
The Role of p53 Mutations in Early and Late Response to Mitotic Aberrations

Abstract
:1. Introduction
2. The Function of p53
The Canonical Model of p53 Activation
3. TP53 Mutations
3.1. Missense Mutations
3.2. Gain of Function Mutation
3.3. TP53 Mutation Dominant-Negative Effect
3.4. The Complexity of Gain-of-Function p53 Mutations
3.5. Isoforms, Frameshift, or Splice Mutations of TP53
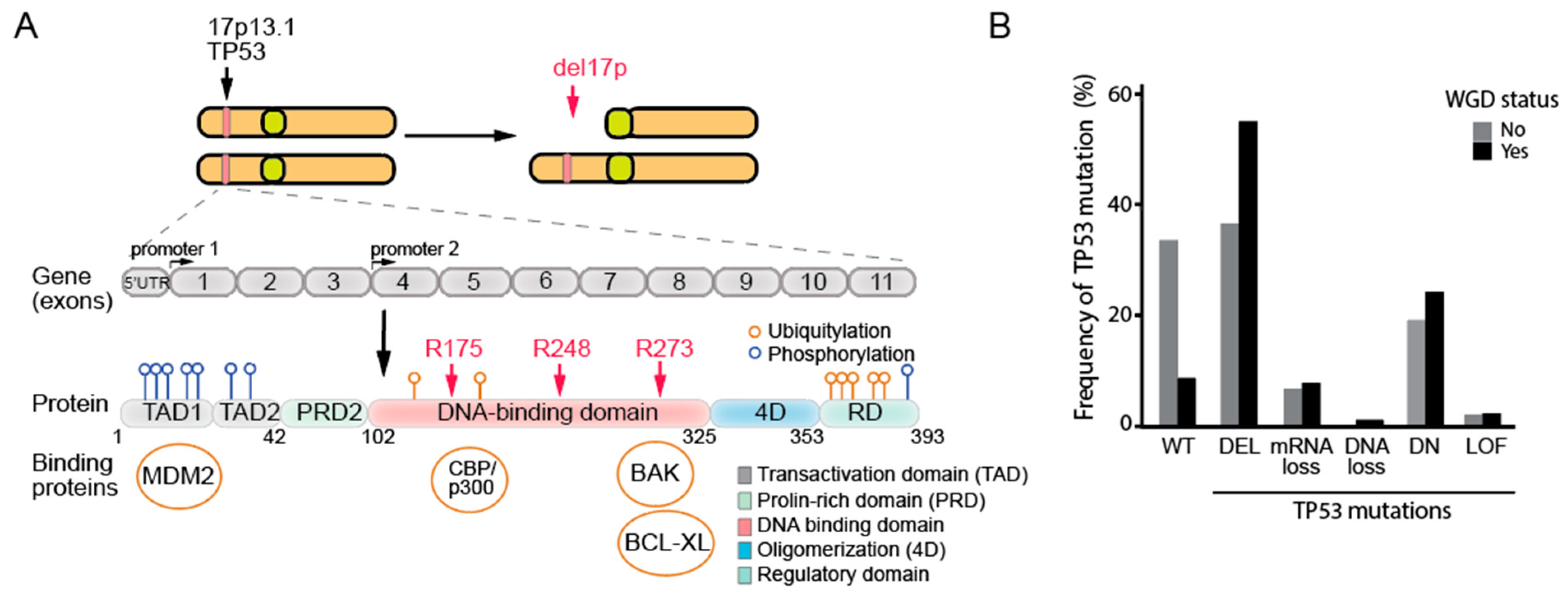

3.6. The Deletion of the 17p Arm and Its Relevance to the Loss of p53 in Human Cells
4. TP53 Mutations Are Strongly Linked to Aneuploidy and Whole-Genome Doubling in Cancer Genomes
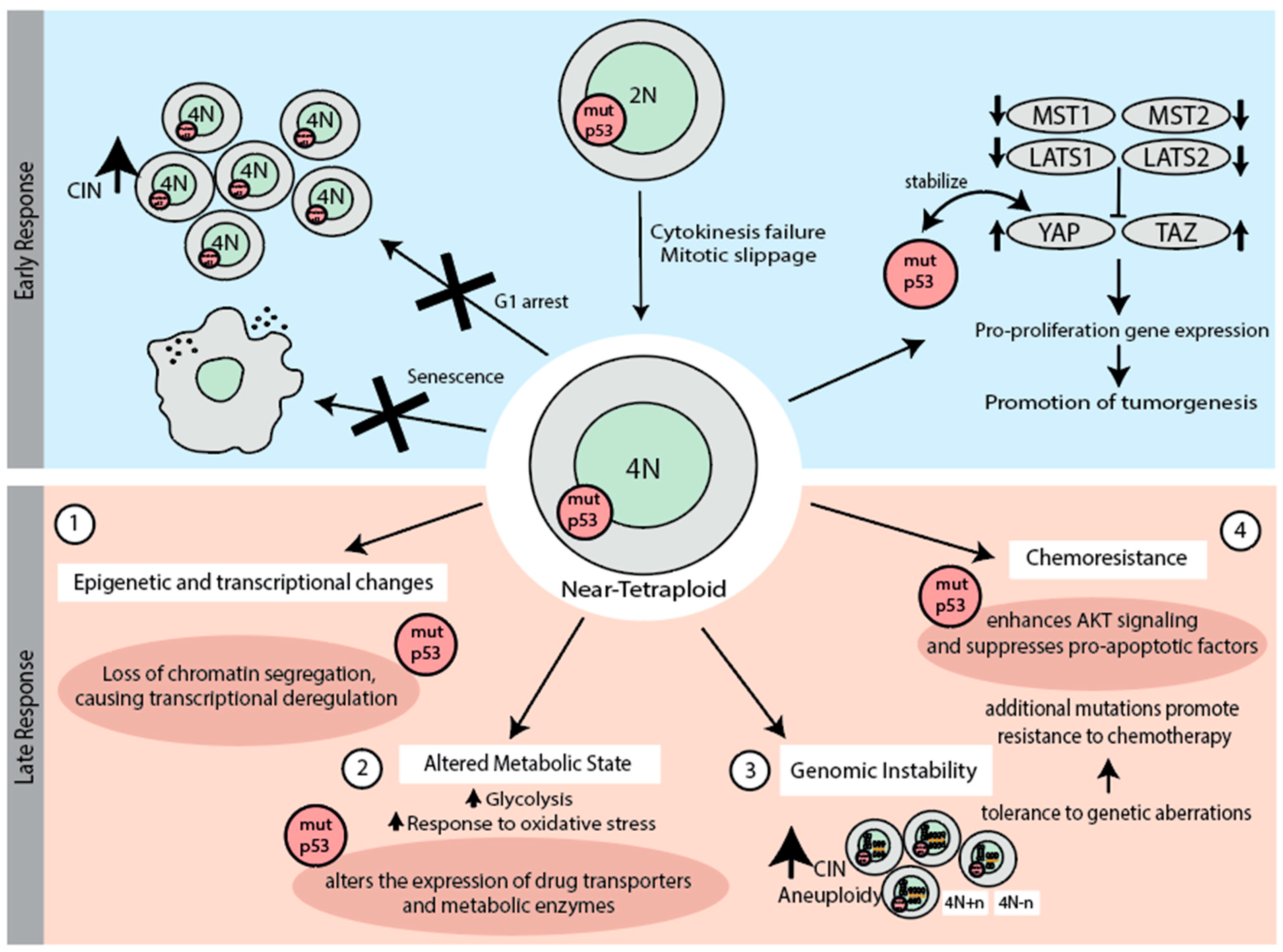
5. Early Response to Chromosome Missegregation and Whole-Genome Duplication
5.1. p53 in the Early Response to Chromosome Missegregation
5.2. p53 in the Early Response to Whole-Genome Doubling
6. Late Response to Whole-Genome Duplication (WGD) and the Role of p53 Mutations
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weinberg, R.A. How TP53 (almost) became an oncogene. J. Mol. Cell Biol. 2019, 11, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Stiewe, T.; Soussi, T. TP53: The unluckiest of genes? Cell Death Differ. 2024. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. The most popular genes in the human genome. Nature 2017, 551, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Baslan, T.; Morris, J.P.; Zhao, Z.; Reyes, J.; Ho, Y.J.; Tsanov, K.M.; Bermeo, J.; Tian, S.; Zhang, S.; Askan, G.; et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature 2022, 608, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Hwang, B.J.; Ford, J.M.; Hanawalt, P.C.; Chu, G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. USA 1999, 96, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef]
- Yu, X.; Riley, T.; Levine, A.J. The regulation of the endosomal compartment by p53 the tumor suppressor gene. FEBS J. 2009, 276, 2201–2212. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell 2009, 136, 378. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Hasty, P.; Sharp, Z.D.; Curiel, T.J.; Campisi, J. mTORC1 and p53: Clash of the gods? Cell Cycle 2013, 12, 20–25. [Google Scholar] [CrossRef]
- Kulawiec, M.; Ayyasamy, V.; Singh, K.K. p53 regulates mtDNA copy number and mitocheckpoint pathway. J. Carcinog. 2009, 8, 8. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Fischer, M.; Sammons, M.A. Determinants of p53 DNA binding, gene regulation, and cell fate decisions. Cell Death Differ. 2024, 31, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Meulmeester, E.; Pereg, Y.; Shiloh, Y.; Jochemsen, A.G. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle 2005, 4, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Appella, E.; Anderson, C.W. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 2001, 268, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Earle, J.; Saito, S.; Anderson, C.W.; Appella, E.; Xu, Y. Mutation of mouse p53 Ser23 and the response to DNA damage. Mol. Cell Biol. 2002, 22, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Serra, F.; Nieto-Aliseda, A.; Fanlo-Escudero, L.; Rovirosa, L.; Cabrera-Pasadas, M.; Lazarenkov, A.; Urmeneta, B.; Alcalde-Merino, A.; Nola, E.M.; Okorokov, A.L.; et al. p53 rapidly restructures 3D chromatin organization to trigger a transcriptional response. Nat. Commun. 2024, 15, 2821. [Google Scholar] [CrossRef]
- Kim, H.M.; Zheng, X.; Lee, E. Experimental Insights into the Interplay between Histone Modifiers and p53 in Regulating Gene Expression. Int. J. Mol. Sci. 2023, 24, 11032. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kim, J.W.; Seo, T.; Hwang, S.G.; Choi, E.J.; Choe, J. SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J. Biol. Chem. 2002, 277, 22330–22337. [Google Scholar] [CrossRef]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Kim, J.; Roeder, R.G. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 2004, 117, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.A.; Andrysik, Z.; Dengler, V.L.; Mellert, H.S.; Guarnieri, A.; Freeman, J.A.; Sullivan, K.D.; Galbraith, M.D.; Luo, X.; Kraus, W.L.; et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife 2014, 3, e02200. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Wang, X.; Hu, X.; Fang, S.; Jin, G.; Liu, K.; Dong, Z. Mutations found in cancer patients compromise DNA binding of the winged helix protein STK19. Sci. Rep. 2024, 14, 14098. [Google Scholar] [CrossRef]
- Laptenko, O.; Prives, C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; Zhu, Y.; Dwight, T.; Yu, B.; Dickson, K.A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Gill, A.J.; Clifton-Bligh, R.; et al. Comprehensive analyses of somatic TP53 mutation in tumors with variable mutant allele frequency. Sci. Data 2017, 4, 170120. [Google Scholar] [CrossRef]
- Kratz, C.P.; Freycon, C.; Maxwell, K.N.; Nichols, K.E.; Schiffman, J.D.; Evans, D.G.; Achatz, M.I.; Savage, S.A.; Weitzel, J.N.; Garber, J.E.; et al. Analysis of the Li-Fraumeni Spectrum Based on an International Germline TP53 Variant Data Set: An International Agency for Research on Cancer TP53 Database Analysis. JAMA Oncol. 2021, 7, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, H.; Tal, P.; Goldfinger, N.; Chattopadhyay, E.; Malkin, D.; Rotter, V.; Attery, A. Mutant p53 reactivation restricts the protumorigenic consequences of wild type p53 loss of heterozygosity in Li-Fraumeni syndrome patient-derived fibroblasts. Cell Death Differ. 2024, 31, 855–867. [Google Scholar] [CrossRef]
- Shetzer, Y.; Kagan, S.; Koifman, G.; Sarig, R.; Kogan-Sakin, I.; Charni, M.; Kaufman, T.; Zapatka, M.; Molchadsky, A.; Rivlin, N.; et al. The onset of p53 loss of heterozygosity is differentially induced in various stem cell types and may involve the loss of either allele. Cell Death Differ. 2014, 21, 1419–1431. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, M.; Amin, M.K.; Daver, N.G.; Shah, M.V.; Hiwase, D.; Arber, D.A.; Kharfan-Dabaja, M.A.; Badar, T. What have we learned about TP53-mutated acute myeloid leukemia? Blood Cancer J. 2024, 14, 202. [Google Scholar] [CrossRef]
- Shajani-Yi, Z.; de Abreu, F.B.; Peterson, J.D.; Tsongalis, G.J. Frequency of Somatic TP53 Mutations in Combination with Known Pathogenic Mutations in Colon Adenocarcinoma, Non-Small Cell Lung Carcinoma, and Gliomas as Identified by Next-Generation Sequencing. Neoplasia 2018, 20, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar] [CrossRef]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Roszkowska, K.A.; Gizinski, S.; Sady, M.; Gajewski, Z.; Olszewski, M.B. Gain-of-Function Mutations in p53 in Cancer Invasiveness and Metastasis. Int. J. Mol. Sci. 2020, 21, 1334. [Google Scholar] [CrossRef] [PubMed]
- Lepre, M.G.; Omar, S.I.; Grasso, G.; Morbiducci, U.; Deriu, M.A.; Tuszynski, J.A. Insights into the Effect of the G245S Single Point Mutation on the Structure of p53 and the Binding of the Protein to DNA. Molecules 2017, 22, 1358. [Google Scholar] [CrossRef]
- Chiang, Y.T.; Chien, Y.C.; Lin, Y.H.; Wu, H.H.; Lee, D.F.; Yu, Y.L. The Function of the Mutant p53-R175H in Cancer. Cancers 2021, 13, 4088. [Google Scholar] [CrossRef]
- Dong, P.; Xu, Z.; Jia, N.; Li, D.; Feng, Y. Elevated expression of p53 gain-of-function mutation R175H in endometrial cancer cells can increase the invasive phenotypes by activation of the EGFR/PI3K/AKT pathway. Mol. Cancer 2009, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Annor, G.K.; Elshabassy, N.; Lundine, D.; Conde, D.G.; Xiao, G.; Ellison, V.; Bargonetti, J. Oligomerization of Mutant p53 R273H is not Required for Gain-of-Function Chromatin Associated Activities. Front. Cell Dev. Biol. 2021, 9, 772315. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, B.; Miao, L.; Mei, Y.; Wu, M. Mutant p53-R273H gains new function in sustained activation of EGFR signaling via suppressing miR-27a expression. Cell Death Dis. 2013, 4, e574. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Lundine, D.; Annor, G.K.; Canar, J.; Ellison, V.; Polotskaia, A.; Donabedian, P.L.; Reiner, T.; Khramtsova, G.F.; Olopade, O.I.; et al. Gain-of-Function Mutant p53 R273H Interacts with Replicating DNA and PARP1 in Breast Cancer. Cancer Res. 2020, 80, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.Y.; Tsai, K.Y.; Chang, S.J.; Chuang, Y.J. Gain-of-Function Mutant TP53 R248Q Overexpressed in Epithelial Ovarian Carcinoma Alters AKT-Dependent Regulation of Intercellular Trafficking in Responses to EGFR/MDM2 Inhibitor. Int. J. Mol. Sci. 2021, 22, 8784. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, M.B.; Pruszko, M.; Snaar-Jagalska, E.; Zylicz, A.; Zylicz, M. Diverse and cancer type-specific roles of the p53 R248Q gain-of-function mutation in cancer migration and invasiveness. Int. J. Oncol. 2019, 54, 1168–1182. [Google Scholar] [CrossRef]
- Nakazawa, S.; Sakata, K.I.; Liang, S.; Yoshikawa, K.; Iizasa, H.; Tada, M.; Hamada, J.I.; Kashiwazaki, H.; Kitagawa, Y.; Yamazaki, Y. Dominant-negative p53 mutant R248Q increases the motile and invasive activities of oral squamous cell carcinoma cells. Biomed. Res. 2019, 40, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Hamada, J.; Tada, M.; Kameyama, T.; Nakagawa, K.; Suzuki, Y.; Ikawa, M.; Hassan, N.M.; Kitagawa, Y.; Moriuchi, T. Mutant p53 R248Q but not R248W enhances in vitro invasiveness of human lung cancer NCI-H1299 cells. Biomed. Res. 2010, 31, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Gouas, D.A.; Shi, H.; Hautefeuille, A.H.; Ortiz-Cuaran, S.L.; Legros, P.C.; Szymanska, K.J.; Galy, O.; Egevad, L.A.; Abedi-Ardekani, B.; Wiman, K.G.; et al. Effects of the TP53 p.R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: Interaction with hepatitis B virus X protein. Carcinogenesis 2010, 31, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liao, P.; Zeng, S.X.; Lu, H. It takes a team: A gain-of-function story of p53-R249S. J. Mol. Cell Biol. 2019, 11, 277–283. [Google Scholar] [CrossRef]
- Scian, M.J.; Stagliano, K.E.; Ellis, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Modulation of gene expression by tumor-derived p53 mutants. Cancer Res. 2004, 64, 7447–7454. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, T.; Gleber-Netto, F.O.; Chen, Z.; McGrail, D.J.; Gomez, J.A.; Ju, W.; Gadhikar, M.A.; Ma, W.; Shen, L.; et al. Mutant p53 gains oncogenic functions through a chromosomal instability-induced cytosolic DNA response. Nat. Commun. 2024, 15, 180. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Vaughan, C.A.; Frum, R.A.; Grossman, S.R.; Deb, S.; Palit Deb, S. Mutant p53 establishes targetable tumor dependency by promoting unscheduled replication. J. Clin. Investig. 2017, 127, 1839–1855. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.; Medcalf, E.A. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991, 65, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Sorrell, A.D.; Espenschied, C.R.; Culver, J.O.; Weitzel, J.N. Tumor protein p53 (TP53) testing and Li-Fraumeni syndrome: Current status of clinical applications and future directions. Mol. Diagn. Ther. 2013, 17, 31–47. [Google Scholar] [CrossRef] [PubMed]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant p53 oncogenicity: Dominant-negative or gain-of-function? Carcinogenesis 2020, 41, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef]
- Aschauer, L.; Muller, P.A. Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem. Soc. Trans. 2016, 44, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Bargonetti, J.; Prives, C. Gain-of-function mutant p53: History and speculation. J. Mol. Cell Biol. 2019, 11, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Bougeard, G.; Sesboue, R.; Baert-Desurmont, S.; Vasseur, S.; Martin, C.; Tinat, J.; Brugieres, L.; Chompret, A.; de Paillerets, B.B.; Stoppa-Lyonnet, D.; et al. Molecular basis of the Li-Fraumeni syndrome: An update from the French LFS families. J. Med. Genet. 2008, 45, 535–538. [Google Scholar] [CrossRef]
- Doyle, B.; Morton, J.P.; Delaney, D.W.; Ridgway, R.A.; Wilkins, J.A.; Sansom, O.J. p53 mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J. Pathol. 2010, 222, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.L.; Olive, K.P.; Tuveson, D.A.; Bronson, R.; Crowley, D.; Brown, M.; Jacks, T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005, 65, 10280–10288. [Google Scholar] [CrossRef] [PubMed]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef]
- Lee, M.K.; Teoh, W.W.; Phang, B.H.; Tong, W.M.; Wang, Z.Q.; Sabapathy, K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell 2012, 22, 751–764. [Google Scholar] [CrossRef]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef]
- Redman-Rivera, L.N.; Shaver, T.M.; Jin, H.; Marshall, C.B.; Schafer, J.M.; Sheng, Q.; Hongo, R.A.; Beckermann, K.E.; Wheeler, F.C.; Lehmann, B.D.; et al. Acquisition of aneuploidy drives mutant p53-associated gain-of-function phenotypes. Nat. Commun. 2021, 12, 5184. [Google Scholar] [CrossRef]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Soussi, T.; Leroy, B.; Taschner, P.E. Recommendations for analyzing and reporting TP53 gene variants in the high-throughput sequencing era. Hum. Mutat. 2014, 35, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Marchini, S.; Marabese, M.; Marrazzo, E.; Mariani, P.; Cattaneo, D.; Fossati, R.; Compagnoni, A.; Fruscio, R.; Lissoni, A.A.; Broggini, M. DeltaNp63 expression is associated with poor survival in ovarian cancer. Ann. Oncol. 2008, 19, 501–507. [Google Scholar] [CrossRef]
- Hofstetter, G.; Berger, A.; Fiegl, H.; Slade, N.; Zoric, A.; Holzer, B.; Schuster, E.; Mobus, V.J.; Reimer, D.; Daxenbichler, G.; et al. Alternative splicing of p53 and p73: The novel p53 splice variant p53delta is an independent prognostic marker in ovarian cancer. Oncogene 2010, 29, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R.J. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.A.; Ruano, D.; Joruiz, S.M.; Stroosma, J.; Glavak, N.; Montali, A.; Pinto, L.M.; Rodriguez-Girondo, M.; Barge-Schaapveld, D.; Nielsen, M.; et al. Germline variant affecting p53beta isoforms predisposes to familial cancer. Nat. Commun. 2024, 15, 8208. [Google Scholar] [CrossRef]
- Kikutake, C.; Suyama, M. Possible involvement of silent mutations in cancer pathogenesis and evolution. Sci. Rep. 2023, 13, 7593. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef]
- Butera, A.; Amelio, I. Deciphering the significance of p53 mutant proteins. Trends Cell Biol. 2024. [Google Scholar] [CrossRef]
- Broad DepMap. DepMap 24Q4 Public. 2024. Available online: https://plus.figshare.com/articles/dataset/DepMap_24Q4_Public/27993248/1 (accessed on 30 January 2025).
- Sonkin, D.; Hassan, M.; Murphy, D.J.; Tatarinova, T.V. Tumor suppressors status in cancer cell line Encyclopedia. Mol. Oncol. 2013, 7, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef]
- Folkins, A.K.; Jarboe, E.A.; Saleemuddin, A.; Lee, Y.; Callahan, M.J.; Drapkin, R.; Garber, J.E.; Muto, M.G.; Tworoger, S.; Crum, C.P. A candidate precursor to pelvic serous cancer (p53 signature) and its prevalence in ovaries and fallopian tubes from women with BRCA mutations. Gynecol. Oncol. 2008, 109, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, X.; Li, S.; Pan, X.; Gong, Y.; Zheng, J.; Xu, J.; Zhao, C.; Zhang, Q.; Zhang, S.; et al. An Epigenetic Mechanism Underlying Chromosome 17p Deletion-Driven Tumorigenesis. Cancer Discov. 2021, 11, 194–207. [Google Scholar] [CrossRef]
- Kumari, L.; Sreedharanunni, S.; Dahiya, D.; Dey, P.; Bhatia, A. High prevalence of chromosome 17 in breast cancer micronuclei: A means to get rid of tumor suppressors? Hum. Cell 2024, 38, 5. [Google Scholar] [CrossRef]
- Daver, N.G.; Iqbal, S.; Huang, J.; Renard, C.; Lin, J.; Pan, Y.; Williamson, M.; Ramsingh, G. Clinical characteristics and overall survival among acute myeloid leukemia patients with TP53 gene mutation or chromosome 17p deletion. Am. J. Hematol. 2023, 98, 1176–1184. [Google Scholar] [CrossRef]
- Froudarakis, M.E.; Sourvinos, G.; Fournel, P.; Bouros, D.; Vergnon, J.M.; Spandidos, D.A.; Siafakas, N.M. Microsatellite instability and loss of heterozygosity at chromosomes 9 and 17 in non-small cell lung cancer. Chest 1998, 113, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.M.; Kida, Y.; Zimmerman, P.V.; Ikenaga, M.; Smith, P.J. Loss of heterozygosity frequently affects chromosome 17q in non-small cell lung cancer. Cancer Res. 1995, 55, 4268–4272. [Google Scholar] [PubMed]
- Campo, E.; Cymbalista, F.; Ghia, P.; Jager, U.; Pospisilova, S.; Rosenquist, R.; Schuh, A.; Stilgenbauer, S. TP53 aberrations in chronic lymphocytic leukemia: An overview of the clinical implications of improved diagnostics. Haematologica 2018, 103, 1956–1968. [Google Scholar] [CrossRef] [PubMed]
- Bomben, R.; Rossi, F.M.; Vit, F.; Bittolo, T.; Zucchetto, A.; Papotti, R.; Tissino, E.; Pozzo, F.; Degan, M.; Polesel, J.; et al. Clinical impact of TP53 disruption in chronic lymphocytic leukemia patients treated with ibrutinib: A campus CLL study. Leukemia 2023, 37, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Pavlova, S.; Baliakas, P.; Chatzikonstantinou, T.; Tausch, E.; Catherwood, M.; Rossi, D.; Soussi, T.; Tichy, B.; Kater, A.P.; et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-2024 update. Leukemia 2024, 38, 1455–1468. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Xu, H.; Jiang, G.; Van der Jeught, K.; Fang, Y.; Zhou, Z.; Zhang, L.; Frieden, M.; Wang, L.; et al. Heterozygous deletion of chromosome 17p renders prostate cancer vulnerable to inhibition of RNA polymerase II. Nat. Commun. 2018, 9, 4394. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e673. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Sen, O.; Harrison, J.U.; Burroughs, N.J.; McAinsh, A.D. Kinetochore life histories reveal an Aurora-B-dependent error correction mechanism in anaphase. Dev. Cell 2021, 56, 3082–3099.E5. [Google Scholar] [CrossRef] [PubMed]
- Sadagopan, A.; Garaffo, N.; Chang, H.-J.; Schreiber, S.L.; Meyerson, M.; Gibson, W.J. p53 protein abundance is a therapeutic window across TP53 mutant cancers and is targetable with proximity inducing small molecules. bioRxiv 2024. [Google Scholar] [CrossRef]
- Stiewe, T.; Haran, T.E. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist. Updat. 2018, 38, 27–43. [Google Scholar] [CrossRef]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Narkar, A.; Johnson, B.A.; Bharne, P.; Zhu, J.; Padmanaban, V.; Biswas, D.; Fraser, A.; Iglesias, P.A.; Ewald, A.J.; Li, R. On the role of p53 in the cellular response to aneuploidy. Cell Rep. 2021, 34, 108892. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, S.; Richardson, A.; Iyer, D.R.; M’Saad, O.; Zasadil, L.; Knouse, K.A.; Wong, Y.L.; Rhind, N.; Desai, A.; Amon, A. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev. Cell 2017, 41, 638–651.E5. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.; Raaijmakers, J.A.; Bakker, B.; Spierings, D.C.J.; Lansdorp, P.M.; Foijer, F.; Medema, R.H. p53 Prohibits Propagation of Chromosome Segregation Errors that Produce Structural Aneuploidies. Cell Rep. 2017, 19, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Meitinger, F.; Belal, H.; Davis, R.L.; Martinez, M.B.; Shiau, A.K.; Oegema, K.; Desai, A. Control of cell proliferation by memories of mitosis. Science 2024, 383, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.S.; Mazo, G.; Das, T.; Goodman, J.; Kim, M.; O’Rourke, B.P.; Izquierdo, D.; Tsou, M.F. 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 2016, 5, e16270. [Google Scholar] [CrossRef]
- Marques, J.F.; Kops, G. Permission to pass: On the role of p53 as a gatekeeper for aneuploidy. Chromosome Res. 2023, 31, 31. [Google Scholar] [CrossRef]
- Hinchcliffe, E.H.; Day, C.A.; Karanjeet, K.B.; Fadness, S.; Langfald, A.; Vaughan, K.T.; Dong, Z. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat. Cell Biol. 2016, 18, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Giunta, S.; Belotserkovskaya, R.; Jackson, S.P. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 2010, 190, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Kabeche, L.; Murnane, J.P.; Zaki, B.I.; Compton, D.A. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 2014, 4, 1281–1289. [Google Scholar] [CrossRef]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Vaddavalli, P.L.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022, 38, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Dalton, W.B.; Yu, B.; Yang, V.W. p53 suppresses structural chromosome instability after mitotic arrest in human cells. Oncogene 2010, 29, 1929–1940. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Baek, K.H.; Shin, H.J.; Yoo, J.K.; Cho, J.H.; Choi, Y.H.; Sung, Y.C.; McKeon, F.; Lee, C.W. p53 deficiency and defective mitotic checkpoint in proliferating T lymphocytes increase chromosomal instability through aberrant exit from mitotic arrest. J. Leukoc. Biol. 2003, 73, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Hixon, M.L.; Flores, A.; Wagner, M.; Gualberto, A. Gain of function properties of mutant p53 proteins at the mitotic spindle cell cycle checkpoint. Histol. Histopathol. 2000, 15, 551–556. [Google Scholar] [CrossRef]
- Chen, G.; Li, Z.; Iemura, K.; Tanaka, K. Oxidative stress induces chromosomal instability through replication stress in fibroblasts from aged mice. J. Cell Sci. 2023, 136, jcs260688. [Google Scholar] [CrossRef] [PubMed]
- Andrs, M.; Stoy, H.; Boleslavska, B.; Chappidi, N.; Kanagaraj, R.; Nascakova, Z.; Menon, S.; Rao, S.; Oravetzova, A.; Dobrovolna, J.; et al. Excessive reactive oxygen species induce transcription-dependent replication stress. Nat. Commun. 2023, 14, 1791. [Google Scholar] [CrossRef]
- Mihalas, B.P.; Marston, A.L.; Wu, L.E.; Gilchrist, R.B. Reproductive Ageing: Metabolic contribution to age-related chromosome missegregation in mammalian oocytes. Reproduction 2024, 168, e230510. [Google Scholar] [CrossRef]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol. Cell Biol. 2017, 37, e00328-17. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Gronroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef]
- Kim, J.E.; Choi, J.; Sung, C.O.; Hong, Y.S.; Kim, S.Y.; Lee, H.; Kim, T.W.; Kim, J.I. High prevalence of TP53 loss and whole-genome doubling in early-onset colorectal cancer. Exp. Mol. Med. 2021, 53, 446–456. [Google Scholar] [CrossRef]
- Davoli, T.; Denchi, E.L.; de Lange, T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef]
- Zeng, J.; Hills, S.A.; Ozono, E.; Diffley, J.F.X. Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication. Cell 2023, 186, 528–542.E14. [Google Scholar] [CrossRef] [PubMed]
- Funk, L.C.; Wan, J.; Ryan, S.D.; Kaur, C.; Sullivan, R.; Roopra, A.; Weaver, B.A. p53 Is Not Required for High CIN to Induce Tumor Suppression. Mol. Cancer Res. 2021, 19, 112–123. [Google Scholar] [CrossRef]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Kienitz, A.; Hofmann, I.; Muller, R.; Bastians, H. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene 2004, 23, 6845–6853. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9819–9824. [Google Scholar] [CrossRef]
- Ganem, N.J.; Cornils, H.; Chiu, S.Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Thery, M.; Camargo, F.D.; Pellman, D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef]
- Fava, L.L.; Schuler, F.; Sladky, V.; Haschka, M.D.; Soratroi, C.; Eiterer, L.; Demetz, E.; Weiss, G.; Geley, S.; Nigg, E.A.; et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017, 31, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Wilsker, D.; Chung, J.H.; Bunz, F. Chk1 suppresses bypass of mitosis and tetraploidization in p53-deficient cancer cells. Cell Cycle 2012, 11, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Fauth, C.; Speicher, M.R.; Dutriaux, A.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res. 2002, 62, 1129–1133. [Google Scholar] [PubMed]
- De Santis Puzzonia, M.; Gonzalez, L.; Ascenzi, S.; Cundari, E.; Degrassi, F. Tetraploid cells produced by absence of substrate adhesion during cytokinesis are limited in their proliferation and enter senescence after DNA replication. Cell Cycle 2016, 15, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, I.; Park, K.Y.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Delta133p53 represses p53-inducible senescence genes and enhances the generation of human induced pluripotent stem cells. Cell Death Differ. 2017, 24, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Potapova, T.A.; Seidel, C.W.; Box, A.C.; Rancati, G.; Li, R. Transcriptome analysis of tetraploid cells identifies cyclin D2 as a facilitator of adaptation to genome doubling in the presence of p53. Mol. Biol. Cell 2016, 27, 3065–3084. [Google Scholar] [CrossRef]
- Bernhard, S.V.; Seget-Trzensiok, K.; Kuffer, C.; Krastev, D.B.; Stautmeister, L.M.; Theis, M.; Keuper, K.; Boekenkamp, J.E.; Kschischo, M.; Buchholz, F.; et al. Loss of USP28 and SPINT2 expression promotes cancer cell survival after whole genome doubling. Cell. Oncol. 2022, 45, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Bloomfield, M.; Levi, H.; Keuper, K.; Bernhard, S.V.; Baudoin, N.C.; Leor, G.; Eliezer, Y.; Giam, M.; Wong, C.K.; et al. Whole-Genome Duplication Shapes the Aneuploidy Landscape of Human Cancers. Cancer Res. 2022, 82, 1736–1752. [Google Scholar] [CrossRef] [PubMed]
- Lambuta, R.A.; Nanni, L.; Liu, Y.; Diaz-Miyar, J.; Iyer, A.; Tavernari, D.; Katanayeva, N.; Ciriello, G.; Oricchio, E. Whole-genome doubling drives oncogenic loss of chromatin segregation. Nature 2023, 615, 925–933. [Google Scholar] [CrossRef]
- Gemble, S.; Wardenaar, R.; Keuper, K.; Srivastava, N.; Nano, M.; Mace, A.S.; Tijhuis, A.E.; Bernhard, S.V.; Spierings, D.C.J.; Simon, A.; et al. Genetic instability from a single S phase after whole-genome duplication. Nature 2022, 604, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Sidana, S.; Jevremovic, D.; Ketterling, R.P.; Tandon, N.; Greipp, P.T.; Baughn, L.B.; Dispenzieri, A.; Gertz, M.A.; Rajkumar, S.V.; Kumar, S.K. Tetraploidy is associated with poor prognosis at diagnosis in multiple myeloma. Am. J. Hematol. 2019, 94, E117–E120. [Google Scholar] [CrossRef]
- Locher, M.; Jukic, E.; Vogi, V.; Keller, M.A.; Kroll, T.; Schwendinger, S.; Oberhuber, K.; Verdorfer, I.; Muhlegger, B.E.; Witsch-Baumgartner, M.; et al. Amp(1q) and tetraploidy are commonly acquired chromosomal abnormalities in relapsed multiple myeloma. Eur. J. Haematol. 2023, 110, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef]
- Senovilla, L.; Vitale, I.; Galluzzi, L.; Vivet, S.; Joza, N.; Younes, A.B.; Rello-Varona, S.; Castedo, M.; Kroemer, G. p53 represses the polyploidization of primary mammary epithelial cells by activating apoptosis. Cell Cycle 2009, 8, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Scian, M.J.; Stagliano, K.E.; Anderson, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol. Cell Biol. 2005, 25, 10097–10110. [Google Scholar] [CrossRef]
- Masciarelli, S.; Fontemaggi, G.; Di Agostino, S.; Donzelli, S.; Carcarino, E.; Strano, S.; Blandino, G. Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells. Oncogene 2014, 33, 1601–1608. [Google Scholar] [CrossRef]
- Zhao, Y.; Coloff, J.L.; Ferguson, E.C.; Jacobs, S.R.; Cui, K.; Rathmell, J.C. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J. Biol. Chem. 2008, 283, 36344–36353. [Google Scholar] [CrossRef] [PubMed]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; Hu, Y.; Qian, J.; Xu, B.; Chen, H.; Zou, W.; Fang, J.Y. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 2014, 5, e1108. [Google Scholar] [CrossRef]
- Li, J.; Yang, L.; Gaur, S.; Zhang, K.; Wu, X.; Yuan, Y.C.; Li, H.; Hu, S.; Weng, Y.; Yen, Y. Mutants TP53 p.R273H and p.R273C but not p.R273G enhance cancer cell malignancy. Hum. Mutat. 2014, 35, 575–584. [Google Scholar] [CrossRef]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S. Integrins as novel drug targets for overcoming innate drug resistance. Curr. Cancer Drug Targets 2002, 2, 37–43. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Locus | Activity Status | Activity Change | Phenotype | Associated Cancer Type | References |
|---|---|---|---|---|---|
| G245S | LOF | Destabilizes p53 protein and impairs DNA-binding ability | Exhibits altered conformational dynamics, reduced flexibility in loop L3, increased genomic instability, and resistance to chemotherapeutic agents. | Not specified | [47] |
| R175H | GOF | Modulates gene expression by binding to promoters, either independently or with transcription factor assistance. | Increases tumorigenicity by driving oncogenic pathways, proliferation, drug resistance, inflammation, angiogenesis, and metabolic reprogramming. | Key cancer types: Colorectal, Breast, Lung, Gastric, Endometrial, Pancreatic | [48,49] |
| R273H | GOF | Loss of tumor-suppressive functions while acquiring new oncogenic properties. | Enhances survival, migration, invasion, and chemoresistance by suppressing miR-27a, increasing EGFR, and activating ERK1/2 to drive proliferation and tumor growth. | Key cancer types: Colorectal, Breast, Lung, Head and Neck Squamous Cell Carcinoma | [50,51,52] |
| R248Q | DN | Interferes with wild-type p53 functions through dominant-negative effects. | Increased motility and invasiveness, coupled with inhibition of macro autophagy. | Key cancer types: High-Grade Serous Ovarian Carcinoma, Colorectal, Breast, Lung, Pancreatic | [53,54,55,56] |
| R249S | GOF LOF | Promotes proliferation via c-MYC-dependent ribosomal biogenesis but fails to bind p53 response elements. | Increased genomic instability, including interchromosomal translocations and aneuploidy. | Key cancer types: Hepatocellular carcinoma (HCC) (30% of all TP53 mutations in HCC), Lung, Head and Neck Squamous Cell Carcinoma, Colorectal | [57,58] |
| D281G | GOF | Activates cell cycle and survival genes while inducing p53 instability and aggregation. | Enhanced growth, survival, and angiogenesis, with increased metastasis, therapy resistance, and potential structural instability. | Key cancer types: Lung, Breast, Colorectal | [54,59,60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hertel, A.; Storchová, Z. The Role of p53 Mutations in Early and Late Response to Mitotic Aberrations. Biomolecules 2025, 15, 244. https://doi.org/10.3390/biom15020244
Hertel A, Storchová Z. The Role of p53 Mutations in Early and Late Response to Mitotic Aberrations. Biomolecules. 2025; 15(2):244. https://doi.org/10.3390/biom15020244
Chicago/Turabian StyleHertel, Anna, and Zuzana Storchová. 2025. "The Role of p53 Mutations in Early and Late Response to Mitotic Aberrations" Biomolecules 15, no. 2: 244. https://doi.org/10.3390/biom15020244
APA StyleHertel, A., & Storchová, Z. (2025). The Role of p53 Mutations in Early and Late Response to Mitotic Aberrations. Biomolecules, 15(2), 244. https://doi.org/10.3390/biom15020244