mutation in a nonsyndromic intellectual disability family with multiple affected male siblings. Am. J. Med. Genet. Part A 2013, 164, 164–169. [Google Scholar] [CrossRef]
- Esposito, T.; De Stefano, G.; Reccia, M.G.; Di Lorenzo, I.; Napolitano, F.; Scalabrì, F.; Lombardi, A.; Saleem, M.A.; Griffiths, L.R.; Gianfrancesco, F. Dysregulation of the Expression of Asparagine-Linked Glycosylation 13 Short Isoform 2 Affects Nephrin Function by Altering Its N-Linked Glycosylation. Nephron 2017, 136, 143–150. [Google Scholar] [CrossRef]
- Charych, E.I.; Akum, B.F.; Goldberg, J.S.; Jörnsten, R.J.; Rongo, C.; Zheng, J.Q.; Firestein, B.L. Activity-Independent Regulation of Dendrite Patterning by Postsynaptic Density Protein PSD-95. J. Neurosci. 2006, 26, 10164–10176. [Google Scholar] [CrossRef]
- Matsuo, N.; Reijmers, L.; Mayford, M. Spine-Type-Specific Recruitment of Newly Synthesized AMPA Receptors with Learning. Science 2008, 319, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Arikkath, J. Molecular mechanisms of dendrite morphogenesis. Front. Cell Neurosci. 2012, 6, 37943. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Xia, Y.; Wang, C.; Wang, F.; Zhang, C.; Xiao, L.; Zhang, X.; Meng, Y.; Wang, Y.; Ding, J.; et al. Decreased cognitive function of ALG13KO female mice may be related to the decreased plasticity of hippocampal neurons. Neuropeptides 2022, 96, 102290. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Proia, R.L.; Sandhoff, K. Combinatorial Ganglioside Biosynthesis. J. Biol. Chem. 2002, 277, 25859–25862. [Google Scholar] [CrossRef]
- Wang, B. Sialic Acid Is an Essential Nutrient for Brain Development and Cognition. Annu. Rev. Nutr. 2009, 29, 177–222. [Google Scholar] [CrossRef]
- Audry, M.; Jeanneau, C.; Imberty, A.; Harduin-Lepers, A.; Delannoy, P.; Breton, C. Current trends in the structure-activity relationships of sialyltransferases. Glycobiology 2010, 21, 716–726. [Google Scholar] [CrossRef]
- Yoo, S.; Motari, M.G.; Susuki, K.; Prendergast, J.; Mountney, A.; Hurtado, A.; Schnaar, R.L. Sialylation regulates brain structure and function. FASEB J. 2015, 29, 3040–3053. [Google Scholar] [CrossRef]
- Rivero, O.; Alhama-Riba, J.; Ku, H.-P.; Fischer, M.; Ortega, G.; Álmos, P.; Diouf, D.; Hove, D.v.D.; Lesch, K.-P. Haploinsufficiency of the Attention-Deficit/Hyperactivity Disorder Risk Gene St3gal3 in Mice Causes Alterations in Cognition and Expression of Genes Involved in Myelination and Sialylation. Front. Genet. 2021, 12, 688488. [Google Scholar] [CrossRef]
- Hu, H.; Eggers, K.; Chen, W.; Garshasbi, M.; Motazacker, M.M.; Wrogemann, K.; Kahrizi, K.; Tzschach, A.; Hosseini, M.; Bahman, I.; et al. ST3GAL3 Mutations Impair the Development of Higher Cognitive Functions. Am. J. Hum. Genet. 2011, 89, 407–414. [Google Scholar] [CrossRef]
- Taniguchi, N.; Honke, K.; Fukuda, M.; Narimatsu, H.; Yamaguchi, Y.; Angata, T. Handbook of Glycosyltransferases and Related Genes, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2014; Volumes 1–2. [Google Scholar]
- Edvardson, S.; Baumann, A.; Mühlenhoff, M.; Stephan, O.; Kuss, A.W.; Shaag, A.; He, L.; Zenvirt, S.; Tanzi, R.; Gerardy-Schahn, R.; et al. West syndrome caused by ST3Gal-III deficiency. Epilepsia 2012, 54, e24–e27. [Google Scholar] [CrossRef]
- Schnaar, R.L.; Gerardy-Schahn, R.; Hildebrandt, H. Sialic Acids in the Brain: Gangliosides and Polysialic Acid in Nervous System Development, Stability, Disease, and Regeneration. Physiol. Rev. 2014, 94, 461–518. [Google Scholar] [CrossRef] [PubMed]
- Indellicato, R.; Domenighini, R.; Malagolini, N.; Cereda, A.; Mamoli, D.; Pezzani, L.; Iascone, M.; Dall’olio, F.; Trinchera, M. A novel nonsense and inactivating variant of ST3GAL3 in two infant siblings suffering severe epilepsy and expressing circulating CA19. Glycobiology 2019, 30, 95–104. [Google Scholar] [CrossRef] [PubMed]




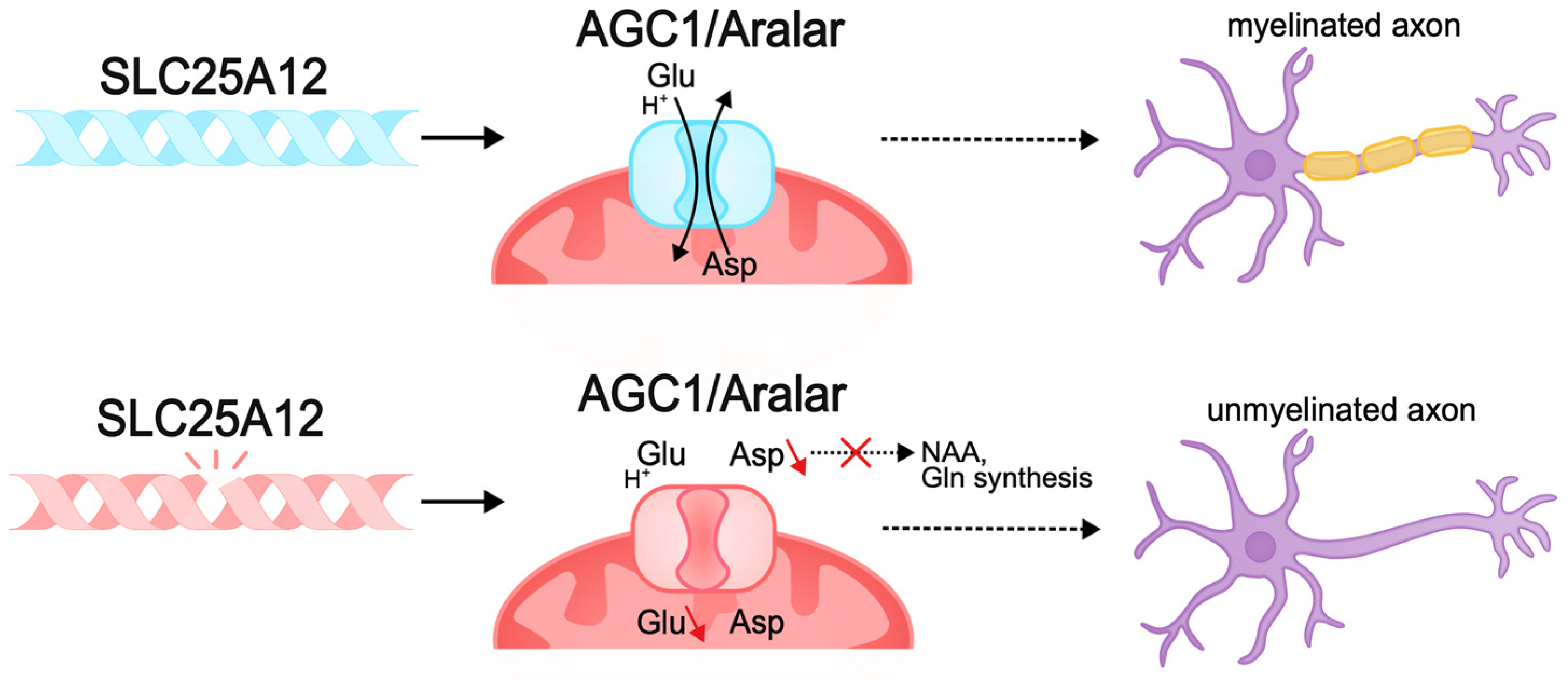


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | Subgroup | Gene Name | Type of Inheritance |
|---|---|---|---|
| Malformation of cortical development | Impaired differentiation and proliferation | ACTL6B | Autosomal recessive; autosomal dominant; de novo [46] |
| Differentiation of inhibitory interneurons | CNTNAP2 | Autosomal recessive [47] | |
| Impaired differentiation and proliferation | CUX2 | Autosomal dominant; de novo [48] | |
| Disruption of dendrito- and axonogenesis | CYFIP2 | Autosomal dominant; de novo [49] | |
| Impaired migration | DCX | X-linked recessive; de novo [50] | |
| Disruption of dendrito- and axonogenesis | DYNC1H1 | Autosomal dominant; de novo [51] | |
| Disruption of dendrito- and axonogenesis | EEF1A2 | Autosomal dominant; de novo [52] | |
| Impaired proliferation | GEMIN5 | Autosomal recessive [53] | |
| Impaired differentiation | GNAO1 | Autosomal dominant; de novo [54] | |
| Impaired differentiation and proliferation | HNRNPU | Autosomal dominant; de novo [55] | |
| Impaired proliferation | INPP4A | Autosomal recessive [56] | |
| Disruption of dendritogenesis | RHOBTB2 | Autosomal dominant; autosomal recessive; de novo [57] | |
| Impaired proliferation | SMC1A | X-linked dominant [58] | |
| Impaired proliferation and migration | SP9 | Autosomal dominant; de novo [59] | |
| Impaired differentiation | SPTAN1 | Autosomal dominant; de novo [60] | |
| Impaired migration | TUBA1A | Autosomal dominant [61] | |
| Synaptopathies | Inhibitory synaptic transmission | ARHGEF9 | X-linked recessive; de novo [62] |
| Effects on dendritic spines | CAMK2 | Autosomal dominant; de novo [63] | |
| Disruptions in synaptic endocytosis | DMXL2 | Autosomal dominant [64] | |
| Dysfunction of glutamate receptors | GRIN2A/B | Autosomal dominant; de novo [65] | |
| Disruption of disassembly and utilization of SNARE complex proteins | NAPB | Autosomal recessive [66] | |
| Disruption of synaptic vesicle fusion | STX1B | Autosomal dominant; de novo [67] | |
| Enhanced glutamate receptor activity | SYNGAP1 | de novo [68] | |
| Dysregulation of synaptic vesicles | TBC1D24 | Autosomal recessive [69] | |
| Metabolic disorders | Membrane transporter dysfunction | AGC1 | Autosomal recessive [70] |
| Congenital disorders of glycosylation | ALG13 | X-linked recessive; de novo [71] | |
| Accumulation of metabolites | HK1 | Autosomal recessive; de novo [72] | |
| Membrane transporter dysfunction | SLC25A12 | Autosomal recessive [73] | |
| Decreased levels of sialoglycans | ST3GAL3 | de novo [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medyanik, A.D.; Anisimova, P.E.; Kustova, A.O.; Tarabykin, V.S.; Kondakova, E.V. Developmental and Epileptic Encephalopathy: Pathogenesis of Intellectual Disability Beyond Channelopathies. Biomolecules 2025, 15, 133. https://doi.org/10.3390/biom15010133
Medyanik AD, Anisimova PE, Kustova AO, Tarabykin VS, Kondakova EV. Developmental and Epileptic Encephalopathy: Pathogenesis of Intellectual Disability Beyond Channelopathies. Biomolecules. 2025; 15(1):133. https://doi.org/10.3390/biom15010133
Chicago/Turabian StyleMedyanik, Alexandra D., Polina E. Anisimova, Angelina O. Kustova, Victor S. Tarabykin, and Elena V. Kondakova. 2025. "Developmental and Epileptic Encephalopathy: Pathogenesis of Intellectual Disability Beyond Channelopathies" Biomolecules 15, no. 1: 133. https://doi.org/10.3390/biom15010133
APA StyleMedyanik, A. D., Anisimova, P. E., Kustova, A. O., Tarabykin, V. S., & Kondakova, E. V. (2025). Developmental and Epileptic Encephalopathy: Pathogenesis of Intellectual Disability Beyond Channelopathies. Biomolecules, 15(1), 133. https://doi.org/10.3390/biom15010133