
Exploring the CNOT1(800–999) HEAT Domain and Its Interactions with Tristetraprolin (TTP) as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry


Abstract
:
1. Introduction

2. Materials and Methods
2.1. DNA Constructs
2.2. Protein Expression and Purification
2.3. Chemicals
2.4. Absorption Spectroscopy
2.5. Hydrogen–Deuterium Exchange Experiments
2.6. Mass Spectrometry Measurements
2.7. Numerical Data Analysis
2.8. Bioinformatics
3. Results
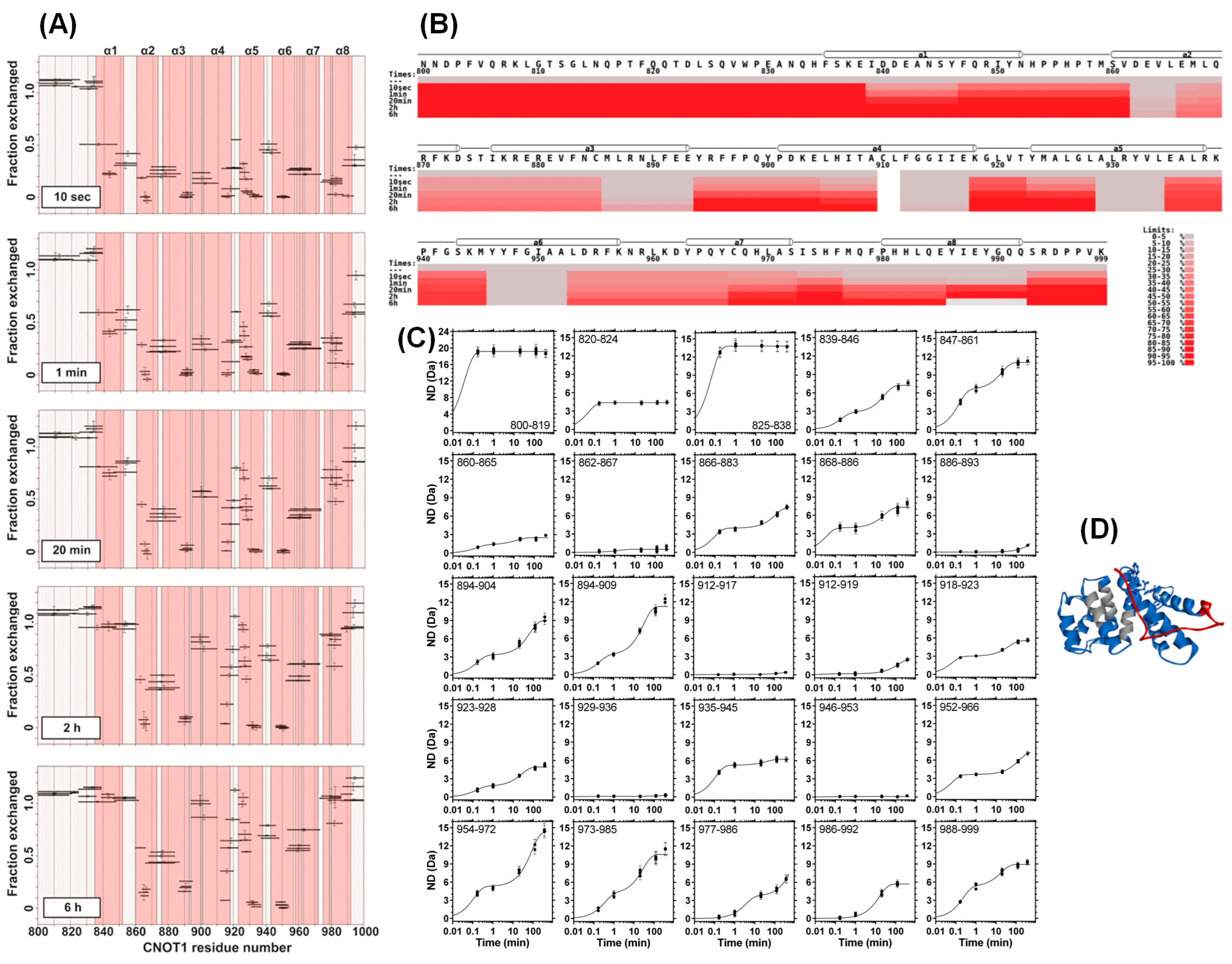
3.1. Structural Dynamics Properties of the CNOT1(800–999) HEAT Domain
3.2. Kinetic Parameters of Hydrogen–Deuterium Exchange of the CNOT1(800–999) HEAT Domain
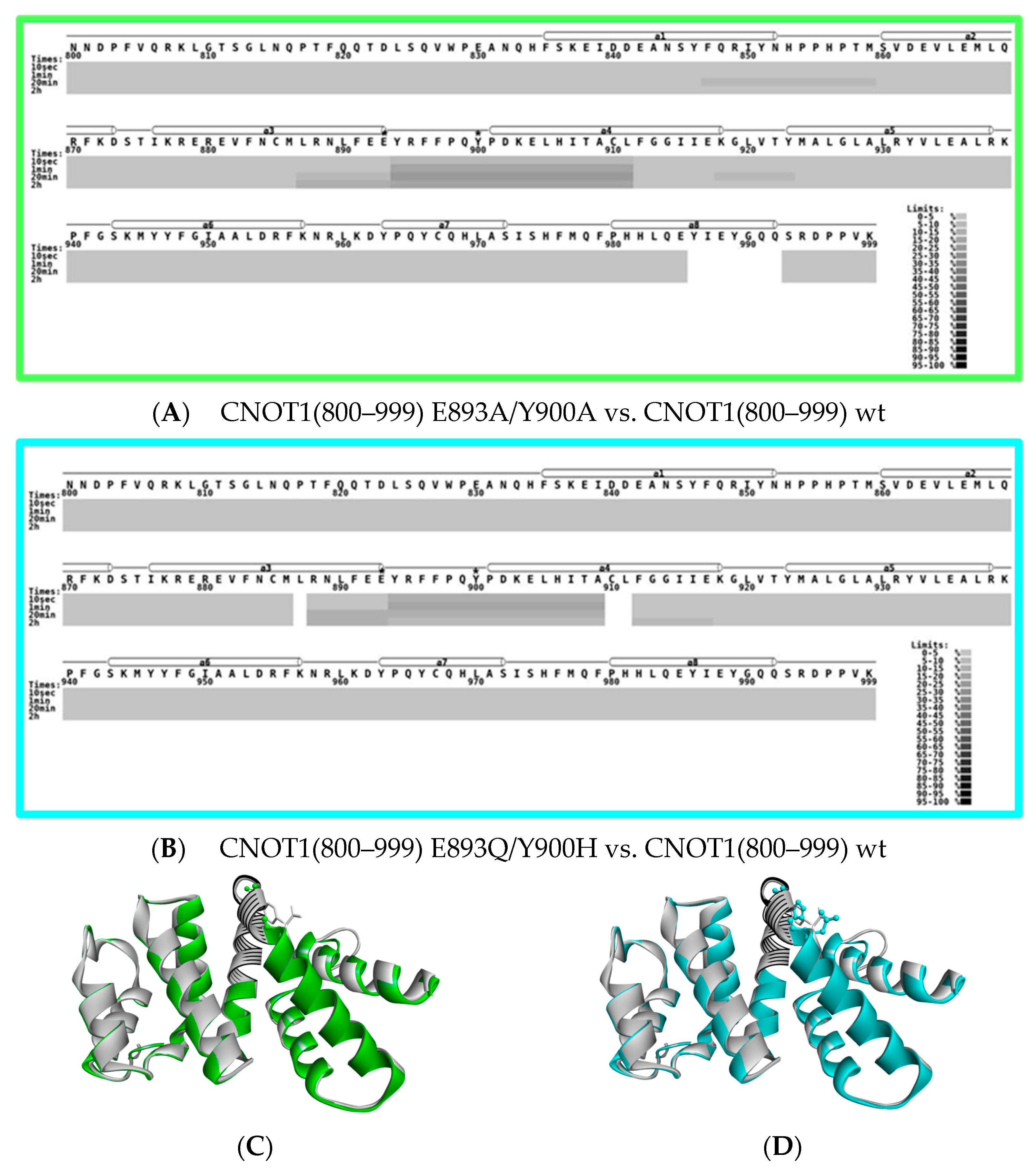
3.3. Protein Changes Caused by Point Mutations Monitored by Pepsin Digestion Patterns
3.4. Protein Changes Caused by Point Mutations Monitored by HDX
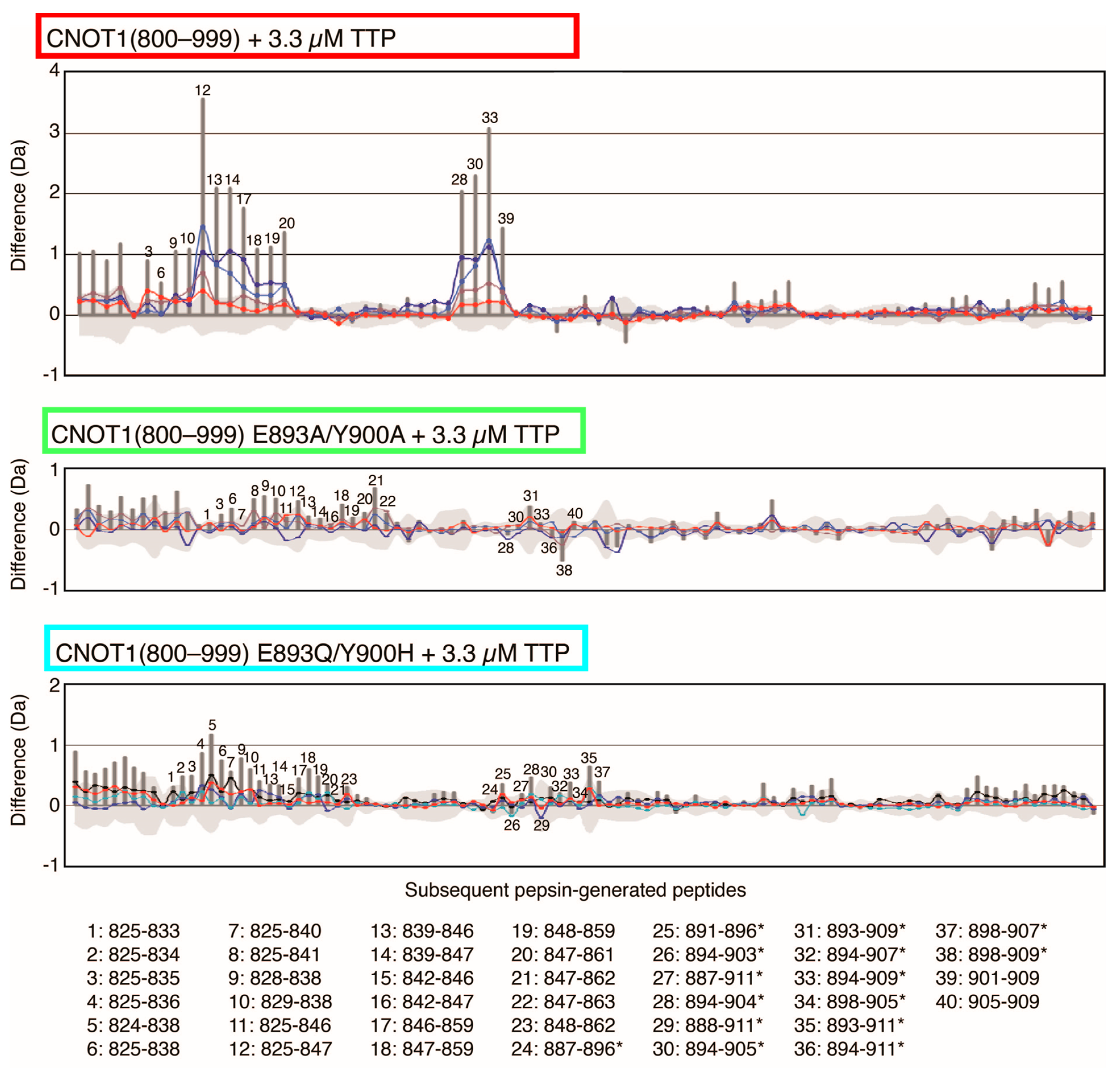
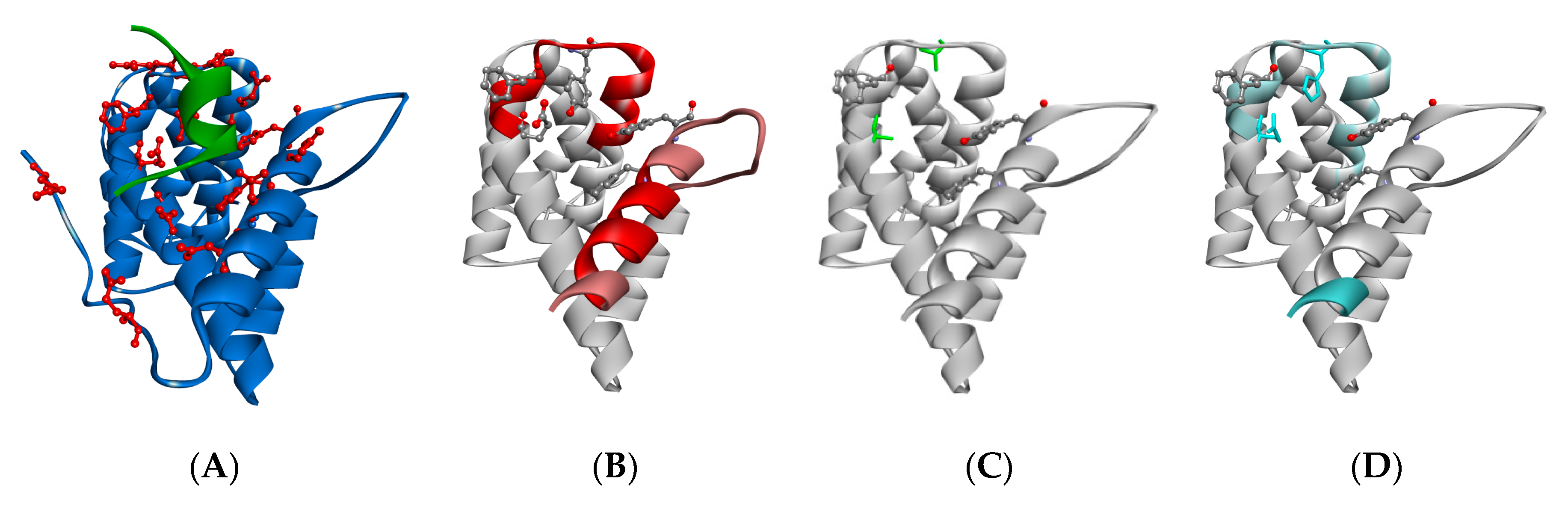
3.5. HDX MS Reveals Structural Impact of CNOT1(800–999) Mutations on TTP Binding
4. Discussion
4.1. HDX Signature of the CNOT1(800–999) HEAT Domain
4.2. Control of the Protein Fold Versus Point Mutations
4.3. TTP Binding Site of CNOT1(800–999) in Solution
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berendsen, H.J.; Hayward, S. Collective protein dynamics in relation to function. Curr. Opin. Struct. Biol. 2000, 10, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Liberles, D.A.; Teichmann, S.A.; Bahar, I.; Bastolla, U.; Bloom, J.; Bornberg-Bauer, E.; Colwell, L.J.; de Koning, A.P.J.; Dokholyan, N.V.; Echave, J.; et al. The interface of protein structure, protein biophysics, and molecular evolution. Protein Sci. 2012, 21, 769–785. [Google Scholar] [CrossRef]
- Hensen, U.; Meyer, T.; Haas, J.; Rex, R.; Vriend, G.; Grubmüller, H. Exploring protein dynamics space: The dynasome as the missing link between protein structure and function. PLoS ONE 2012, 7, e33931. [Google Scholar] [CrossRef]
- Smith, C.A.; Ban, D.; Pratihar, S.; Giller, K.; Paulat, M.; Becker, S.; Griesinger, C.; Lee, D.; de Groot, B.L. Allosteric switch regulates protein-protein binding through collective motion. Proc. Natl. Acad. Sci. USA 2016, 113, 3269–3274. [Google Scholar] [CrossRef]
- Englander, J.J.; Del Mar, C.; Li, W.; Englander, S.W.; Kim, J.S.; Stranz, D.D.; Hamuro, Y.; Woods, V.L., Jr. Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc. Natl. Acad. Sci. USA 2003, 100, 7057–7062. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, D.; Komives, E.A. Hydrogen-exchange mass spectrometry for the study of intrinsic disorder in proteins. Biochim. Biophys. Acta—Proteins Proteom. 2013, 1834, 1202–1209. [Google Scholar] [CrossRef]
- Zheng, J.; Yong, H.Y.; Panutdaporn, N.; Liu, C.; Tang, K.; Luo, D. High-resolution HDX-MS reveals distinct mechanisms of RNA recognition and activation by RIG-I and MDA5. Nucleic Acids Res. 2015, 43, 1216–1230. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, R.; Migas, L.G.; Payne, K.A.P.; Scrutton, N.S.; Leys, D.; Barran, P.E. Mass spectrometry locates local and allosteric conformational changes that occur on cofactor binding. Nat. Commun. 2016, 7, 12163. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, L.; DeColli, A.; Freel Meyers, C.; Nemeria, N.S.; Jordan, F. Conformational dynamics of 1-deoxy-D-xylulose 5-phosphate synthase on ligand binding revealed by H/D exchange MS. Proc. Natl. Acad. Sci. USA 2017, 114, 9355–9360. [Google Scholar] [CrossRef]
- Chen, R.; Glauninger, H.; Kahan, D.N.; Shangguan, J.; Sachleben, J.R.; Riback, J.A.; Drummond, D.A.; Sosnick, T.R. HDX–MS finds that partial unfolding with sequential domain activation controls condensation of a cellular stress marker. Proc. Natl. Acad. Sci. USA 2024, 121, e2321606121. [Google Scholar] [CrossRef]
- Ye, X.; Kotaru, S.; Lopes, R.; Cravens, S.; Lasagna, M.; Wand, A.J. Cooperative Substructure and Energetics of Allosteric Regulation of the Catalytic Core of the E3 Ubiquitin Ligase Parkin by Phosphorylated Ubiquitin. Biomolecules 2024, 14, 1338. [Google Scholar] [CrossRef] [PubMed]
- Markwick, P.R.L.; Peacock, R.B.; Komives, E.A. Accurate Prediction of Amide Exchange in the Fast Limit Reveals Thrombin Allostery. Biophys. J. 2019, 116, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Engen, J.R.; Komives, E.A. Complementarity of Hydrogen/Deuterium Exchange Mass Spectrometry and Cryo-Electron Microscopy. Trends Biochem. Sci. 2020, 45, 906–918. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Resing, K.A.; Goldsmith, E.J.; Ahn, N.G. Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. USA 2001, 98, 956–961. [Google Scholar] [CrossRef]
- Maher, S.; Jjunju, F.P.M.; Taylor, S. Colloquium: 100 years of mass spectrometry: Perspectives and future trends. Rev. Mod. Phys. 2015, 87, 113–135. [Google Scholar] [CrossRef]
- James, E.I.; Murphree, T.A.; Vorauer, C.; Engen, J.R.; Guttman, M. Advances in Hydrogen/Deuterium Exchange Mass Spectrometry and the Pursuit of Challenging Biological Systems. Chem. Rev. 2022, 122, 7562–7623. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Dill, K.A. A statistical mechanical model for hydrogen exchange in globular proteins. Protein Sci. 1995, 4, 1860–1873. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Resing, K.A.; Ahn, N.G. Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 1–25. [Google Scholar] [CrossRef]
- Skinner, J.J.; Lim, W.K.; Bédard, S.; Black, B.E.; Englander, S.W. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012, 21, 996–1005. [Google Scholar] [CrossRef]
- Cieplak-Rotowska, M.K.; Tarnowski, K.; Rubin, M.; Fabian, M.R.; Sonenberg, N.; Dadlez, M.; Niedzwiecka, A. Structural Dynamics of the GW182 Silencing Domain Including its RNA Recognition motif (RRM) Revealed by Hydrogen-Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 158–173. [Google Scholar] [CrossRef]
- Tsutsui, Y.; Wintrode, P.L. Hydrogen/deuterium exchange-mass spectrometry: A powerful tool for probing protein structure, dynamics and interactions. Curr. Med. Chem. 2007, 14, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Marcsisin, S.R.; Engen, J.R. Hydrogen exchange mass spectrometry: What is it and what can it tell us? Anal. Bioanal. Chem. 2010, 397, 967–972. [Google Scholar] [CrossRef]
- Iacob, R.E.; Engen, J.R. Hydrogen exchange mass spectrometry: Are we out of the quicksand? J. Am. Soc. Mass Spectrom. 2012, 23, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Wand, A.J.; Englander, S.W. Protein complexes studied by NMR spectroscopy. Curr. Opin. Biotechnol. 1996, 7, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.S.; Mayne, L.; Roder, H.; Wand, A.J.; Englander, S.W. Determinants of protein hydrogen exchange studied in equine cytochrome c. Protein Sci. 1998, 7, 739–745. [Google Scholar] [CrossRef]
- Morgan, C.R.; Engen, J.R. Investigating solution-phase protein structure and dynamics by hydrogen exchange mass spectrometry. Curr. Protoc. Protein Sci. 2009, 17.6, 1–17. [Google Scholar] [CrossRef]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef]
- Drewniak, P.; Xiao, P.; Ladizhansky, V.; Bondar, A.N.; Brown, L.S. A conserved H-bond network in human aquaporin-1 is necessary for native folding and oligomerization. Biophys. J. 2024, 123, 4285–4303. [Google Scholar] [CrossRef]
- Selinger, M.; Novotný, R.; Sýs, J.; Roby, J.A.; Tykalová, H.; Ranjani, G.S.; Vancová, M.; Jaklová, K.; Kaufman, F.; Bloom, M.E.; et al. Tick-borne encephalitis virus capsid protein induces translational shutoff as revealed by its structural–biological analysis. J. Biol. Chem. 2022, 298, 102585. [Google Scholar] [CrossRef]
- Wahle, E.; Winkler, G.S. RNA decay machines: Deadenylation by the Ccr4-not and Pan2-Pan3 complexes. Biochim. Biophys. Acta 2013, 1829, 561–570. [Google Scholar] [CrossRef]
- Collart, M.A. Global control of gene expression in yeast by the Ccr4-Not complex. Gene 2003, 313, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Inada, T.; Makino, S. Novel roles of the multi-functional CCR4-NOT complex in post-transcriptional regulation. Front. Genet. 2014, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.-T.; Suzuki, T.; Morita, M.; Takahashi, A.; Yamamoto, T. Multifunctional roles of the mammalian CCR4-NOT complex in physiological phenomena. Front. Genet. 2014, 5, 286. [Google Scholar] [CrossRef]
- Collart, M.A.; Panasenko, O.O. The Ccr4-not complex. Gene 2012, 492, 42–53. [Google Scholar] [CrossRef]
- Wang, H.; Morita, M.; Yang, X.; Suzuki, T.; Yang, W.; Wang, J.; Ito, K.; Wang, Q.; Zhao, C.; Bartlam, M.; et al. Crystal structure of the human CNOT6L nuclease domain reveals strict poly(A) substrate specificity. EMBO J. 2010, 29, 2566–2576. [Google Scholar] [CrossRef]
- Zhang, Q.; Yan, D.; Guo, E.; Ding, B.; Yang, W.; Liu, R.; Yamamoto, T.; Bartlam, M. Structural basis for inhibition of the deadenylase activity of human CNOT6L. FEBS Lett. 2016, 590, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Aslam, A.; Doidge, R.; Medica, R.; Winkler, G.S. The Ccr4a (CNOT6) and Ccr4b (CNOT6L) deadenylase subunits of the human Ccr4-Not complex contribute to the prevention of cell death and senescence. Mol. Biol. Cell 2011, 22, 748–758. [Google Scholar] [CrossRef]
- Mostafa, D.; Takahashi, A.; Yanagiya, A.; Yamaguchi, T.; Abe, T.; Kureha, T.; Kuba, K.; Kanegae, Y.; Furuta, Y.; Yamamoto, T.; et al. Essential functions of the CNOT7/8 catalytic subunits of the CCR4-NOT complex in mRNA regulation and cell viability. RNA Biol. 2020, 17, 403–416. [Google Scholar] [CrossRef]
- Maillet, L.; Tu, C.; Hong, Y.K.; Shuster, E.O.; Collart, M.A. The essential function of Not1 lies within the Ccr4-Not complex. J. Mol. Biol. 2000, 303, 131–143. [Google Scholar] [CrossRef]
- Ito, K.; Takahashi, A.; Morita, M.; Suzuki, T.; Yamamoto, T. The role of the CNOT1 subunit of the CCR4-NOT complex in mRNA deadenylation and cell viability. Protein Cell 2011, 2, 755–763. [Google Scholar] [CrossRef]
- Sandler, H.; Kreth, J.; Timmers, H.T.M.; Stoecklin, G. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res. 2011, 49, 4373–4386. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Blackshear, P.J. Tristetraprolin (TTP): Interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim. Biophys. Acta 2013, 1829, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.; Raisch, T.; Weichenrieder, O.; Jonas, S.; Izaurralde, E. Structural basis for the Nanos-mediated recruitment of the CCR4-NOT complex and translational repression. Genes Dev. 2014, 28, 888–901. [Google Scholar] [CrossRef]
- Fabian, M.R.; Cieplak, M.K.; Frank, F.; Morita, M.; Green, J.; Srikumar, T.; Nagar, B.; Yamamoto, T.; Raught, B.; Duchaine, T.F.; et al. miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat. Struct. Mol. Biol. 2011, 18, 1211–1217. [Google Scholar] [CrossRef]
- Chekulaeva, M.; Mathys, H.; Zipprich, J.T.; Attig, J.; Colic, M.; Parker, R.; Filipowicz, W. miRNA repression involves GW182-mediated recruitment of CCR4–NOT through conserved W-containing motifs. Nat. Struct. Mol. Biol. 2011, 18, 1218–1226. [Google Scholar] [CrossRef]
- Braun, J.E.; Huntzinger, E.; Fauser, M.; Izaurralde, E. GW182 proteins directly recruit cytoplasmic deadenylase complexes to miRNA targets. Mol. Cell 2011, 44, 120–133. [Google Scholar] [CrossRef]
- Mathonnet, G.; Fabian, M.R.; Svitkin, Y.V.; Parsyan, A.; Huck, L.; Murata, T.; Biffo, S.; Merrick, W.C.; Darzynkiewicz, E.; Pillai, R.S.; et al. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science 2007, 317, 1764–1767. [Google Scholar] [CrossRef]
- Fabian, M.R.; Mathonnet, G.; Sundermeier, T.; Mathys, H.; Zipprich, J.T.; Svitkin, Y.V.; Rivas, F.; Jinke, M.; Wohlschlegel, J.; Doudna, J.A.; et al. Mammalian miRNA RISC recruits CAF1 and PABP to affect PABP-dependent deadenylation. Mol. Cell 2009, 35, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Bazzini, A.A.; Lee, M.T.; Giraldez, A.J. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 2012, 336, 233–237. [Google Scholar] [CrossRef]
- Béthune, J.; Artus-Revel, C.G.; Filipowicz, W. Kinetic analysis reveals successive steps leading to miRNA-mediated silencing in mammalian cells. EMBO Rep. 2012, 13, 716–723. [Google Scholar] [CrossRef]
- Djuranovic, S.; Nahvi, A.; Green, R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.S.; Bhattacharyya, S.N.; Artus, C.G.; Zoller, T.; Cougot, N.; Basyuk, E.; Bertrand, E.; Filipowicz, W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 2005, 309, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Thermann, R.; Hentze, M.W. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature 2007, 447, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Meijer, H.A.; Kong, Y.W.; Lu, W.T.; Wilczynska, A.; Spriggs, R.V.; Robinson, S.W.; Godfrey, J.D.; Willis, A.E.; Bushell, M. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 2013, 340, 82–85. [Google Scholar] [CrossRef]
- Chen, C.-Y.A.; Zheng, D.; Xia, Z.; Shyu, A.-B. Ago-TNRC6 triggers microRNA-mediated decay by promoting two deadenylation steps. Nat. Struct. Mol. Biol. 2009, 16, 1160–1166. [Google Scholar] [CrossRef]
- Piao, X.; Zhang, X.; Wu, L.; Belasco, J.G. CCR4-NOT deadenylates mRNA associated with RNA-induced silencing complexes in human cells. Mol. Cell. Biol. 2010, 30, 1486–1494. [Google Scholar] [CrossRef]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [CrossRef]
- Mathys, H.; Basquin, J.; Ozgur, S.; Czarnocki-Cieciura, M.; Bonneau, F.; Aartse, A.; Dziembowski, A.; Nowotny, M.; Conti, E.; Filipowicz, W. Structural and Biochemical Insights to the Role of the CCR4-NOT Complex and DDX6 ATPase in MicroRNA Repression. Mol. Cell 2014, 9, 751–765. [Google Scholar] [CrossRef]
- Chen, Y.; Boland, A.; Kuzuoğlu-Öztürk, D.; Bawankar, P.; Loh, B.; Chang, C.-T.; Weichenrieder, O.; Izaurralde, E. A DDX6-CNOT1 Complex and W-Binding Pockets in CNOT9 Reveal Direct Links between miRNA Target Recognition and Silencing. Mol. Cell 2014, 54, 737–750. [Google Scholar] [CrossRef]
- Eulalio, A.; Rehwinkel, J.; Stricker, M.; Huntzinger, E.; Yang, S.-F.; Doerks, T.; Dorner, S.; Bork, P.; Boutros, M.; Izaurralde, E. Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing. Genes Dev. 2007, 21, 2558–2570. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Behm-Ansmant, I.; Gatfield, D.; Izaurralde, E. A crucial role for GW182 and the DCP1: DCP2 decapping complex in miRNA-mediated gene silencing. RNA 2005, 11, 1640–1647. [Google Scholar] [CrossRef]
- Pratt, A.J.; MacRae, I.J. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, H.-O.; Tomari, Y. Life of RISC: Formation, action, and degradation of RNA-induced silencing complex. Mol. Cell 2022, 82, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Nasertorabi, F.; Batisse, C.; Diepholz, M.; Suck, D.; Böttcher, B. Insights into the structure of the CCR4-NOT complex by electron microscopy. FEBS Lett. 2011, 585, 2182–2186. [Google Scholar] [CrossRef] [PubMed]
- Bawankar, P.; Loh, B.; Wohlbold, L.; Schmidt, S.; Izaurralde, E. NOT10 and C2orf29/NOT11 form a conserved module of the CCR4-NOT complex that docks onto the NOT1 N-terminal domain. RNA Biol. 2013, 10, 228–244. [Google Scholar] [CrossRef]
- Ukleja, M.; Cuellar, J.; Siwaszek, A.; Kasprzak, J.M.; Czarnocki-Cieciura, M.; Bujnicki, J.M.; Dziembowski, A.; Valpuesta, J.M. The architecture of the Schizosaccharomyces pombe CCR4-NOT complex. Nat. Commun. 2016, 7, 10433. [Google Scholar] [CrossRef]
- Fabian, M.R.; Frank, F.; Rouya, C.; Siddiqui, N.; Lai, W.S.; Karetnikov, A.; Blackshear, P.J.; Nagar, B.; Sonenberg, N. Structural basis for the recruitment of the human CCR4-NOT deadenylase complex by tristetraprolin. Nat. Struct. Mol. Biol. 2013, 20, 735–739. [Google Scholar] [CrossRef]
- Chen, C.-Y.A.; Shyu, A.-B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Mohamed, H.M.A.; Takahashi, A.; Nishijima, S.; Adachi, S.; Murai, I.; Okamura, H.; Yamamoto, T. CNOT1 regulates circadian behaviour through Per2 mRNA decay in a deadenylation-dependent manner. RNA Biol. 2022, 19, 703–718. [Google Scholar] [CrossRef]
- Carballo, E.; Lai, W.S.; Blackshear, P.J. Feedback Inhibition of Macrophage Tumor Necrosis Factor- Production by Tristetraprolin. Science 1998, 281, 1001–1005. [Google Scholar] [CrossRef]
- Carreño, A.; Lykke-Andersen, J. The Conserved CNOT1 Interaction Motif of Tristetraprolin Regulates ARE-mRNA Decay Independently of the p38 MAPK-MK2 Kinase Pathway. Mol. Cell. Biol. 2022, 42, e00055-22. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.S.; Wells, M.L.; Perera, L.; Blackshear, P.J. The tandem zinc finger RNA binding domain of members of the tristetraprolin protein family. Wiley Interdiscip. Rev. RNA 2019, 10, e1531. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.L.; Scheckel, C.; Stoecklin, G.; Lykke-Andersen, J. Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol. Cell. Biol. 2011, 31, 256–266. [Google Scholar] [CrossRef]
- Chrestensen, C.A.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F.; Pelo, J.W.; Worthington, M.T.; Sturgill, T.W. MAPKAP Kinase 2 Phosphorylates Tristetraprolin on in Vivo Sites Including Ser178, a Site Required for 14-3-3 Binding. J. Biol. Chem. 2004, 279, 10176–10184. [Google Scholar] [CrossRef]
- Tyagi, S.; Tyagi, S.; Singh, A.; Gautam, A.; Singh, A.; Jindal, S.; Singh, R.P.; Chaturvedi, R.; Kushwaha, H.R. Linking COVID-19 and cancer: Underlying mechanism. Biochim. Biophys. Acta—Mol. Basis Dis. 2025, 1871, 167563. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Maus, M.V.; Hinrichs, C.S. CAR T Cells and T-Cell Therapies for Cancer: A Translational Science Review. JAMA 2024, 332, 1924–1935. [Google Scholar] [CrossRef]
- Snyder, B.L.; Blackshear, P.J. Clinical implications of tristetraprolin (TTP) modulation in the treatment of inflammatory diseases. Pharmacol. Ther. 2022, 239, 108198. [Google Scholar] [CrossRef]
- Andrade, M.A.; Bork, P. HEAT repeats in the Huntington’s disease protein. Nat. Genet. 1995, 11, 115–116. [Google Scholar] [CrossRef]
- Petit, A.-P.; Wohlbold, L.; Bawankar, P.; Huntzinger, E.; Schmidt, S.; Izaurralde, E.; Weichenrieder, O. The structural basis for the interaction between the CAF1 nuclease and the NOT1 scaffold of the human CCR4-NOT deadenylase complex. Nucleic Acids Res. 2012, 40, 11058–11072. [Google Scholar] [CrossRef]
- Boland, A.; Chen, Y.; Raisch, T.; Jonas, S.; Kuzuoğlu-Öztürk, D.; Wohlbold, L.; Weichenrider, O.; Izaurralde, E. Structure and assembly of the NOT module of the human CCR4-NOT complex. Nat. Struct. Mol. Biol. 2013, 20, 1289–1297. [Google Scholar] [CrossRef]
- Mauxion, F.; Basquin, J.; Ozgur, S.; Rame, M.; Albrecht, J.; Schafer, I.; Seraphin, B.; Conti, E. The human CNOT1-CNOT10-CNOT11 complex forms a structural platform for protein-protein interactions. Cell Rep. 2023, 42, 111902. [Google Scholar] [CrossRef]
- Yoshimura, S.H.; Hirano, T. HEAT repeats—Versatile arrays of amphiphilic helices working in crowded environments? J. Cell Sci. 2016, 129, 3963–3970. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, D.; Marintchev, A.; Arthanari, H. The metaphorical swiss army knife: The multitude and diverse roles of HEAT domains in eukaryotic translation initiation. Nucleic Acids Res. 2022, 50, 5424–5442. [Google Scholar] [CrossRef]
- Conti, E.; Müller, C.W.; Stewart, M. Karyopherin flexibility in nucleocytoplasmic transport. Curr. Opin. Struct. Biol. 2006, 16, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.; Chang, C.-W.; Róna, G.; Smith, K.M.; Stewart, A.G.; Takeda, A.A.S.; Fontes, M.R.M.; Stewart, M.; Vértessy, B.G.; Forwood, J.K.; et al. Structural Biology and Regulation of Protein Import into the Nucleus. J. Mol. Biol. 2016, 428, 2060–2090. [Google Scholar] [CrossRef]
- Marcotrigiano, J.; Lomakin, I.B.; Sonenberg, N.; Pestova, T.V.; Hellen, C.U.T. A Conserved HEAT Domain within eIF4G Directs Assembly of the Translation Initiation Machinery. Mol. Cell 2001, 7, 193–203. [Google Scholar] [CrossRef] [PubMed]
- He, H.; von der Haar, T.; Singh, C.R.; Ii, M.; Li, B.; Hinnebusch, A.G.; McCarthy, J.E.G.; Asano, K. The Yeast Eukaryotic Initiation Factor 4G (eIF4G) HEAT Domain Interacts with eIF1 and eIF5 and Is Involved in Stringent AUG Selection. Mol. Cell. Biol. 2003, 23, 5431–5445. [Google Scholar] [CrossRef]
- Rimmer, M.A.; Nadeau, O.W.; Yang, J.; Artigues, A.; Zhang, Y.; Carlson, G.M. The structure of the large regulatory α subunit of phosphorylase kinase examined by modeling and hydrogen-deuterium exchange. Protein Sci. 2018, 27, 472–484. [Google Scholar] [CrossRef]
- Jung, T.; Shin, B.; Tamo, G.; Kim, H.; Vijayvargia, R.; Leitner, A.; Marcaida, M.J.; Astorga-Wells, J.; Jung, R.; Aebersold, R.; et al. The Polyglutamine Expansion at the N-Terminal of Huntingtin Protein Modulates the Dynamic Configuration and Phosphorylation of the C-Terminal HEAT Domain. Structure 2020, 28, 1035–1050.e8. [Google Scholar] [CrossRef]
- Shaffer, J.M.; Jiou, J.; Tripathi, K.; Olaluwoye, O.S.; Fung, H.Y.J.; Chook, Y.M.; D’Arcy, S. Molecular basis of RanGTP-activated release of Histones H2A-H2B from Importin-9. Structure 2023, 31, 903–911.e3. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Rutkowska-Wlodarczyk, I.; Stepinski, J.; Dadlez, M.; Darzynkiewicz, E.; Stolarski, R.; Niedzwiecka, A. Structural changes of eIF4E upon binding to the mRNA 5′ monomethylguanosine and trimethylguanosine Cap. Biochemistry 2008, 47, 2710–2720. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, K.; Fituch, K.; Szczepanowski, R.H.; Dadlez, M.; Kaus-Drobek, M. Patterns of structural dynamics in RACK1 protein retained throughout evolution: A hydrogen-deuterium exchange study of three orthologs. Protein Sci. 2014, 23, 639–651. [Google Scholar] [CrossRef]
- Mandell, J.G.; Baerga-Ortiz, A.; Akashi, S.; Takio, K.; Komives, E.A. Solvent accessibility of the thrombin-thrombomodulin interface. J. Mol. Biol. 2001, 306, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Gemmecker, G.; Jahnke, W.; Kessler, H. Measurement of Fast Proton Exchange Rates in Isotopically Labeled Compounds. J. Am. Chem. Soc. 1993, 115, 11620–11621. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Automat. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Beyer, W.H. CRC Standard Mathematical Tables, 28th ed.; CRC Press: Boca Raton, FL, USA, 1987. [Google Scholar]
- Zhang, Y.-Z. Protein and Peptide Structure and Interactions Studied by Hydrogen Exchange and NMR. Ph.D. Thesis, Structural Biology and Molecular Biophysics, University of Pennsylvania, Philadelphia, PA, USA, 1995. [Google Scholar]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins 1993, 17, 75–86. [Google Scholar] [CrossRef]
- Connelly, G.P.; Bai, Y.; Jeng, M.F.; Englander, S.W. Isotope effects in peptide group hydrogen exchange. Proteins 1993, 17, 87–92. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Kavan, D.; Man, P. MSTools—Web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 2011, 302, 53–58. [Google Scholar] [CrossRef]
- Nielsen, S.D.H.; Liang, N.; Rathish, H.; Kim, B.J.; Lueangsakulthai, J.; Koh, J.; Qu, Y.; Schulz, H.-J.; Dallas, D.C. Bioactive milk peptides: An updated comprehensive overview and database. Crit. Rev. Food Sci. Nutr. 2023, 64, 11510–11529. [Google Scholar] [CrossRef]
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Englander, S.W.; Kallen, R.G. Primary structure effects on peptide group hydrogen exchange. Biochemistry 1972, 11, 150–158. [Google Scholar] [CrossRef]
- Fruton, J.S. The Specificity and Mechanism of Pepsin Action. In Advances in Enzymology and Related Areas of Molecular Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1970; pp. 401–443. [Google Scholar]
- Ahn, J.; Cao, M.-J.J.; Yu, Y.Q.; Engen, J.R. Accessing the reproducibility and specificity of pepsin and other aspartic proteases. Biochim. Biophys. Acta 2013, 1834, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Moussong, É.; Wien, F.; Boros, E.; Vadászi, H.; Murvai, N.; Lee, Y.-H.; Molnár, T.; Réfrégiers, M.; Goto, Y.; et al. BeStSel: Webserver for secondary structure and fold prediction for protein CD spectroscopy. Nucleic Acids Res. 2022, 50, W90–W98. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CNOT1 | ||||||
|---|---|---|---|---|---|---|
| Res. no. | 840–846 | 847 | 849–859 | 895–901 | 902–904 | 905–909 |
| Sequence | DDEANSY | F | RIYNHPPHPTM | RFFPQYP | DKE | LHITA |
| ΔND (Da) | 1.6 | 1 | 1.5 | 1.4 | 0.5 | 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cieplak-Rotowska, M.K.; Dadlez, M.; Niedzwiecka, A. Exploring the CNOT1(800–999) HEAT Domain and Its Interactions with Tristetraprolin (TTP) as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Biomolecules 2025, 15, 403. https://doi.org/10.3390/biom15030403
Cieplak-Rotowska MK, Dadlez M, Niedzwiecka A. Exploring the CNOT1(800–999) HEAT Domain and Its Interactions with Tristetraprolin (TTP) as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Biomolecules. 2025; 15(3):403. https://doi.org/10.3390/biom15030403
Chicago/Turabian StyleCieplak-Rotowska, Maja K., Michał Dadlez, and Anna Niedzwiecka. 2025. "Exploring the CNOT1(800–999) HEAT Domain and Its Interactions with Tristetraprolin (TTP) as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry" Biomolecules 15, no. 3: 403. https://doi.org/10.3390/biom15030403
APA StyleCieplak-Rotowska, M. K., Dadlez, M., & Niedzwiecka, A. (2025). Exploring the CNOT1(800–999) HEAT Domain and Its Interactions with Tristetraprolin (TTP) as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Biomolecules, 15(3), 403. https://doi.org/10.3390/biom15030403