
The Intersection of Epigenetics and Senolytics in Mechanisms of Aging and Therapeutic Approaches



Abstract
1. Introduction
2. Materials and Methods
3. Epigenetics in Aging
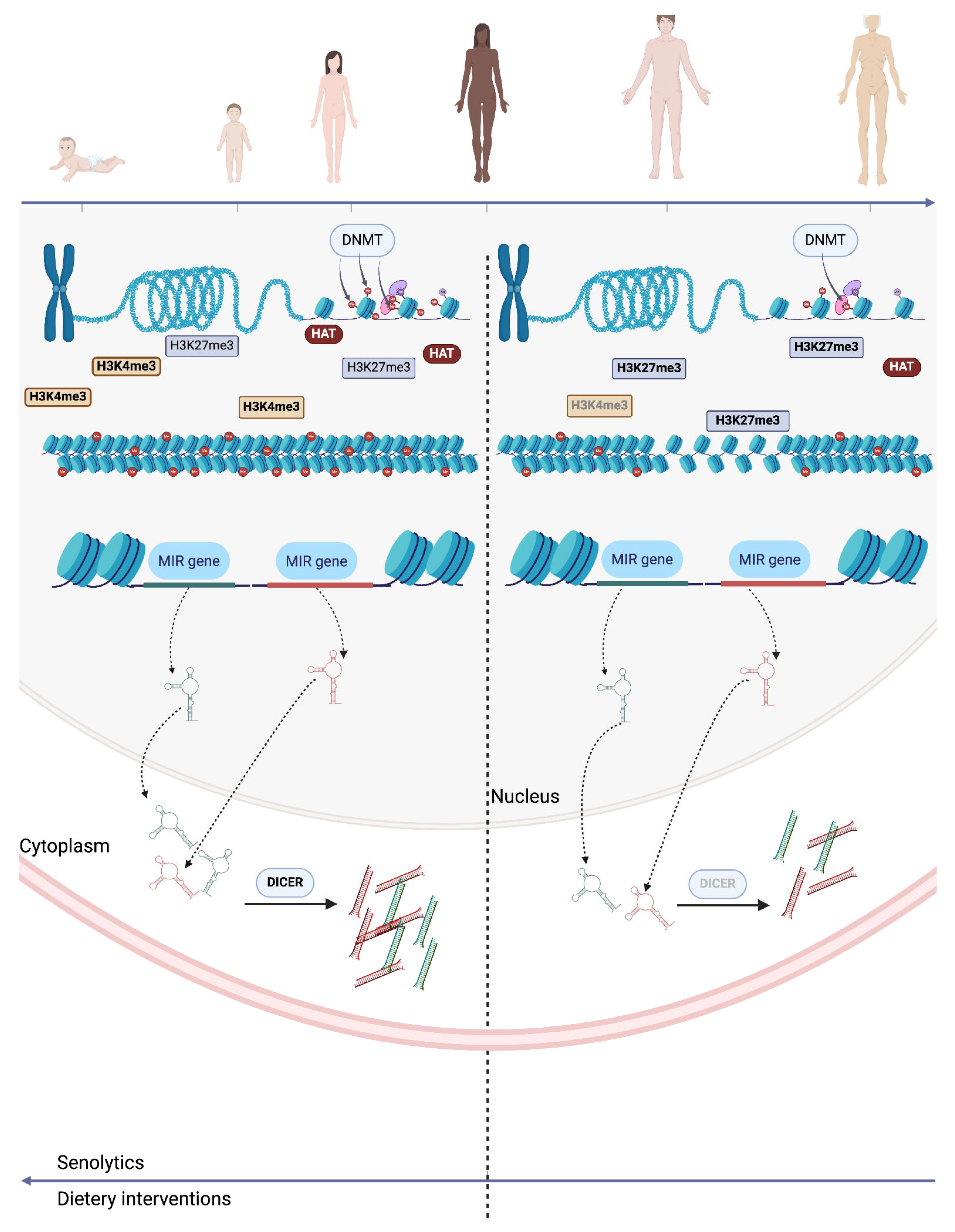
3.1. DNA Methylation
3.2. Histone Modifications
3.3. Chromatin Remodelling
3.4. Non-Coding RNAs
3.5. Epigenetic and Telomerase Clocks and Circadian Rhythm in Aging
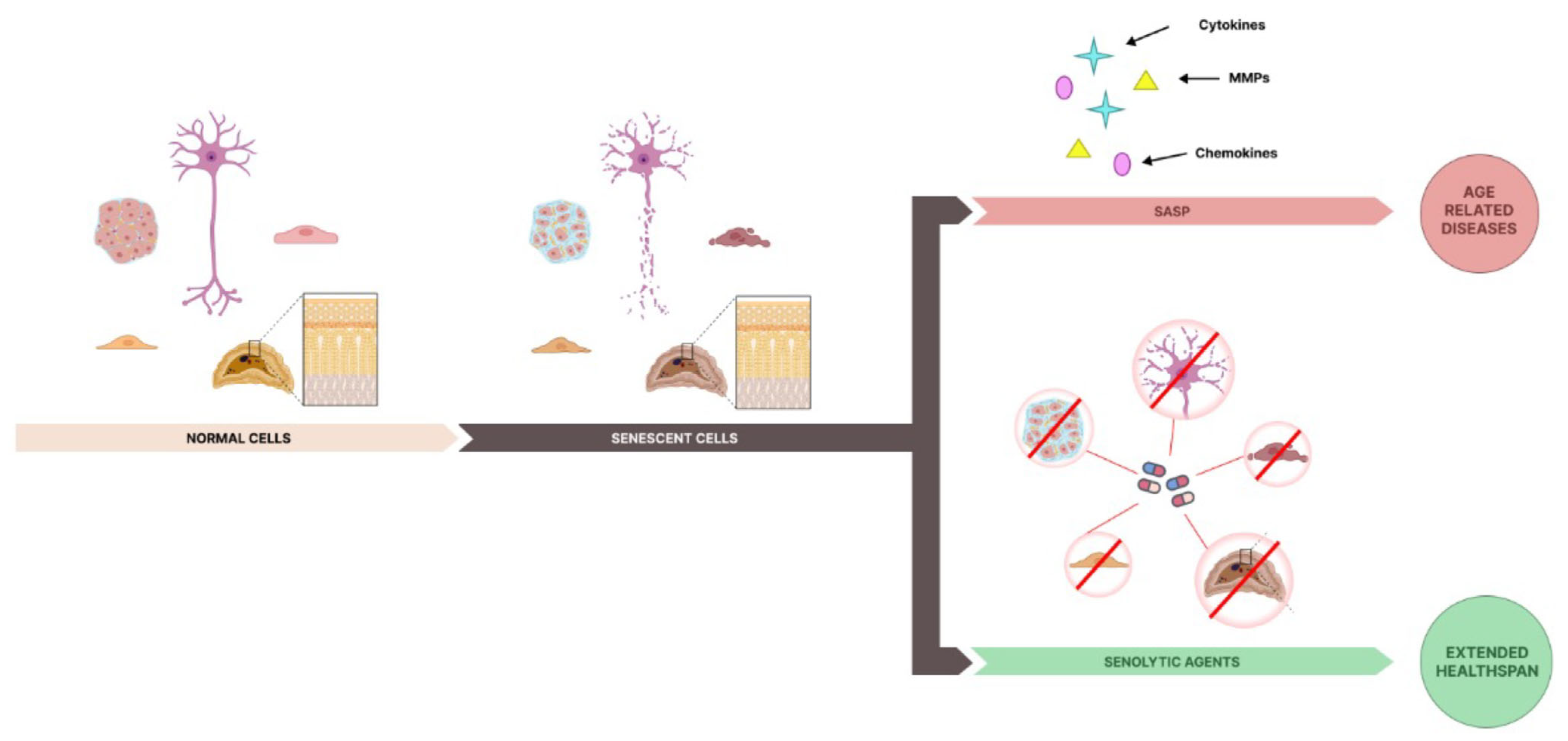
4. The Future of Senolytics
5. The Interplay Between Epigenetics and Senolytics
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Kosinsky, R.L. Epigenetics of Aging and Aging-Associated Diseases. Int. J. Mol. Sci. 2021, 22, 401. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef]
- Naylor, R.M.; Baker, D.J.; Van Deursen, J.M. Senescent Cells: A Novel Therapeutic Target for Aging and Age-Related Diseases. Clin. Pharmacol. Ther. 2012, 93, 105–116. [Google Scholar] [CrossRef]
- Yang, J.; Petty, C.A.; Dixon-McDougall, T.; Lopez, M.V.; Tyshkovskiy, A.; Maybury-Lewis, S.; Tian, X.; Ibrahim, N.; Chen, Z.; Griffin, P.T.; et al. Chemically induced reprogramming to reverse cellular aging. Aging 2023, 15, 5966–5989. [Google Scholar] [CrossRef]
- Fahy, G.M.; Brooke, R.T.; Watson, J.P.; Good, Z.; Vasanawala, S.S.; Maecker, H.; Leipold, M.D.; Lin, D.T.S.; Kobor, M.S.; Horvath, S. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 2019, 18, e13028. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Recent advances in the discovery of senolytics. Mech. Ageing Dev. 2021, 200, 111587. [Google Scholar] [CrossRef]
- Kaur, J.; Farr, J.N. Cellular senescence in age-related disorders. Transl. Res. 2020, 226, 96–104. [Google Scholar] [CrossRef]
- Coppé, J.; Desprez, P.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Capell, B.C. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2017, 217, 65–77. [Google Scholar] [CrossRef]
- Campisi, J. Aging and Cancer: The Double-Edged Sword of Replicative Senescence. J. Am. Geriatr. Soc. 1997, 45, 482–488. [Google Scholar] [CrossRef]
- Zhao, S.; Qiao, Z.; Pfeifer, R.; Pape, H.; Mao, K.; Tang, H.; Meng, B.; Chen, S.; Liu, H. Modulation of fracture healing by senescence-associated secretory phenotype (SASP): A narrative review of the current literature. Eur. J. Med. Res. 2024, 29, 38. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Coryell, P.R.; Diekman, B.O.; Loeser, R.F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 2020, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Matta, C.; Zákány, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2014, 80, 237–244. [Google Scholar] [CrossRef]
- Galkin, F.; Kovalchuk, O.; Koldasbayeva, D.; Zhavoronkov, A.; Bischof, E. Stress, diet, exercise: Common environmental factors and their impact on epigenetic age. Ageing Res. Rev. 2023, 88, 101956. [Google Scholar] [CrossRef]
- Soto-Palma, C.; Niedernhofer, L.J.; Faulk, C.D.; Dong, X. Epigenetics, DNA damage, and aging. J. Clin. Investig. 2022, 132, e158446. [Google Scholar] [CrossRef]
- López-Gil, L.; Pascual-Ahuir, A.; Proft, M. Genomic Instability and Epigenetic Changes during Aging. Int. J. Mol. Sci. 2023, 24, 14279. [Google Scholar] [CrossRef]
- Zwamborn, R.; Slieker, R.C.; Mulder, P.C.A.; Zoetemelk, I.; Verschuren, L.; Suchiman, H.E.D.; Toet, K.H.; Droog, S.; Slagboom, P.E.; Kooistra, T.; et al. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Sci. Rep. 2017, 7, 43261. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiao, X.; Zheng, J.; Li, M.; Yu, M.; Ping, F.; Wang, T.; Wang, X. A Maternal High-Fat Diet Induces DNA Methylation Changes That Contribute to Glucose Intolerance in Offspring. Front. Endocrinol. 2019, 10, 871. [Google Scholar] [CrossRef]
- Parrillo, L.; Spinelli, R.; Nicolò, A.; Longo, M.; Mirra, P.; Raciti, G.A.; Miele, C.; Beguinot, F. Nutritional Factors, DNA Methylation, and Risk of Type 2 Diabetes and Obesity: Perspectives and Challenges. Int. J. Mol. Sci. 2019, 20, 2983. [Google Scholar] [CrossRef]
- Kenanoglu, S.; Gokce, N.; Akalin, H.; Ergoren, M.C.; Beccari, T.; Bertelli, M.; Dundar, M. Implication of the Mediterranean diet on the human epigenome. J. Prev. Med. Hyg. 2022, 63 Pt 2 (Suppl. 3), E44–E55. [Google Scholar] [CrossRef]
- Chen, Q.; Fan, R.; Song, L.; Wang, S.; You, M.; Cai, M.; Wu, Y.; Li, Y.; Xu, M. Association of Methyl Donor Nutrients’ Dietary Intake and Cognitive Impairment in the Elderly Based on the Intestinal Microbiome. Nutrients 2024, 16, 2061. [Google Scholar] [CrossRef] [PubMed]
- Samodien, E.; Pheiffer, C.; Erasmus, M.; Mabasa, L.; Louw, J.; Johnson, R. Diet-induced DNA methylation within the hypothalamic arcuate nucleus and dysregulated leptin and insulin signaling in the pathophysiology of obesity. Food Sci. Nutr. 2019, 7, 3131–3145. [Google Scholar] [CrossRef]
- Zong, D.; Liu, X.; Li, J.; Ouyang, R.; Chen, P. The role of cigarette smoke-induced epigenetic alterations in inflammation. Epigenet. Chromatin 2019, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Talikka, M.; Sierro, N.; Ivanov, N.V.; Chaudhary, N.; Peck, M.J.; Hoeng, J.; Coggins, C.R.E.; Peitsch, M.C. Genomic impact of cigarette smoke, with application to three smoking-related diseases. Crit. Rev. Toxicol. 2012, 42, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Gaskins, A.J.; Hood, R.B.; Ford, J.B.; Hauser, R.; Smith, A.K.; Everson, T.M. Former smoking associated with epigenetic modifications in human granulosa cells among women undergoing assisted reproduction. Sci. Rep. 2024, 14, 5009. [Google Scholar] [CrossRef]
- Skov-Jeppesen, S.M.; Kobylecki, C.J.; Jacobsen, K.K.; Bojesen, S.E. Changing Smoking Behavior and Epigenetics. Chest J. 2023, 163, 1565–1575. [Google Scholar] [CrossRef]
- McCartney, D.L.; Stevenson, A.J.; Hillary, R.F.; Walker, R.M.; Bermingham, M.L.; Morris, S.W.; Clarke, T.; Campbell, A.; Murray, A.D.; Whalley, H.C.; et al. Epigenetic signatures of starting and stopping smoking. EBioMedicine 2018, 37, 214–220. [Google Scholar] [CrossRef]
- Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Circadian disruption: Potential implications in inflammatory and metabolic diseases associated with alcohol. Alcohol Res. 2013, 35, 87–96. [Google Scholar]
- Potter, G.D.M.; Skene, D.J.; Arendt, J.; Cade, J.E.; Grant, P.J.; Hardie, L.J. Circadian Rhythm and Sleep Disruption: Causes, Metabolic Consequences, and Countermeasures. Endocr. Rev. 2016, 37, 584–608. [Google Scholar] [CrossRef]
- Vieira, E.; Mirizio, G.G.; Barin, G.R.; De Andrade, R.V.; Nimer, N.F.S.; La Sala, L. Clock Genes, Inflammation and the Immune System—Implications for Diabetes, Obesity and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9743. [Google Scholar] [CrossRef]
- Zeng, Y.; Guo, Z.; Wu, M.; Chen, F.; Chen, L. Circadian rhythm regulates the function of immune cells and participates in the development of tumors. Cell Death Discov. 2024, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wolff, S.E.C.; Korpel, N.; Milanova, I.; Sandu, C.; Rensen, P.C.N.; Kooijman, S.; Cassel, J.; Kalsbeek, A.; Boutillier, A.; et al. Deficiency of the Circadian Clock Gene Bmal1 Reduces Microglial Immunometabolism. Front. Immunol. 2020, 11, 586399. [Google Scholar] [CrossRef] [PubMed]
- Rasmi, Y.; Shokati, A.; Hassan, A.; Aziz, S.G.; Bastani, S.; Jalali, L.; Moradi, F.; Alipour, S. The role of DNA methylation in progression of neurological disorders and neurodegenerative diseases as well as the prospect of using DNA methylation inhibitors as therapeutic agents for such disorders. IBRO Neurosci. Rep. 2022, 14, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Sabot, D.; Lovegrove, R.; Stapleton, P. The association between sleep quality and telomere length: A systematic literature review. Brain Behav. Immun.–Health 2023, 28, 100577. [Google Scholar] [CrossRef]
- Fagiani, F.; Di Marino, D.; Romagnoli, A.; Travelli, C.; Voltan, D.; Di Cesare Mannelli, L.; Racchi, M.; Govoni, S.; Lanni, C. Molecular regulations of circadian rhythm and implications for physiology and diseases. Signal Transduct. Target. Ther. 2022, 7, 41. [Google Scholar] [CrossRef]
- Jiang, S.; Postovit, L.; Cattaneo, A.; Binder, E.B.; Aitchison, K.J. Epigenetic Modifications in Stress Response Genes Associated with Childhood Trauma. Front. Psychiatry 2019, 10, 808. [Google Scholar] [CrossRef]
- Gatta, E.; Saudagar, V.; Auta, J.; Grayson, D.R.; Guidotti, A. Epigenetic landscape of stress surfeit disorders: Key role for DNA methylation dynamics. Int. Rev. Neurobiol. 2020, 156, 127–183. [Google Scholar] [CrossRef]
- Suresh, S.; Singh, S.A.; Rushendran, R.; Vellapandian, C.; Prajapati, B. Alzheimer’s disease: The role of extrinsic factors in its development, an investigation of the environmental enigma. Front. Neurol. 2023, 14, 1303111. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Deng, G.; Nguyen, A.; Tanaka, H.; Matsuzaki, K.; Bell, I.; Mehta, K.R.; Terdiman, J.P.; Waldman, F.M.; Kakar, S.; Gum, J.; et al. Regional hypermethylation and global hypomethylation are associated with altered chromatin conformation and histone acetylation in colorectal cancer. Int. J. Cancer 2006, 118, 2999–3005. [Google Scholar] [CrossRef]
- Pappalardo, X.G.; Barra, V. Losing DNA methylation at repetitive elements and breaking bad. Epigenet. Chromatin 2021, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lopez, O.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Epigenetic signatures underlying inflammation: An interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 2020, 70, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, S.; Longhitano, L.; Denaro, S.; D’Aprile, S.; Torrisi, F.; La Spina, E.; Giallongo, C.; Mannino, G.; Lo Furno, D.; Zappalà, A.; et al. The Role of Epigenetics in Neuroinflammatory-Driven Diseases. Int. J. Mol. Sci. 2022, 23, 15218. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, C.; Rodrigues, C.E.; Homann, T.; Kleuser, B. The Role of Ten-Eleven Translocation Proteins in Inflammation. Front. Immunol. 2022, 13, 861351. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, R.; Haraguchi, R.; Kitazawa, S. Histone Modification in Histochemistry and Cytochemistry. Acta Histochem. Cytochem. 2023, 56, 41–47. [Google Scholar] [CrossRef]
- Hsu, C.; Lo, Y.; Kao, C. H3K4 Methylation in Aging and Metabolism. Epigenomes 2021, 5, 14. [Google Scholar] [CrossRef]
- Petruk, S.; Cai, J.; Sussman, R.; Sun, G.; Kovermann, S.K.; Mariani, S.A.; Calabretta, B.; McMahon, S.B.; Brock, H.W.; Iacovitti, L.; et al. Delayed Accumulation of H3K27me3 on Nascent DNA Is Essential for Recruitment of Transcription Factors at Early Stages of Stem Cell Differentiation. Mol. Cell 2017, 66, 247–257.e5. [Google Scholar] [CrossRef]
- Della Valle, F.; Reddy, P.; Yamamoto, M.; Liu, P.; Saera-Vila, A.; Bensaddek, D.; Zhang, H.; Martinez, J.P.; Abassi, L.; Celii, M.; et al. LINE-1 RNA causes heterochromatin erosion and is a target for amelioration of senescent phenotypes in progeroid syndromes. Sci. Transl. Med. 2022, 14, eabl6057. [Google Scholar] [CrossRef]
- Gorbunova, V.; Boeke, J.D.; Helfand, S.L.; Sedivy, J.M. Sleeping dogs of the genome. Science 2014, 346, 1187–1188. [Google Scholar] [CrossRef]
- Morrison, A.J. Chromatin-remodeling links metabolic signaling to gene expression. Mol. Metab. 2020, 38, 100973. [Google Scholar] [CrossRef]
- Narlikar, G.J.; Sundaramoorthy, R.; Owen-Hughes, T. Mechanisms and Functions of ATP-Dependent Chromatin-Remodeling Enzymes. Cell 2013, 154, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Weissman, L.; Jo, D.; Sorensen, M.M.; De Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef]
- Sepe, S.; Milanese, C.; Gabriels, S.; Derks, K.W.; Payan-Gomez, C.; Van IJcken, W.F.; Rijksen, Y.M.; Nigg, A.L.; Moreno, S.; Cerri, S.; et al. Inefficient DNA Repair Is an Aging-Related Modifier of Parkinson’s Disease. Cell Rep. 2016, 15, 1866–1875. [Google Scholar] [CrossRef]
- Bucholtz, N.; Demuth, I. DNA-repair in mild cognitive impairment and Alzheimer’s disease. DNA Repair 2013, 12, 811–816. [Google Scholar] [CrossRef]
- Obulesu, M.; Rao, D.M. DNA Damage and Impairment of DNA Repair in Alzheimer’s Disease. Int. J. Neurosci. 2010, 120, 397–403. [Google Scholar] [CrossRef]
- Sepe, S.; Payan-Gomez, C.; Milanese, C.; Hoeijmakers, J.H.; Mastroberardino, P.G. Nucleotide excision repair in chronic neurodegenerative diseases. DNA Repair 2013, 12, 568–577. [Google Scholar] [CrossRef]
- Li, X.; Bie, L.; Wang, Y.; Hong, Y.; Zhou, Z.; Fan, Y.; Yan, X.; Tao, Y.; Huang, C.; Zhang, Y.; et al. LINE-1 transcription activates long-range gene expression. Nat. Genet. 2024, 56, 1494–1502. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Baniahmad, A.; Branicki, W.; Taheri, M.; Eghbali, A. Emerging Role of Non-Coding RNAs in Senescence. Front. Cell Dev. Biol. 2022, 10, 869011. [Google Scholar] [CrossRef]
- Bushati, N.; Cohen, S.M. microRNA Functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef] [PubMed]
- Munkhzul, C.; Yi, S.S.; Kim, J.; Lee, S.; Kim, H.; Moon, J.; Lee, M. The microRNA-mediated gene regulatory network in the hippocampus and hypothalamus of the aging mouse. PLoS ONE 2023, 18, e0291943. [Google Scholar] [CrossRef] [PubMed]
- Karnati, H.K.; Panigrahi, M.K.; Gutti, R.K.; Greig, N.H.; Tamargo, I.A. miRNAs: Key Players in Neurodegenerative Disorders and Epilepsy. J. Alzheimer’s Dis. 2015, 48, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Varghese, L.N.; Schwenke, D.O.; Katare, R. Role of noncoding RNAs in cardiac ageing. Front. Cardiovasc. Med. 2023, 10, 1142575. [Google Scholar] [CrossRef]
- De Lencastre, A.; Pincus, Z.; Zhou, K.; Kato, M.; Lee, S.S.; Slack, F.J. MicroRNAs Both Promote and Antagonize Longevity in C. elegans. Curr. Biol. 2010, 20, 2159–2168. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Fang, L.; Xuan, J.; Li, H.; Khorshidi, A.; Gupta, S.; Li, X.; Yang, B.B. miR-17 extends mouse lifespan by inhibiting senescence signaling mediated by MKP7. Cell Death Dis. 2014, 5, e1355. [Google Scholar] [CrossRef]
- Gombar, S.; Jung, H.J.; Dong, F.; Calder, B.; Atzmon, G.; Barzilai, N.; Tian, X.; Pothof, J.; Hoeijmakers, J.H.; Campisi, J.; et al. Comprehensive microRNA profiling in B-cells of human centenarians by massively parallel sequencing. BMC Genom. 2012, 13, 353. [Google Scholar] [CrossRef]
- Serna, E.; Gambini, J.; Borras, C.; Abdelaziz, K.M.; Belenguer, A.; Sanchis, P.; Avellana, J.A.; Rodriguez-Mañas, L.; Viña, J. Centenarians, but not octogenarians, up-regulate the expression of microRNAs. Sci. Rep. 2012, 2, 961. [Google Scholar] [CrossRef]
- Schraml, E.; Grillari, J. From cellular senescence to age-associated diseases: The miRNA connection. Longev. Health 2012, 1, 10. [Google Scholar] [CrossRef]
- He, R.; Luo, D.; Mo, Y. Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes. Dis. 2019, 6, 6–15. [Google Scholar] [CrossRef]
- Bink, D.; Lozano-Vidal, N.; Boon, R. Long Non-Coding RNA in Vascular Disease and Aging. Non-Coding RNA 2019, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Ou, C.; Xiao, Y.; Han, Q.; Li, H.; Zhou, S. LncRNAs: Key players and novel insights into diabetes mellitus. Oncotarget 2017, 8, 71325–71341. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.P.; Bitar, M.; Jacobs, F.; Barry, G. Long Non-Coding RNAs in Neuronal Aging. Non-Coding RNA 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.T.E.; Pessoa, J.; Nóbrega-Pereira, S.; De Jesus, B.B. The Impact of Long Noncoding RNAs in Tissue Regeneration and Senescence. Cells 2024, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, J.; Xu, S.; Wang, J. Circulating noncoding RNAs: Promising biomarkers in liquid biopsy for the diagnosis, prognosis, and therapy of NSCLC. Discov. Oncol. 2023, 14, 142. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef]
- Logan, R.W.; McClung, C.A. Rhythms of life: Circadian disruption and brain disorders across the lifespan. Nat. Rev. Neurosci. 2018, 20, 49–65. [Google Scholar] [CrossRef]
- Calabrò, A.; Accardi, G.; Aiello, A.; Caruso, C.; Galimberti, D.; Candore, G. Senotherapeutics to Counteract Senescent Cells Are Prominent Topics in the Context of Anti-Ageing Strategies. Int. J. Mol. Sci. 2024, 25, 1792. [Google Scholar] [CrossRef]
- Ovadya, Y.; Krizhanovsky, V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 2014, 15, 627–642. [Google Scholar] [CrossRef]
- Aghababaei, F.; Hadidi, M. Recent Advances in Potential Health Benefits of Quercetin. Pharmaceuticals 2023, 16, 1020. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Mahmood, S.; Hilles, A.R.; Ali, A.; Khan, M.Z.; Zaidi, S.A.A.; Iqbal, Z.; Ge, Y. Quercetin as a Therapeutic Product: Evaluation of Its Pharmacological Action and Clinical Applications—A Review. Pharmaceuticals 2023, 16, 1631. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Tavenier, J.; Nehlin, J.O.; Houlind, M.B.; Rasmussen, L.J.; Tchkonia, T.; Kirkland, J.L.; Andersen, O.; Rasmussen, L.J.H. Fisetin as a Senotherapeutic Agent: Evidence and Perspectives for Age-Related Diseases. Mech. Ageing Dev. 2024, 222, 111995. [Google Scholar] [CrossRef]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2015, 15, 428–435. [Google Scholar] [CrossRef]
- Riessland, M.; Orr, M. Translating the Biology of Aging into New Therapeutics for Alzheimer’s Disease: Senolytics. J. Prev. Alzheimer’s Dis. 2023, 10, 633–646. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2015, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2022, 290, 1362–1383. [Google Scholar] [CrossRef] [PubMed]
- Al-Mansour, F.; Alraddadi, A.; He, B.; Saleh, A.; Poblocka, M.; Alzahrani, W.; Cowley, S.; Macip, S. Characterization of the HDAC/PI3K inhibitor CUDC-907 as a novel senolytic. Aging 2023, 15, 2373–2394. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Correia, F.P.; Alves, I.A.; Costa, M.; Gameiro, M.; Martins, A.P.; Saraiva, J.A. Epigenetic Reprogramming as a Key to Reverse Ageing and Increase Longevity. Ageing Res. Rev. 2024, 95, 102204. [Google Scholar] [CrossRef]
- Cipriano, A.; Moqri, M.; Maybury-Lewis, S.Y.; Rogers-Hammond, R.; De Jong, T.A.; Parker, A.; Rasouli, S.; Schöler, H.R.; Sinclair, D.A.; Sebastiano, V. Mechanisms, pathways and strategies for rejuvenation through epigenetic reprogramming. Nat. Aging 2023, 4, 14–26. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Suda, M.; Katsuumi, G.; Tchkonia, T.; Kirkland, J.L.; Minamino, T. Potential Clinical Implications of Senotherapies for Cardiovascular Disease. Circ. J. 2023, 88, 277–284. [Google Scholar] [CrossRef]
- Franzin, R.; Stasi, A.; Ranieri, E.; Netti, G.S.; Cantaluppi, V.; Gesualdo, L.; Stallone, G.; Castellano, G. Targeting Premature Renal Aging: From Molecular Mechanisms of Cellular Senescence to Senolytic Trials. Front. Pharmacol. 2021, 12, 630419. [Google Scholar] [CrossRef]
- Palmer, A.K.; Tchkonia, T.; Kirkland, J.L. Senolytics: Potential for Alleviating Diabetes and Its Complications. Endocrinology 2021, 162, bqab058. [Google Scholar] [CrossRef]
- Doolittle, M.L.; Monroe, D.G.; Farr, J.N.; Khosla, S. The role of senolytics in osteoporosis and other skeletal pathologies. Mech. Ageing Dev. 2021, 199, 111565. [Google Scholar] [CrossRef] [PubMed]
- Alsuraih, M.; O’Hara, S.P.; Woodrum, J.E.; Pirius, N.E.; LaRusso, N.F. Genetic or pharmacological reduction of cholangiocyte senescence improves inflammation and fibrosis in the Mdr2 mouse. JHEP Rep. 2021, 3, 100250. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, E.O.W.; Misra, A.; Netto, J.M.E.; Tchkonia, T.; Kirkland, J.L. Strategies for late phase preclinical and early clinical trials of senolytics. Mech. Ageing Dev. 2021, 200, 111591. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Prata, L.G.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Lee, P.J.; Benz, C.C.; Blood, P.; Börner, K.; Campisi, J.; Chen, F.; Daldrup-Link, H.; De Jager, P.; Ding, L.; Duncan, F.E.; et al. NIH SenNet Consortium to map senescent cells throughout the human lifespan to understand physiological health. Nat. Aging 2022, 2, 1090–1100. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Q.; Kirkland, J.L. Targeting senescent cells for a healthier longevity: The roadmap for an era of global aging. Life Med. 2022, 1, 103–119. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2019, 20, 89–106. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Drug | Mechanism of Action | Key Findings | Study Reference |
|---|---|---|---|
| Dasatinib | Pan-tyrosine kinase inhibitor | Reduces senescent cell burden; improved physical function in aged mice | [89] |
| Quercetin | Inhibits BCL-2 family proteins | Enhances apoptosis in senescent cells; improved healthspan in murine models | [91,92] |
| Fisetin | Flavonoid that targets multiple pathways | Induces apoptosis in endothelial cells; reduces markers of senescence | [94,95,96] |
| Navitoclax | BCL-2 family inhibitor | Induces apoptosis in specific senescent cell types; effective in models of osteoarthritis | [98] |
| FOXO4-DRI | Disrupts FOXO4-p53 interaction | Triggers apoptosis in senescent cells by releasing p53 into the cytosol | [99] |
| Piperlongumine | Induces reactive oxygen species (ROS) in senescent cells | Selectively induces senescent cell death via ROS-mediated pathways; reduces senescent markers in vivo. | [99] |
| ABT-263 | BCL-2/BCL-xL inhibitor | Targets BCL-2/BCL-xL pathways to induce apoptosis in senescent cells; effective in reducing senescence in hematopoietic stem cells. | [100] |
| D + Q (Dasatinib + Quercetin) | Combination therapy targeting multiple senescent pathways | Improved cardiovascular function and extended healthspan in preclinical models of aged mice. | [89,93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burdusel, D.; Doeppner, T.R.; Surugiu, R.; Hermann, D.M.; Olaru, D.G.; Popa-Wagner, A. The Intersection of Epigenetics and Senolytics in Mechanisms of Aging and Therapeutic Approaches. Biomolecules 2025, 15, 18. https://doi.org/10.3390/biom15010018
Burdusel D, Doeppner TR, Surugiu R, Hermann DM, Olaru DG, Popa-Wagner A. The Intersection of Epigenetics and Senolytics in Mechanisms of Aging and Therapeutic Approaches. Biomolecules. 2025; 15(1):18. https://doi.org/10.3390/biom15010018
Chicago/Turabian StyleBurdusel, Daiana, Thorsten R. Doeppner, Roxana Surugiu, Dirk M. Hermann, Denissa Greta Olaru, and Aurel Popa-Wagner. 2025. "The Intersection of Epigenetics and Senolytics in Mechanisms of Aging and Therapeutic Approaches" Biomolecules 15, no. 1: 18. https://doi.org/10.3390/biom15010018
APA StyleBurdusel, D., Doeppner, T. R., Surugiu, R., Hermann, D. M., Olaru, D. G., & Popa-Wagner, A. (2025). The Intersection of Epigenetics and Senolytics in Mechanisms of Aging and Therapeutic Approaches. Biomolecules, 15(1), 18. https://doi.org/10.3390/biom15010018