
Ursolic Acid Inhibits Collagen Production and Promotes Collagen Degradation in Skin Dermal Fibroblasts: Potential Antifibrotic Effects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. RNA Isolation and Quantitative Real-Time RT-PCR
2.4. Western Blot Analysis
2.5. Transient Transfection and Luciferase Assays
2.6. Mouse Model of Skin Fibrosis
2.7. Charts and Statistics
3. Results
3.1. UA Suppresses Type I Collagen Expression and Stimulates MMP-1 Expression
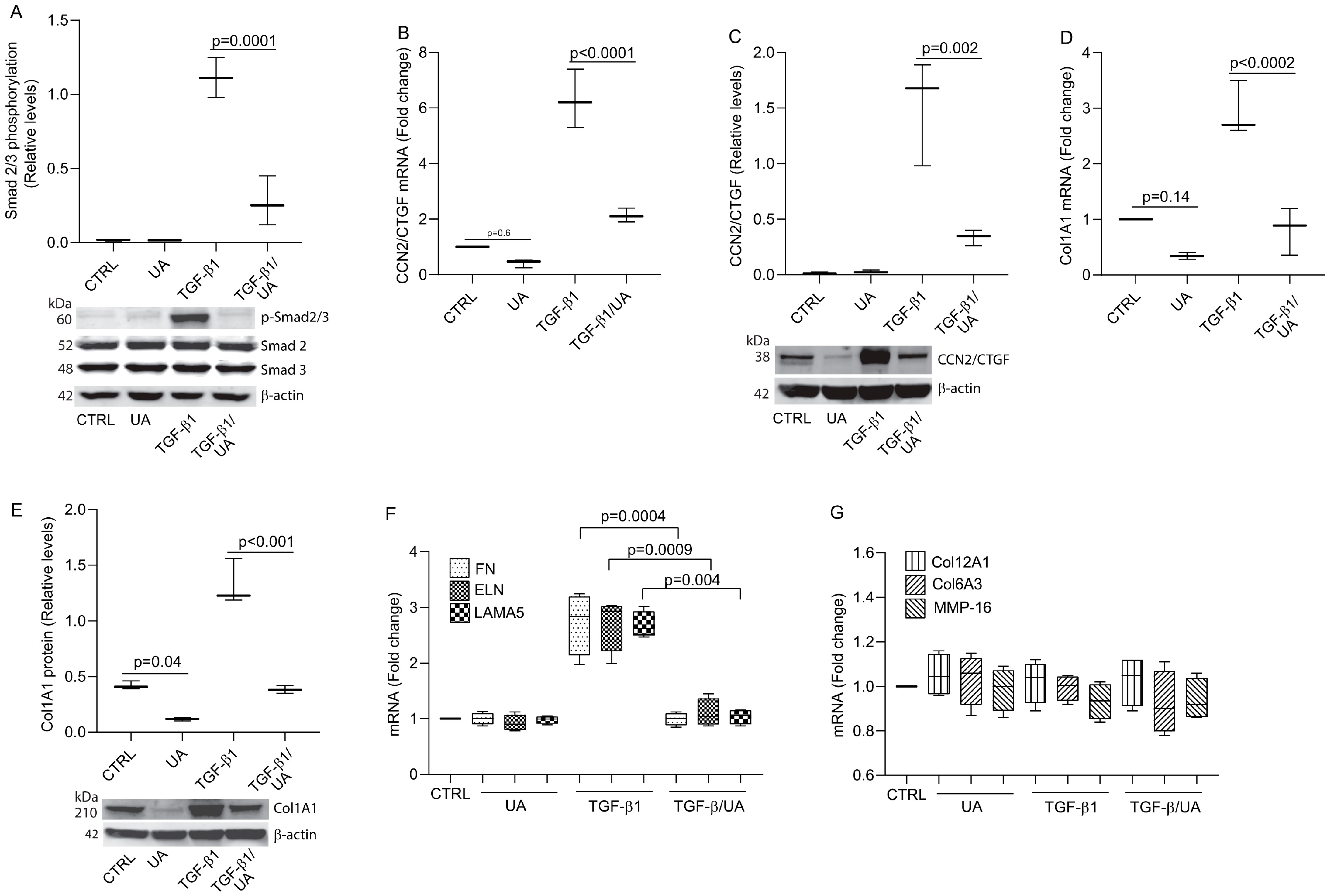
3.2. UA Inhibits Type I Collagen Expression by Impairing TGF-β/Smad Signaling
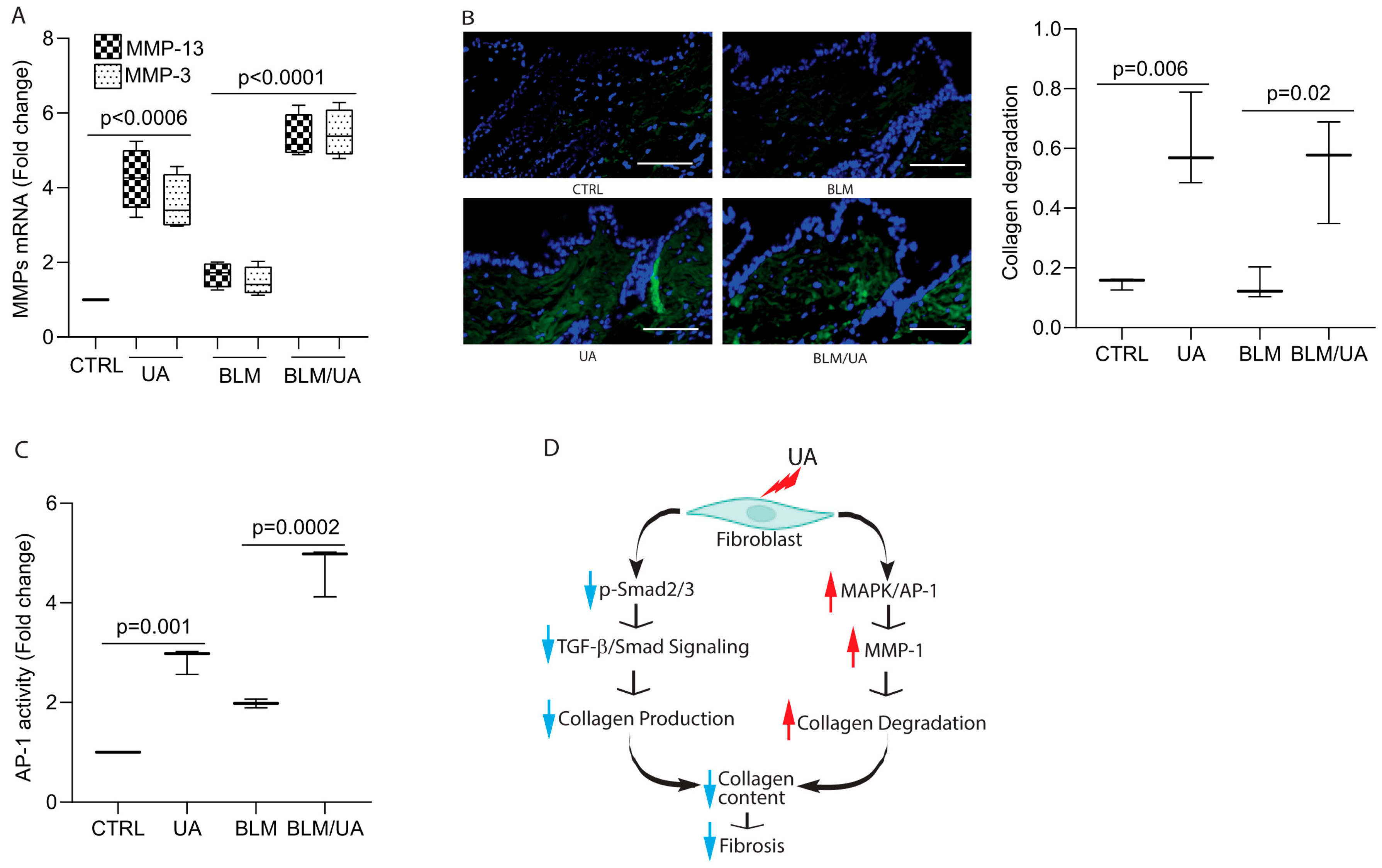
3.3. UA Induces MMP-1 by Activation of MAPK Pathways
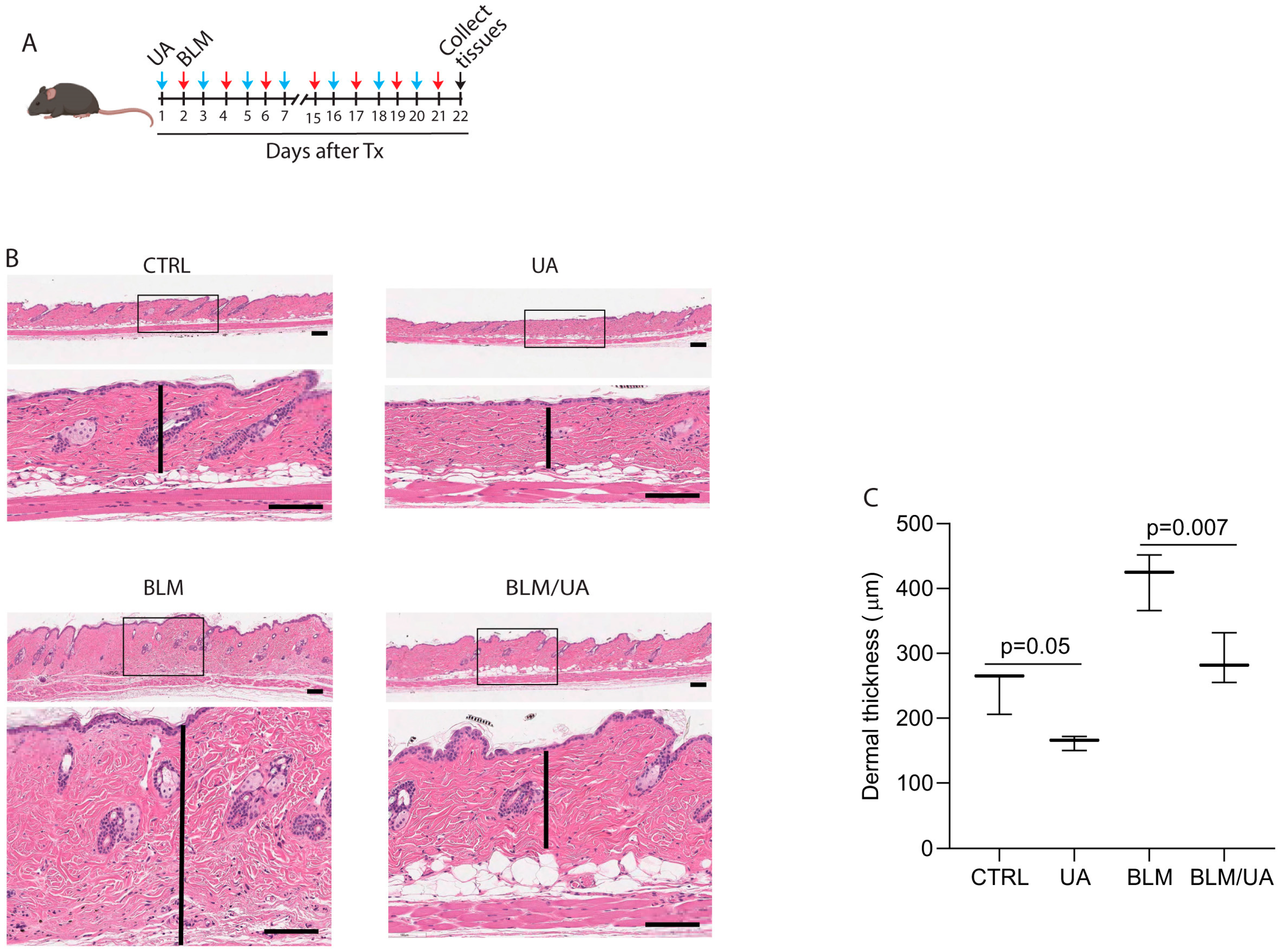
3.4. UA Inhibits Skin Thickness in Bleomycin-Induced Mouse Model of Fibrosis
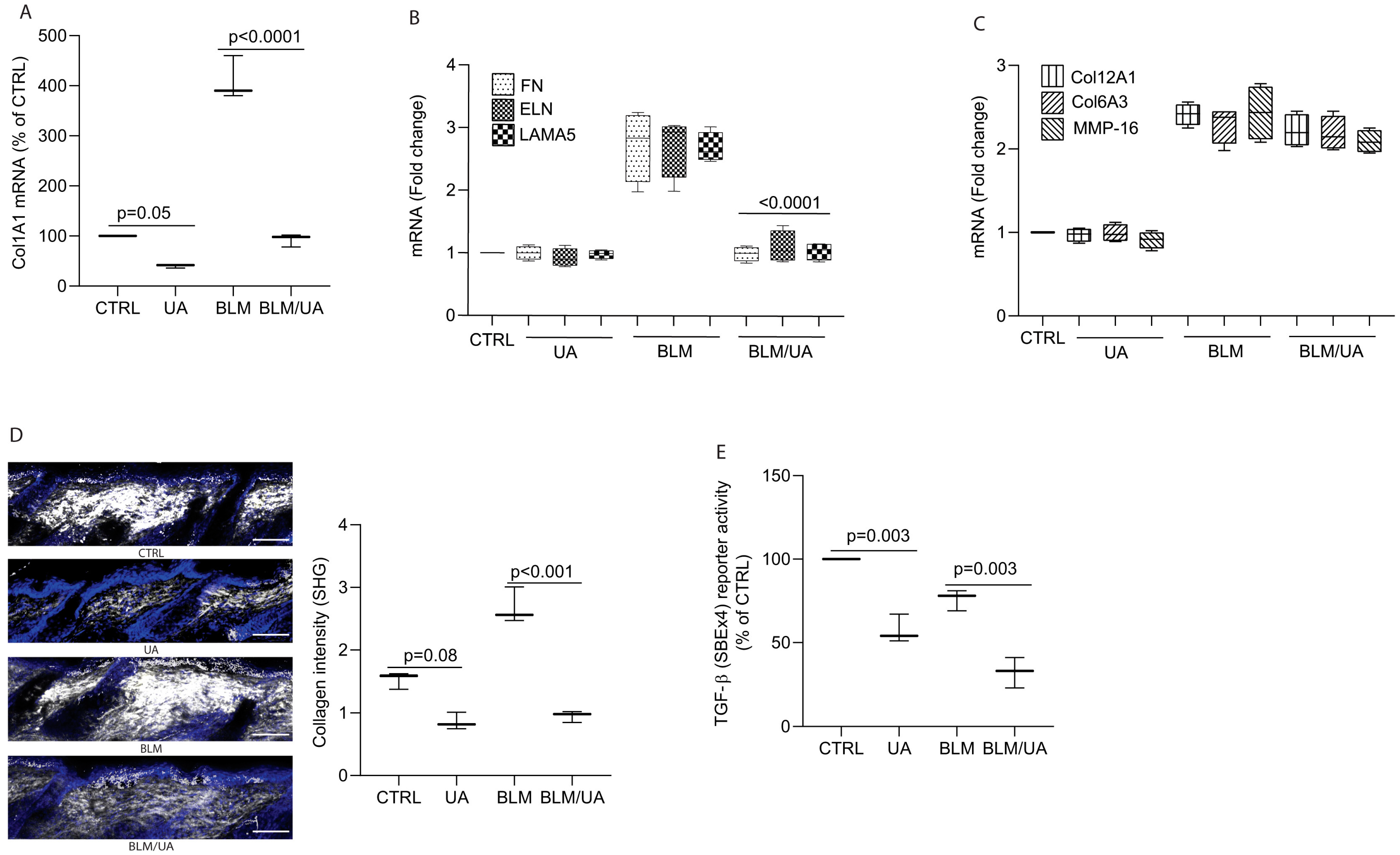
3.5. UA Inhibits Collagen Production by Impairing TGF-β/Smad Signaling in Bleomycin-Induced Mouse Model of Fibrosis
3.6. UA Promotes Collagen Degradation by Activating AP-1 in Bleomycin-Induced Mouse Model of Fibrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 373, 96. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair. Regen. 2009, 17, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, H.A.M.; Merrild, C.; Norregaard, R.; Plana-Ripoll, O. The impact of fibrotic diseases on global mortality from 1990 to 2019. J. Transl. Med. 2023, 21, 818. [Google Scholar] [CrossRef]
- Rockey, D.C.; Friedman, S.L. Fibrosis Regression After Eradication of Hepatitis C Virus: From Bench to Bedside. Gastroenterology 2021, 160, 1502–1520.e1. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Tsumanuma, R.; Aizawa, K.; Osakabe, M.; Maeda, K.; Omoto, E. Early Improvement in Marrow Fibrosis Following Haploidentical Stem Cell Transplantation for a Patient with Myelodysplastic Syndrome with Bone Marrow Fibrosis. Intern. Med. 2016, 55, 3351–3356. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sridhar, V.S.; Lovblom, L.E.; Lytvyn, Y.; Burger, D.; Burns, K.; Brinc, D.; Lawler, P.R.; Cherney, D.Z.I. Markers of Kidney Injury, Inflammation, and Fibrosis Associated with Ertugliflozin in Patients with CKD and Diabetes. Kidney Int. Rep. 2021, 6, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wu, F.; Tang, Z.; Yang, X.; Liu, Y.; Wang, F.; Chen, B. Anti-inflammatory and antioxidant activity of ursolic acid: A systematic review and meta-analysis. Front. Pharmacol. 2023, 14, 1256946. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, L.; Skapska, S.; Marszalek, K. Ursolic Acid—A Pentacyclic Triterpenoid with a Wide Spectrum of Pharmacological Activities. Molecules 2015, 20, 20614–20641. [Google Scholar] [CrossRef]
- Seo, D.Y.; Lee, S.R.; Heo, J.W.; No, M.H.; Rhee, B.D.; Ko, K.S.; Kwak, H.B.; Han, J. Ursolic acid in health and disease. Korean J. Physiol. Pharmacol. 2018, 22, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, L.; Niu, Y.; Mao, L.; Du, X.; Zhang, P.; Li, Z.; Li, H.; Li, N. A review of edible plant-derived natural compounds for the therapy of liver fibrosis. Eur. J. Gastroenterol. Hepatol. 2023, 35, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ikejima, K.; Kon, K.; Arai, K.; Aoyama, T.; Okumura, K.; Abe, W.; Sato, N.; Watanabe, S. Ursolic acid ameliorates hepatic fibrosis in the rat by specific induction of apoptosis in hepatic stellate cells. J. Hepatol. 2011, 55, 379–387. [Google Scholar] [CrossRef]
- Kwon, E.Y.; Shin, S.K.; Choi, M.S. Ursolic Acid Attenuates Hepatic Steatosis, Fibrosis, and Insulin Resistance by Modulating the Circadian Rhythm Pathway in Diet-Induced Obese Mice. Nutrients 2018, 10, 1719. [Google Scholar] [CrossRef]
- Ma, J.Q.; Ding, J.; Zhang, L.; Liu, C.M. Protective effects of ursolic acid in an experimental model of liver fibrosis through Nrf2/ARE pathway. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Gan, D.; Zhang, W.; Huang, C.; Chen, J.; He, W.; Wang, A.; Li, B.; Zhu, X. Ursolic acid ameliorates CCl4-induced liver fibrosis through the NOXs/ROS pathway. J. Cell Physiol. 2018, 233, 6799–6813. [Google Scholar] [CrossRef]
- Zhou, X.; Ye, H.; Wang, X.; Sun, J.; Tu, J.; Lv, J. Ursolic acid inhibits human dermal fibroblasts hyperproliferation, migration, and collagen deposition induced by TGF-β via regulating the Smad2/3 pathway. Gene 2023, 867, 147367. [Google Scholar] [CrossRef] [PubMed]
- Radhiga, T.; Senthil, S.; Sundaresan, A.; Pugalendi, K.V. Ursolic acid modulates MMPs, collagen-I, alpha-SMA, and TGF-β expression in isoproterenol-induced myocardial infarction in rats. Hum. Exp. Toxicol. 2019, 38, 785–793. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Shi, F.; Zhou, Z.W.; Li, B.; Zhang, K.; Zhang, X.; Ouyang, C.; Zhou, S.F.; Zhu, X. A bioinformatic and mechanistic study elicits the antifibrotic effect of ursolic acid through the attenuation of oxidative stress with the involvement of ERK, PI3K/Akt, and p38 MAPK signaling pathways in human hepatic stellate cells and rat liver. Drug Des. Devel. Ther. 2015, 9, 3989–4104. [Google Scholar]
- Yarosh, D.B.; Both, D.; Brown, D. Liposomal ursolic acid (merotaine) increases ceramides and collagen in human skin. Horm. Res. 2000, 54, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Both, D.M.; Goodtzova, K.; Yarosh, D.B.; Brown, D.A. Liposome-encapsulated ursolic acid increases ceramides and collagen in human skin cells. Arch. Dermatol. Res. 2002, 293, 569–575. [Google Scholar] [CrossRef]
- Lopez-Hortas, L.; Perez-Larran, P.; Gonzalez-Munoz, M.J.; Falque, E.; Dominguez, H. Recent developments on the extraction and application of ursolic acid. A review. Food Res. Int. 2018, 103, 130–149. [Google Scholar] [CrossRef]
- Farwick, M.; Kohler, T.; Schild, J.; Mentel, M.; Maczkiewitz, U.; Pagani, V.; Bonfigli, A.; Rigano, L.; Bureik, D.; Gauglitz, G.G. Pentacyclic triterpenes from Terminalia arjuna show multiple benefits on aged and dry skin. Skin. Pharmacol. Physiol. 2014, 27, 71–81. [Google Scholar] [CrossRef]
- Tan, H.; Sonam, T.; Shimizu, K. The Potential of Triterpenoids from Loquat Leaves (Eriobotrya japonica) for Prevention and Treatment of Skin Disorder. Int. J. Mol. Sci. 2017, 18, 1030. [Google Scholar] [CrossRef]
- Mascharak, S.; desJardins-Park, H.E.; Davitt, M.F.; Griffin, M.; Borrelli, M.R.; Moore, A.L.; Chen, K.; Duoto, B.; Chinta, M.; Foster, D.S.; et al. Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science 2021, 372, eaba2374. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Fisher, G.J. Role of Age-Associated Alterations of the Dermal Extracellular Matrix Microenvironment in Human Skin Aging: A Mini-Review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.; Fisher, G. Solar ultraviolet irradiation reduces collagen in photoaged human skin by blocking transforming growth factor-ß type II receptor/Smad signaling. Am. J. Pathol. 2004, 165, 741–751. [Google Scholar] [CrossRef]
- Quan, T.; Xu, Y.; Qin, Z.; Robichaud, P.; Betcher, S.; Calderone, K.; He, T.; Johnson, T.M.; Voorhees, J.J.; Fisher, G.J. Elevated YAP and its downstream targets CCN1 and CCN2 in basal cell carcinoma: Impact on keratinocyte proliferation and stromal cell activation. Am. J. Pathol. 2014, 184, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Shao, Y.; He, T.; Voorhees, J.J.; Fisher, G.J. Reduced expression of connective tissue growth factor (CTGF/CCN2) mediates collagen loss in chronologically aged human skin. J. Investig. Dermatol. 2010, 130, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Zawel, L.; Dai, J.L.; Buckhaults, P.; Zhou, S.; Kinzler, K.W.; Vogelstein, B.; Kern, S.E. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1998, 1, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Quan, T.; Purohit, T.; Shao, Y.; Cho, M.K.; He, T.; Varani, J.; Kang, S.; Voorhees, J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am. J. Pathol. 2009, 174, 101–114. [Google Scholar] [CrossRef]
- Fields, G.B. A model for interstitial collagen catabolism by mammalian collagenases. J. Theor. Biol. 1991, 153, 585–602. [Google Scholar] [CrossRef]
- Gross, J.; Harper, E.; Harris, E.D.; McCroskery, P.A.; Highberger, J.H.; Corbett, C.; Kang, A.H. Animal collagenases: Specificity of action, and structures of the substrate cleavage site. Biochem. Biophys. Res. Commun. 1974, 61, 605–612. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed]
- Prunier, C.; Baker, D.; Ten Dijke, P.; Ritsma, L. TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5, 66–78. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.C.; Hill, R.C.; Calderone, K.; Cui, Y.; Yan, Y.; Quan, T.; Fisher, G.J.; Hansen, K.C. Alterations in extracellular matrix composition during aging and photoaging of the skin. Matrix Biol. Plus 2020, 8, 100041. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Quan, H.; Guo, C.; Qin, Z.; Quan, T. Alterations of Matrisome Gene Expression in Naturally Aged and Photoaged Human Skin In Vivo. Biomolecules 2024, 14, 900. [Google Scholar] [CrossRef]
- Hall, M.C.; Young, D.A.; Waters, J.G.; Rowan, A.D.; Chantry, A.; Edwards, D.R.; Clark, I.M. The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-β 1. J. Biol. Chem. 2003, 278, 10304–10313. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Ask, K.; Warburton, D.; Gauldie, J.; Kolb, M. The bleomycin animal model: A useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 2008, 40, 362–382. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Parapuram, S.K.; Shi-Wen, X.; Abraham, D.J. Connective tissue growth factor (CTGF, CCN2) gene regulation: A potent clinical bio-marker of fibroproliferative disease? J. Cell Commun. Signal 2009, 3, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Usinger, W.; Nichols, B.; Gray, J.; Xu, L.; Seeley, T.W.; Brenner, M.; Guo, G.; Zhang, W.; Oliver, N.; et al. Cooperative interaction of CTGF and TGF-β in animal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011, 4, 4. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Luan, Z. Oleanolic Acid Attenuates Renal Fibrosis through TGF-β/Smad Pathway in a Rat Model of Unilateral Ureteral Obstruction. Evid. Based Complement. Alternat Med. 2020, 2020, 2085303. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.; Deng, R.; Gao, S.; Jiang, Q.; Liu, R.; Li, H.; Miao, Y.; Zhai, Y.; Zhang, S.; et al. Betulinic acid attenuated bleomycin-induced pulmonary fibrosis by effectively intervening Wnt/β-catenin signaling. Phytomedicine 2021, 81, 153428. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, T.; Xiang, Y.; Quan, H.; Liu, Y.; Guo, C.; Quan, T. Ursolic Acid Inhibits Collagen Production and Promotes Collagen Degradation in Skin Dermal Fibroblasts: Potential Antifibrotic Effects. Biomolecules 2025, 15, 365. https://doi.org/10.3390/biom15030365
He T, Xiang Y, Quan H, Liu Y, Guo C, Quan T. Ursolic Acid Inhibits Collagen Production and Promotes Collagen Degradation in Skin Dermal Fibroblasts: Potential Antifibrotic Effects. Biomolecules. 2025; 15(3):365. https://doi.org/10.3390/biom15030365
Chicago/Turabian StyleHe, Tianyuan, Yaping Xiang, Hehui Quan, Yingchun Liu, Chunfang Guo, and Taihao Quan. 2025. "Ursolic Acid Inhibits Collagen Production and Promotes Collagen Degradation in Skin Dermal Fibroblasts: Potential Antifibrotic Effects" Biomolecules 15, no. 3: 365. https://doi.org/10.3390/biom15030365
APA StyleHe, T., Xiang, Y., Quan, H., Liu, Y., Guo, C., & Quan, T. (2025). Ursolic Acid Inhibits Collagen Production and Promotes Collagen Degradation in Skin Dermal Fibroblasts: Potential Antifibrotic Effects. Biomolecules, 15(3), 365. https://doi.org/10.3390/biom15030365