
The Pathogenesis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency


Abstract
1. Mitochondrial Metabolism
2. Oxidative Phosphorylation
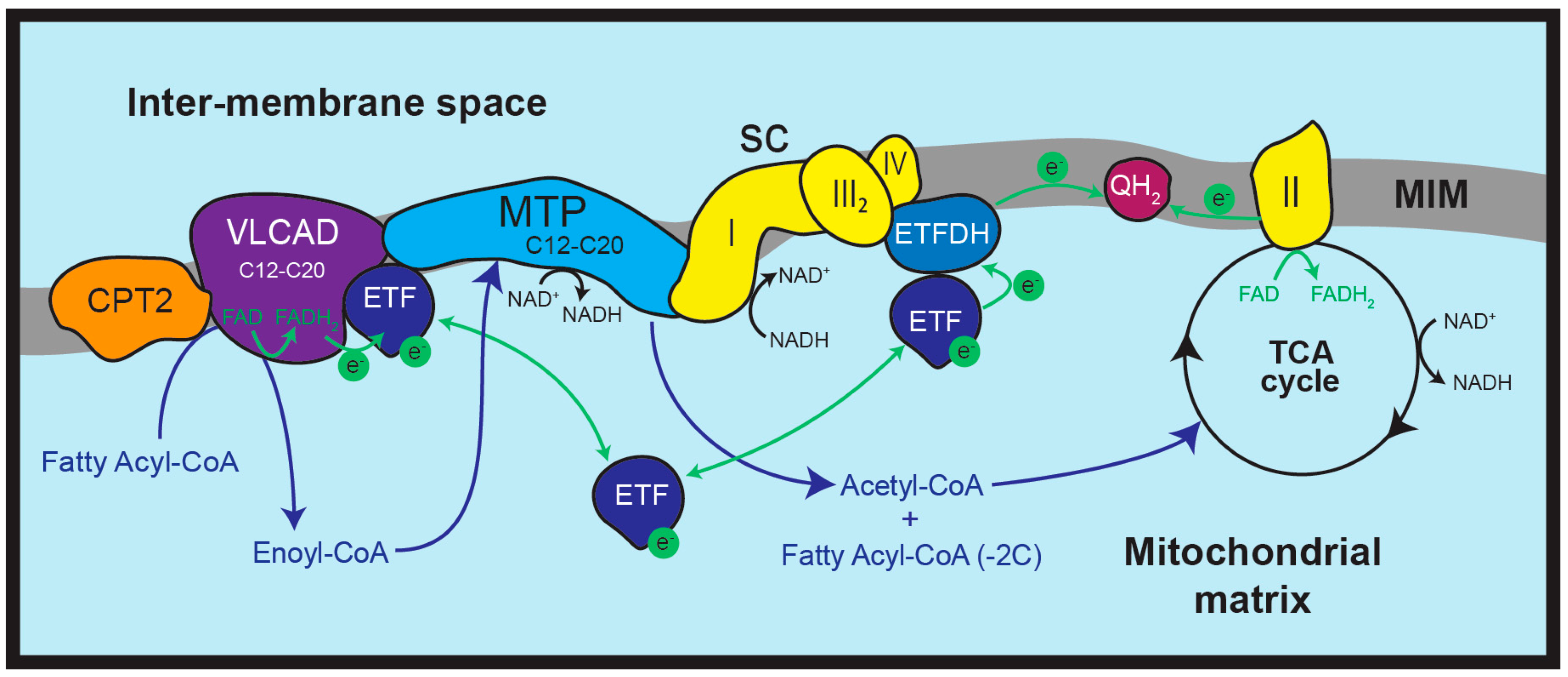
3. Mitochondrial Fatty Acid β-Oxidation (FAO)
4. FAO Disorders
5. VLCAD Deficiency (VLCADD)
VLCADD Treatment
6. Mouse Models of VLCADD
7. Interactions Between FAO and OXPHOS Proteins
8. FAO Deficiency and Secondary OXPHOS Defects
9. Relationship Between VLCADD and OXPHOS Defects
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef]
- Schägger, H. Respiratory chain supercomplexes. IUBMB Life 2001, 52, 119–128. [Google Scholar] [CrossRef]
- Acín-Pérez, R.; Fernández-Silva, P.; Peleato, M.L.; Pérez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef]
- Fedor, J.G.; Hirst, J. Mitochondrial Supercomplexes Do Not Enhance Catalysis by Quinone Channeling. Cell Metab. 2018, 28, 525–531.e524. [Google Scholar] [CrossRef]
- Moreno-Lastres, D.; Fontanesi, F.; Garcia-Consuegra, I.; Martin, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 2012, 15, 324–335. [Google Scholar] [CrossRef]
- Cogliati, S.; Calvo, E.; Loureiro, M.; Guaras, A.M.; Nieto-Arellano, R.; Garcia-Poyatos, C.; Ezkurdia, I.; Mercader, N.; Vázquez, J.; Enriquez, J.A. Mechanism of super-assembly of respiratory complexes III and IV. Nature 2016, 539, 579–582. [Google Scholar] [CrossRef]
- Sun, D.; Li, B.; Qiu, R.; Fang, H.; Lyu, J. Cell Type-Specific Modulation of Respiratory Chain Supercomplex Organization. Int. J. Mol. Sci. 2016, 17, 926. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Chapa-Dubocq, X.R.; Khuchua, Z.; Camara, A.K. Mitochondrial respiratory supercomplexes in mammalian cells: Structural versus functional role. J. Mol. Med. 2021, 99, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mohsen, A.W.; Mihalik, S.J.; Goetzman, E.S.; Vockley, J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem. 2010, 285, 29834–29841. [Google Scholar] [CrossRef]
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.; St Croix, C.; et al. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. Biol. Chem. 2019, 294, 12380–12391. [Google Scholar] [CrossRef]
- Panov, A.V. The Structure of the Cardiac Mitochondria Respirasome Is Adapted for the β-Oxidation of Fatty Acids. Int. J. Mol. Sci. 2024, 25, 2410. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Mannaerts, G.P.; van Veldhoven, P.P. Functions and organization of peroxisomal beta-oxidation. Ann. New York Acad. Sci. 1996, 804, 99–115. [Google Scholar] [CrossRef]
- Wanders, R.J.; Vreken, P.; Ferdinandusse, S.; Jansen, G.A.; Waterham, H.R.; van Roermund, C.W.; Van Grunsven, E.G. Peroxisomal fatty acid alpha- and beta-oxidation in humans: Enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem. Soc. Trans. 2001, 29, 250–267. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Waterham, H.R. Biochemistry of Mammalian Peroxisomes Revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Kihara, A. Very long-chain fatty acids: Elongation, physiology and related disorders. J. Biochem. 2012, 152, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, J.K.; Qin, Y.-M. β-Oxidation—Strategies for the metabolism of a wide variety of acyl-CoA esters. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2000, 1484, 117–128. [Google Scholar] [CrossRef]
- Ghisla, S.; Thorpe, C. Acyl-CoA dehydrogenases. Eur. J. Biochem. 2004, 271, 494–508. [Google Scholar] [CrossRef]
- Chegary, M.; Brinke, H.t.; Ruiter, J.P.N.; Wijburg, F.A.; Stoll, M.S.K.; Minkler, P.E.; van Weeghel, M.; Schulz, H.; Hoppel, C.L.; Wanders, R.J.A.; et al. Mitochondrial long chain fatty acid β-oxidation in man and mouse. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2009, 1791, 806–815. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Beck, M.E.; Goetzman, E.S. The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol. 2019, 26, 101253. [Google Scholar] [CrossRef]
- Ensenauer, R.; He, M.; Willard, J.-M.; Goetzman, E.S.; Corydon, T.J.; Vandahl, B.B.; Mohsen, A.-W.; Isaya, G.; Vockley, J. Human Acyl-CoA Dehydrogenase-9 Plays a Novel Role in the Mitochondrial β-Oxidation of Unsaturated Fatty Acids. J. Biol. Chem. 2005, 280, 32309–32316. [Google Scholar] [CrossRef]
- He, M.; Pei, Z.; Mohsen, A.W.; Watkins, P.; Murdoch, G.; Van Veldhoven, P.P.; Ensenauer, R.; Vockley, J. Identification and characterization of new long chain acyl-CoA dehydrogenases. Mol. Genet. Metab. 2011, 102, 418–429. [Google Scholar] [CrossRef]
- Nouws, J.; Nijtmans, L.; Houten, S.M.; van den Brand, M.; Huynen, M.; Venselaar, H.; Hoefs, S.; Gloerich, J.; Kronick, J.; Hutchin, T.; et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010, 12, 283–294. [Google Scholar] [CrossRef]
- Morant-Ferrando, B.; Jimenez-Blasco, D.; Alonso-Batan, P.; Agulla, J.; Lapresa, R.; Garcia-Rodriguez, D.; Yunta-Sanchez, S.; Lopez-Fabuel, I.; Fernandez, E.; Carmeliet, P.; et al. Fatty acid oxidation organizes mitochondrial supercomplexes to sustain astrocytic ROS and cognition. Nat. Metab. 2023, 5, 1290–1302. [Google Scholar] [CrossRef]
- Barber, C.N.; Raben, D.M. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front. Cell. Neurosci. 2019, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Koczwara, J.B.; Gallelli, C.A.; Vergara, D.; Micioni Di Bonaventura, M.V.; Gaetani, S.; Giudetti, A.M. Fats for thoughts: An update on brain fatty acid metabolism. Int. J. Biochem. Cell Biol. 2017, 84, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty Acids in Energy Metabolism of the Central Nervous System. BioMed. Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Izai, K.; Orii, T.; Hashimoto, T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J. Biol. Chem. 1992, 267, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Perevoshchikova, I.V.; Quinlan, C.L.; Orr, A.L.; Gerencser, A.A.; Brand, M.D. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2013, 61, 298–309. [Google Scholar] [CrossRef]
- Panov, A.V.; Mayorov, V.I.; Dikalova, A.E.; Dikalov, S.I. Long-Chain and Medium-Chain Fatty Acids in Energy Metabolism of Murine Kidney Mitochondria. Int. J. Mol. Sci. 2023, 24, 379. [Google Scholar] [CrossRef]
- Kiens, B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol. Rev. 2006, 86, 205–243. [Google Scholar] [CrossRef]
- Wajner, M.; Amaral, A.U. Mitochondrial dysfunction in fatty acid oxidation disorders: Insights from human and animal studies. Biosci. Rep. 2016, 36, e00281. [Google Scholar] [CrossRef]
- Tonin, A.M.; Amaral, A.U.; Busanello, E.N.B.; Grings, M.; Castilho, R.F.; Wajner, M. Long-chain 3-hydroxy fatty acids accumulating in long-chain 3-hydroxyacyl-CoA dehydrogenase and mitochondrial trifunctional protein deficiencies uncouple oxidative phosphorylation in heart mitochondria. J. Bioenerg. Biomembr. 2013, 45, 47–57. [Google Scholar] [CrossRef]
- Chace, D.H.; Kalas, T.A.; Naylor, E.W. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin. Chem. 2003, 49, 1797–1817. [Google Scholar] [CrossRef]
- Violante, S.; Ijlst, L.; Te Brinke, H.; Tavares de Almeida, I.; Wanders, R.J.; Ventura, F.V.; Houten, S.M. Carnitine palmitoyltransferase 2 and carnitine/acylcarnitine translocase are involved in the mitochondrial synthesis and export of acylcarnitines. FASEB J. 2013, 27, 2039–2044. [Google Scholar] [CrossRef]
- Ventura, F.V.; Costa, C.G.; Struys, E.A.; Ruiter, J.; Allers, P.; Ijlst, L.; Tavares de Almeida, I.; Duran, M.; Jakobs, C.; Wanders, R.J.A. Quantitative acylcarnitine profiling in fibroblasts using [U-13C] palmitic acid: An improved tool for the diagnosis of fatty acid oxidation defects. Clin. Chim. Acta 1999, 281, 1–17. [Google Scholar] [CrossRef]
- Landau, Y.E.; Lichter-Konecki, U.; Levy, H.L. Genomics in Newborn Screening. J. Pediatr. 2014, 164, 14–19. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Ruiter, J.P.N.; Ijlst, L.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef]
- Schiff, M.; Mohsen, A.W.; Karunanidhi, A.; McCracken, E.; Yeasted, R.; Vockley, J. Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2013, 109, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Souri, M.; Aoyama, T.; Hoganson, G.; Hashimoto, T. Very-long-chain acyl-CoA dehydrogenase subunit assembles to the dimer form on mitochondrial inner membrane. FEBS Lett. 1998, 426, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Izai, K.; Uchida, Y.; Orii, T.; Yamamoto, S.; Hashimoto, T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. I. Purification and properties of very-long-chain acyl-coenzyme A dehydrogenase. J. Biol. Chem. 1992, 267, 1027–1033. [Google Scholar] [CrossRef]
- Andresen, B.S.; Bross, P.; Vianey-Saban, C.; Divry, P.; Zabot, M.-T.; Roe, C.R.; Nada, M.A.; Byskov, A.; Kruse, T.A.; Neve, S.; et al. Cloning and Characterization of Human Very-Long-Chain Acyl-CoA Dehydrogenase cDNA, Chromosomal Assignment of the Gene and Identification in Four Patients of Nine Different Mutations Within the VLCAD Gene. Hum. Mol. Genet. 1996, 5, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, C.; Largillière, C.; Zabot, M.T.; Mathieu, M.; Vianey-Saban, C. Very long chain acyl-CoA dehydrogenase deficiency: Identification of a new inborn error of mitochondrial fatty acid oxidation in fibroblasts. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1993, 1180, 327–329. [Google Scholar] [CrossRef]
- Marsden, D.; Bedrosian, C.L.; Vockley, J. Impact of newborn screening on the reported incidence and clinical outcomes associated with medium- and long-chain fatty acid oxidation disorders. Genet. Med. 2021, 23, 816–829. [Google Scholar] [CrossRef]
- Spiekerkoetter, U. Mitochondrial fatty acid oxidation disorders: Clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J. Inherit. Metab. Dis. 2010, 33, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Furuta, S.; Mayazawa, S.; Hashimoto, T. Purification and Properties of Rat Liver Acyl-CoA Dehydrogenases and Electron Transfer Flavoprotein1. J. Biochem. 1981, 90, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, L.; Haussmann, U.; Mueller, M.; Spiekerkoetter, U. VLCAD enzyme activity determinations in newborns identified by screening: A valuable tool for risk assessment. J. Inherit. Metab. Dis. 2012, 35, 269–277. [Google Scholar] [CrossRef]
- Obaid, A.; Nashabat, M.; Alfadhel, M.; Alasmari, A.; Al Mutairi, F.; Alswaid, A.; Faqeih, E.; Mushiba, A.; Albanyan, M.; Alalwan, M.; et al. Clinical, Biochemical, and Molecular Features in 37 Saudi Patients with Very Long Chain Acyl CoA Dehydrogenase Deficiency. JIMD Rep. 2018, 40, 47–53. [Google Scholar] [CrossRef]
- Watanabe, H.; Orii, K.E.; Fukao, T.; Song, X.Q.; Aoyama, T.; IJlst, L.; Ruiter, J.; Wanders, R.J.; Kondo, N. Molecular basis of very long chain acyl-CoA dehydrogenase deficiency in three Israeli patients: Identification of a complex mutant allele with P65L and K247Q mutations, the former being an exonic mutation causing exon 3 skipping. Hum. Mutat. 2000, 15, 430–438. [Google Scholar] [CrossRef]
- Evans, M.; Andresen, B.S.; Nation, J.; Boneh, A. VLCAD deficiency: Follow-up and outcome of patients diagnosed through newborn screening in Victoria. Mol. Genet. Metab. 2016, 118, 282–287. [Google Scholar] [CrossRef]
- Fukao, T.; Watanabe, H.; Orii, K.; Takahashi, Y.; Hirano, A.; Kondo, T.; Yamaguchi, S.; Aoyama, T.; Kondo, N. Myopathic form of very-long chain acyl-coa dehydrogenase deficiency: Evidence for temperature-sensitive mild mutations in both mutant alleles in a Japanese girl. Pediatr. Res. 2001, 49, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U.; Sun, B.; Zytkovicz, T.; Wanders, R.; Strauss, A.W.; Wendel, U. MS/MS-based newborn and family screening detects asymptomatic patients with very-long-chain acyl-CoA dehydrogenase deficiency. J. Pediatr. 2003, 143, 335–342. [Google Scholar] [CrossRef]
- Mathur, A.; Sims, H.F.; Gopalakrishnan, D.; Gibson, B.; Rinaldo, P.; Vockley, J.; Hug, G.; Strauss, A.W. Molecular Heterogeneity in Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency Causing Pediatric Cardiomyopathy and Sudden Death. Circulation 1999, 99, 1337–1343. [Google Scholar] [CrossRef]
- Pena, L.D.; van Calcar, S.C.; Hansen, J.; Edick, M.J.; Walsh Vockley, C.; Leslie, N.; Cameron, C.; Mohsen, A.W.; Berry, S.A.; Arnold, G.L.; et al. Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol. Genet. Metab. 2016, 118, 272–281. [Google Scholar] [CrossRef]
- Andresen, B.S.; Olpin, S.; Poorthuis, B.J.H.M.; Scholte, H.R.; Vianey-Saban, C.; Wanders, R.; Ijlst, L.; Morris, A.; Pourfarzam, M.; Bartlett, K.; et al. Clear Correlation of Genotype with Disease Phenotype in Very–Long-Chain Acyl-CoA Dehydrogenase Deficiency. Am. J. Hum. Genet. 1999, 64, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Strauss, A.W.; Powell, C.K.; Hale, D.E.; Anderson, M.M.; Ahuja, A.; Brackett, J.C.; Sims, H.F. Molecular basis of human mitochondrial very-long-chain acyl-CoA dehydrogenase deficiency causing cardiomyopathy and sudden death in childhood. Proc. Natl. Acad. Sci. USA 1995, 92, 10496–10500. [Google Scholar] [CrossRef]
- Brown, A.; Crowe, L.; Andresen, B.S.; Anderson, V.; Boneh, A. Neurodevelopmental profiles of children with very long chain acyl-CoA dehydrogenase deficiency diagnosed by newborn screening. Mol. Genet. Metab. 2014, 113, 278–282. [Google Scholar] [CrossRef]
- Miller, M.J.; Burrage, L.C.; Gibson, J.B.; Strenk, M.E.; Lose, E.J.; Bick, D.P.; Elsea, S.H.; Sutton, V.R.; Sun, Q.; Graham, B.H.; et al. Recurrent ACADVL molecular findings in individuals with a positive newborn screen for very long chain acyl-coA dehydrogenase (VLCAD) deficiency in the United States. Mol. Genet. Metab. 2015, 116, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Ryder, B.; Knoll, D.; Love, D.R.; Shepherd, P.; Love, J.M.; Reed, P.W.; de Hora, M.; Webster, D.; Glamuzina, E.; Wilson, C. The natural history of elevated tetradecenoyl-L-carnitine detected by newborn screening in New Zealand: Implications for very long chain acyl-CoA dehydrogenase deficiency screening and treatment. J. Inherit. Metab. Dis. 2016, 39, 409–414. [Google Scholar] [CrossRef]
- Boneh, A.; Andresen, B.S.; Gregersen, N.; Ibrahim, M.; Tzanakos, N.; Peters, H.; Yaplito-Lee, J.; Pitt, J.J. VLCAD deficiency: Pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol. Genet. Metab. 2006, 88, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Seminotti, B.; Leipnitz, G.; Karunanidhi, A.; Kochersperger, C.; Roginskaya, V.Y.; Basu, S.; Wang, Y.; Wipf, P.; Van Houten, B.; Mohsen, A.W.; et al. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum. Mol. Genet. 2019, 28, 928–941. [Google Scholar] [CrossRef]
- Van Calcar, S.C.; Sowa, M.; Rohr, F.; Beazer, J.; Setlock, T.; Weihe, T.U.; Pendyal, S.; Wallace, L.S.; Hansen, J.G.; Stembridge, A.; et al. Nutrition management guideline for very-long chain acyl-CoA dehydrogenase deficiency (VLCAD): An evidence- and consensus-based approach. Mol. Genet. Metab. 2020, 131, 23–37. [Google Scholar] [CrossRef]
- Vockley, J.; Charrow, J.; Ganesh, J.; Eswara, M.; Diaz, G.A.; McCracken, E.; Conway, R.; Enns, G.M.; Starr, J.; Wang, R.; et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol. Genet. Metab. 2016, 119, 223–231. [Google Scholar] [CrossRef]
- Vockley, J.; Burton, B.K.; Berry, G.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Chapman, K.A.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; et al. Triheptanoin for the treatment of long-chain fatty acid oxidation disorders: Final results of an open-label, long-term extension study. J Inherit Metab Dis 2023, 46, 943–955. [Google Scholar] [CrossRef]
- Tucci, S.; Flögel, U.; Hermann, S.; Sturm, M.; Schäfers, M.; Spiekerkoetter, U. Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD−/−) mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 677–685. [Google Scholar] [CrossRef]
- Chin, H.-L.; Lai, P.S.; Tay, S.K.H. A clinical approach to diagnosis and management of mitochondrial myopathies. Neurotherapeutics 2024, 21, e00304. [Google Scholar] [CrossRef] [PubMed]
- D’Annibale, O.M.; Phua, Y.L.; Van’t Land, C.; Karunanidhi, A.; Dorenbaum, A.; Mohsen, A.-W.; Vockley, J. Treatment of VLCAD-Deficient Patient Fibroblasts with Peroxisome Proliferator-Activated Receptor δ Agonist Improves Cellular Bioenergetics. Cells 2022, 11, 2635. [Google Scholar] [CrossRef] [PubMed]
- Steele, H.; Gomez-Duran, A.; Pyle, A.; Hopton, S.; Newman, J.; Stefanetti, R.J.; Charman, S.J.; Parikh, J.D.; He, L.; Viscomi, C.; et al. Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol. Med. 2020, 12, e11589. [Google Scholar] [CrossRef]
- Djouadi, F.; Aubey, F.; Schlemmer, D.; Ruiter, J.P.; Wanders, R.J.; Strauss, A.W.; Bastin, J. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum. Mol. Genet. 2005, 14, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, H.; Yamada, K.; Egawa, K.; Ishige, M.; Ochi, F.; Watanabe, A.; Kawakami, S.; Kuzume, K.; Watanabe, K.; Sameshima, K.; et al. Efficacy of bezafibrate for preventing myopathic attacks in patients with very long-chain acyl-CoA dehydrogenase deficiency. Brain Dev. 2021, 43, 214–219. [Google Scholar] [CrossRef]
- Lund, M.; Andersen, K.G.; Heaton, R.; Hargreaves, I.P.; Gregersen, N.; Olsen, R.K.J. Bezafibrate activation of PPAR drives disturbances in mitochondrial redox bioenergetics and decreases the viability of cells from patients with VLCAD deficiency. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166100. [Google Scholar] [CrossRef]
- Bastin, J.; Lopes-Costa, A.; Djouadi, F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum. Mol. Genet. 2011, 20, 2048–2057. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Mengarelli, I.; Wüst, R.C.I.; Baartscheer, A.; Bleeker, J.C.; Coronel, R.; Ferdinandusse, S.; Guan, K.; IJlst, L.; Li, W.; et al. Electrophysiological Abnormalities in VLCAD Deficient hiPSC-Cardiomyocytes Can Be Improved by Lowering Accumulation of Fatty Acid Oxidation Intermediates. Int. J. Mol. Sci. 2020, 21, 2589. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Wall, S.R.; Olley, P.M.; Davies, N.J. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ. Res. 1988, 63, 1036–1043. [Google Scholar] [CrossRef]
- Holubarsch, C.J.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. 2007, 113, 205–212. [Google Scholar] [CrossRef]
- Gueguen, N.; Desquiret-Dumas, V.; Leman, G.; Chupin, S.; Baron, S.; Nivet-Antoine, V.; Vessières, E.; Ayer, A.; Henrion, D.; Lenaers, G.; et al. Resveratrol Directly Binds to Mitochondrial Complex I and Increases Oxidative Stress in Brain Mitochondria of Aged Mice. PLoS ONE 2015, 10, e0144290. [Google Scholar] [CrossRef] [PubMed]
- Zieger, M.; Keeler, A.M.; Flotte, T.R.; ElMallah, M.K. AAV9 gene replacement therapy for respiratory insufficiency in very-long chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2019, 42, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-J.; Mohsen, A.I.W.; Mihalik, S.; Solo, K.; Aliu, E.; Shi, H.; Basu, S.; Kochersperger, C.; Van’t Land, C.; Karunanidhi, A.; et al. Synthetic mRNA rescues very long-chain acyl-CoA dehydrogenase deficiency in patient fibroblasts and a murine model. Mol. Genet. Metab. 2023, 138, 106982. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.B.; Hamm, D.A.; Millington, D.S.; Matern, D.; Vockley, J.; Rinaldo, P.; Pinkert, C.A.; Rhead, W.J.; Lindsey, J.R.; Wood, P.A. Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum. Mol. Genet. 2001, 10, 2069–2077. [Google Scholar] [CrossRef]
- Exil, V.J.; Roberts, R.L.; Sims, H.; McLaughlin, J.E.; Malkin, R.A.; Gardner, C.D.; Ni, G.; Rottman, J.N.; Strauss, A.W. Very-long-chain acyl-coenzyme a dehydrogenase deficiency in mice. Circ. Res. 2003, 93, 448–455. [Google Scholar] [CrossRef]
- Babcock, S.J.; Houten, S.M.; Gillingham, M.B. A review of fatty acid oxidation disorder mouse models. Mol. Genet. Metab. 2024, 142, 108351. [Google Scholar] [CrossRef]
- Zytkovicz, T.H.; Fitzgerald, E.F.; Marsden, D.; Larson, C.A.; Shih, V.E.; Johnson, D.M.; Strauss, A.W.; Comeau, A.M.; Eaton, R.B.; Grady, G.F. Tandem Mass Spectrometric Analysis for Amino, Organic, and Fatty Acid Disorders in Newborn Dried Blood Spots: A Two-Year Summary from the New England Newborn Screening Program. Clin. Chem. 2001, 47, 1945–1955. [Google Scholar] [CrossRef]
- Exil, V.J.; Gardner, C.D.; Rottman, J.N.; Sims, H.; Bartelds, B.; Khuchua, Z.; Sindhal, R.; Ni, G.; Strauss, A.W. Abnormal mitochondrial bioenergetics and heart rate dysfunction in mice lacking very-long-chain acyl-CoA dehydrogenase. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H1289–H1297. [Google Scholar] [CrossRef]
- Cox, K.B.; Liu, J.; Tian, L.; Barnes, S.; Yang, Q.; Wood, P.A. Cardiac hypertrophy in mice with long-chain acyl-CoA dehydrogenase or very long-chain acyl-CoA dehydrogenase deficiency. Lab. Investig. 2009, 89, 1348–1354. [Google Scholar] [CrossRef]
- ter Veld, F.; Primassin, S.; Hoffmann, L.; Mayatepek, E.; Spiekerkoetter, U. Corresponding increase in long-chain acyl-CoA and acylcarnitine after exercise in muscle from VLCAD mice. J. Lipid Res. 2009, 50, 1556–1562. [Google Scholar] [CrossRef]
- Riggs, C.E.; Michaelides, M.A.; Parpa, K.M.; Smith-Blair, N.J. The effects of aerobic interval training on the left ventricular morphology and function of VLCAD-deficient mice. Eur. J. Appl. Physiol. 2010, 110, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Bakermans, A.J.; van Weeghel, M.; Denis, S.; Nicolay, K.; Prompers, J.J.; Houten, S.M. Carnitine supplementation attenuates myocardial lipid accumulation in long-chain acyl-CoA dehydrogenase knockout mice. J. Inherit. Metab. Dis. 2013, 36, 973–981. [Google Scholar] [CrossRef]
- Goetzman, E.S.; Tian, L.; Wood, P.A. Differential induction of genes in liver and brown adipose tissue regulated by peroxisome proliferator-activated receptor-α during fasting and cold exposure in acyl-CoA dehydrogenase-deficient mice. Mol. Genet. Metab. 2005, 84, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Diekman, E.F.; van Weeghel, M.; Wanders, R.J.A.; Visser, G.; Houten, S.M. Food withdrawal lowers energy expenditure and induces inactivity in long-chain fatty acid oxidation–deficient mouse models. FASEB J. 2014, 28, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- Sumegi, B.; Srere, P.A. Complex I binds several mitochondrial NAD-coupled dehydrogenases. J. Biol. Chem. 1984, 259, 15040–15045. [Google Scholar] [CrossRef]
- PARKER, A.; ENGEL, P.C. Preliminary evidence for the existence of specific functional assemblies between enzymes of the β-oxidation pathway and the respiratory chain. Biochem. J. 2000, 345, 429–435. [Google Scholar] [CrossRef]
- Qin, C.; Gong, S.; Liang, T.; Zhang, Z.; Thomas, J.; Deng, J.; Liu, Y.; Hu, P.; Zhu, B.; Song, S.; et al. HADHA Regulates Respiratory Complex Assembly and Couples FAO and OXPHOS. Adv. Sci. 2024, 11, 2405147. [Google Scholar] [CrossRef]
- Nouws, J.; Te Brinke, H.; Nijtmans, L.G.; Houten, S.M. ACAD9, a complex I assembly factor with a moonlighting function in fatty acid oxidation deficiencies. Hum. Mol. Genet. 2014, 23, 1311–1319. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef]
- Ventura, F.V.; Ruiter, J.P.N.; IJlst, L.; Tavares de Almeida, I.; Wanders, R.J.A. Lactic acidosis in long-chain fatty acid β-oxidation disorders. J. Inherit. Metab. Dis. 1998, 21, 645–654. [Google Scholar] [CrossRef]
- Tyni, T.; Majander, A.; Kalimo, H.; Rapola, J.; Pihko, H. Pathology of skeletal muscle and impaired respiratory chain function in long-chain 3-hydroxyacyl-coa dehydrogenase deficiency with the G1528C mutation. Neuromuscul. Disord. 1996, 6, 327–337. [Google Scholar] [CrossRef]
- Ribas, G.S.; Vargas, C.R. Evidence that Oxidative Disbalance and Mitochondrial Dysfunction are Involved in the Pathophysiology of Fatty Acid Oxidation Disorders. Cell. Mol. Neurobiol. 2022, 42, 521–532. [Google Scholar] [CrossRef]
- Das, A.M.; Fingerhut, R.; Wanders, R.J.; Ullrich, K. Secondary respiratory chain defect in a boy with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Possible diagnostic pitfalls. Eur. J. Pediatr. 2000, 159, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Cecatto, C.; Godoy, K.d.S.; da Silva, J.C.; Amaral, A.U.; Wajner, M. Disturbance of mitochondrial functions provoked by the major long-chain 3-hydroxylated fatty acids accumulating in MTP and LCHAD deficiencies in skeletal muscle. Toxicol. Vitr. 2016, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tonin, A.M.; Ferreira, G.C.; Grings, M.; Viegas, C.M.; Busanello, E.N.; Amaral, A.U.; Zanatta, Â.; Schuck, P.F.; Wajner, M. Disturbance of mitochondrial energy homeostasis caused by the metabolites accumulating in LCHAD and MTP deficiencies in rat brain. Life Sci. 2010, 86, 825–831. [Google Scholar] [CrossRef]
- Lim, S.C.; Tajika, M.; Shimura, M.; Carey, K.T.; Stroud, D.A.; Murayama, K.; Ohtake, A.; McKenzie, M. Loss of the Mitochondrial Fatty Acid β-Oxidation Protein Medium-Chain Acyl-Coenzyme A Dehydrogenase Disrupts Oxidative Phosphorylation Protein Complex Stability and Function. Sci. Rep. 2018, 8, 153. [Google Scholar] [CrossRef]
- Scaini, G.; Simon, K.R.; Tonin, A.M.; Busanello, E.N.B.; Moura, A.P.; Ferreira, G.C.; Wajner, M.; Streck, E.L.; Schuck, P.F. Toxicity of octanoate and decanoate in rat peripheral tissues: Evidence of bioenergetic dysfunction and oxidative damage induction in liver and skeletal muscle. Mol. Cell. Biochem. 2012, 361, 329–335. [Google Scholar] [CrossRef]
- Amaral, A.U.; Cecatto, C.; da Silva, J.C.; Wajner, A.; Godoy, K.d.S.; Ribeiro, R.T.; Wajner, M. cis-4-Decenoic and decanoic acids impair mitochondrial energy, redox and Ca2+ homeostasis and induce mitochondrial permeability transition pore opening in rat brain and liver: Possible implications for the pathogenesis of MCAD deficiency. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1363–1372. [Google Scholar] [CrossRef]
- Burgin, H.J.; McKenzie, M. Understanding the role of OXPHOS dysfunction in the pathogenesis of ECHS1 deficiency. FEBS Lett. 2020, 594, 590–610. [Google Scholar] [CrossRef]
- Peters, H.; Buck, N.; Wanders, R.; Ruiter, J.; Waterham, H.; Koster, J.; Yaplito-Lee, J.; Ferdinandusse, S.; Pitt, J. ECHS1 mutations in Leigh disease: A new inborn error of metabolism affecting valine metabolism. Brain 2014, 137, 2903–2908. [Google Scholar] [CrossRef] [PubMed]
- Burgin, H.; Sharpe, A.J.; Nie, S.; Ziemann, M.; Crameri, J.J.; Stojanovski, D.; Pitt, J.; Ohtake, A.; Murayama, K.; McKenzie, M. Loss of mitochondrial fatty acid β-oxidation protein short-chain Enoyl-CoA hydratase disrupts oxidative phosphorylation protein complex stability and function. FEBS J. 2023, 290, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Bennett, M.J.; Hoppel, C.L.; Goodman, S.I.; Weisiger, K.; Ohnstad, C.; Golabi, M.; Packman, S. Mitochondrial respiratory chain complex I deficiency with clinical and biochemical features of long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. J. Pediatr. 2000, 136, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Leipnitz, G.; Mohsen, A.-W.; Karunanidhi, A.; Seminotti, B.; Roginskaya, V.Y.; Markantone, D.M.; Grings, M.; Mihalik, S.J.; Wipf, P.; Van Houten, B.; et al. Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex I deficiency. Sci. Rep. 2018, 8, 1165. [Google Scholar] [CrossRef]
- Gargus, J.J.; Boyle, K.; Bocian, M.; Roe, D.S.; Vianey-Saban, C.; Roe, C.R. Respiratory complex II defect in siblings associated with a symptomatic secondary block in fatty acid oxidation. J. Inherit. Metab. Dis. 2003, 26, 659–670. [Google Scholar] [CrossRef]
- Laforêt, P.; Acquaviva-Bourdain, C.; Rigal, O.; Brivet, M.; Penisson-Besnier, I.; Chabrol, B.; Chaigne, D.; Boespflug-Tanguy, O.; Laroche, C.; Bedat-Millet, A.L.; et al. Diagnostic assessment and long-term follow-up of 13 patients with Very Long-Chain Acyl-Coenzyme A dehydrogenase (VLCAD) deficiency. Neuromuscul. Disord. 2009, 19, 324–329. [Google Scholar] [CrossRef]
- Alfares, A.; Alfadhel, M.; Mujamammi, A.; Alotaibi, B.; Albahkali, S.; Al Balwi, M.; Benabdelkamel, H.; Masood, A.; Ali, R.; Almuaysib, A.; et al. Proteomic and Molecular Assessment of the Common Saudi Variant in ACADVL Gene Through Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2020, 7, 365. [Google Scholar] [CrossRef]
- Yang, H.; Zhao, C.; Tang, M.-C.; Wang, Y.; Wang, S.P.; Allard, P.; Furtos, A.; Mitchell, G.A. Inborn errors of mitochondrial acyl-coenzyme a metabolism: Acyl-CoA biology meets the clinic. Mol. Genet. Metab. 2019, 128, 30–44. [Google Scholar] [CrossRef]
- Matsuishi, T.; Stumpf, D.A.; Seliem, M.; Eguren, L.A.; Chrislip, K. Propionate mitochondrial toxicity in liver and skeletal muscle: Acyl CoA levels. Biochem. Med. Metab. Biol. 1991, 45, 244–253. [Google Scholar] [CrossRef]
- McKenzie, M.; Lazarou, M.; Thorburn, D.R.; Ryan, M.T. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J. Mol. Biol. 2006, 361, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Vieira Neto, E.; Wang, M.; Szuminsky, A.J.; Ferraro, L.; Koppes, E.; Wang, Y.; Van’t Land, C.; Mohsen, A.-W.; Zanatta, G.; El-Gharbawy, A.H.; et al. Mitochondrial bioenergetics and cardiolipin remodeling abnormalities in mitochondrial trifunctional protein deficiency. JCI Insight 2024, 9, e176887. [Google Scholar] [CrossRef] [PubMed]
- Cecatto, C.; Amaral, A.U.; Roginski, A.C.; Castilho, R.F.; Wajner, M. Impairment of mitochondrial bioenergetics and permeability transition induction caused by major long-chain fatty acids accumulating in VLCAD deficiency in skeletal muscle as potential pathomechanisms of myopathy. Toxicol. Vitr. 2020, 62, 104665. [Google Scholar] [CrossRef]
- Cecatto, C.; Amaral, A.U.; da Silva, J.C.; Wajner, A.; Schimit, M.O.V.; da Silva, L.H.R.; Wajner, S.M.; Zanatta, Â.; Castilho, R.F.; Wajner, M. Metabolite accumulation in VLCAD deficiency markedly disrupts mitochondrial bioenergetics and Ca(2+) homeostasis in the heart. FEBS J. 2018, 285, 1437–1455. [Google Scholar] [CrossRef]
- Kakimoto, P.A.H.B.; Tamaki, F.K.; Cardoso, A.R.; Marana, S.R.; Kowaltowski, A.J. H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol. 2015, 4, 375–380. [Google Scholar] [CrossRef]
- Ito, F.; Sono, Y.; Ito, T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef]

{kind=link}
| Mutation | Protein | Reference | Presentation | |
|---|---|---|---|---|
| 1 | c.65C>A | p.S22X | [52,53] | Symptomatic |
| 2 | c.134C>A | P.S45X | [52,53] | Symptomatic |
| 3 | c.494T>C | p.F165S | [52] | Symptomatic |
| 4 | c.1349G>A | p.R450H | [52,54,55] | Symptomatic |
| 5 | c.848T>C | p.V243A | [51,56,57] | Hepatic or Asymptomatic |
| 6 | c.1468G>C | p.A450P | [56,57] | Hepatic or Asymptomatic |
| 7 | c.602A>G | p.Y161C | [56] | Asymptomatic |
| 8 | c.865G>A | p.G289R | [56,58] | Asymptomatic |
| 9 | c.1376G>A | R419Q | [56] | Asymptomatic |
| 10 | c.1844G>A | p.R575Q | [51,57,58] | Cardiac |
| 11 | c.779C>T | p.T220M | [51,57] | Cardiac |
| 12 | c.1405C>T | p.R469W | [51,54,59] | Asymptomatic |
| 13 | c.1532G>C | R511P | [51,58] | Asymptomatic |
| 14 | c.1280G>A | W347ter Frame Shift | [57] | Hepatic |
| 15 | c.1600G>A | p.E454K | [57] | Sudden Death |
| 16 | c.1372T>C | p.F418L | [57] | Cardiac |
| 17 | c.739A>G | p.K207E | [57] | Sudden Death |
| 18 | G-1A | Splice site | [57] | Cardiac |
| 19 | A-2C | Splice site | [57] | Cardiac |
| 20 | Δ887-88 | Frame Shift | [57] | Cardiac |
| 21 | c.1322G>A | p.G401D | [46,54,57,59] | Cardiac |
| 22 | c.637G>C | p.A173P | [57] | Sudden Death |
| 23 | G+1A | Splice site | [57] | Cardiac and Hepatic |
| 24 | Δ386-88 | ΔE89 In Frame Deletion | [57] | Cardiac and Hepatic |
| 25 | ΔG-1 | Splice site | [57] | Cardiac and Hepatic |
| 26 | c.1837C>T | p.R573W | [57,60] | Cardiac |
| 27 | 41 bp insertion | Frame Shift | [57] | Cardiac |
| 28 | ΔG1621 | Frame Shift | [57] | Cardiac and Hepatic |
| 29 | Δ891-3 | ΔK258 In Frame deletion | [57] | Hepatic |
| 30 | ΔT932 | Frame Shift | [57] | Cardiac |
| 31 | c.1146GNC | p.K382N | [58] | Asymptomatic |
| 32 | c.1076C>T | p.A359V | [58] | Asymptomatic |
| 33 | c.1504C>G | p.L502V | [58] | Asymptomatic |
| 34 | c.1066A>G | p.I356V | [58] | Asymptomatic |
| 35 | c.622G>A | p.G208R | [58] | Rhabdomyolysis |
| 36 | c.689C>T | p.T230I | [58] | Symptomatic |
| 37 | c.1173_1174insT | Frame Shift | [58] | Rhabdomyolysis |
| 38 | c.1806_1807delCT | Frame Shift | [58] | Hypoglycaemia |
| 39 | c.388_390delGAG | Unstable protein | [58] | Asymptomatic |
| 40 | c.439C>T | p.P147S | [58] | Elevated creatine kinase and liver function test |
| 41 | c.956C>A | stop codon | [58] | Elevated creatine kinase and liver function test |
| 42 | c.1001T>G | p.M334R | [58] | Asymptomatic |
| 43 | c.889-91delGAG | p.E297del | [54,61] | Cardiac and hypoglycaemia |
| 44 | c.1246G>T | p.A416S | [54,61] | Cardiac and hypoglycaemia |
| 45 | c.1097G>A | p.R366H | [54,59] | Elevated creatine kinase, rhabdomyolysis, metabolic acidosis, and hypoglycaemia |
| 46 | c.1019G>T | p.G340V | [54,62] | Asymptomatic |
| 47 | c.559A>G | p.K187E | [54] | Asymptomatic |
| 48 | c.1226C>T | p.T409M | [54,63] | Asymptomatic |
| 49 | c.481G>A | p.A161T | [54,64] | Mildly symptomatic |
| 50 | c.476A>G | p.Q159R | [54,59] | Mildly symptomatic |
| 51 | c.950T>C | p.V317A | [46,54,59] | Mildly symptomatic |
| 52 | c.1117A>T | p.I373F | [54,64] | Mildly symptomatic |
| 53 | c.1153C>T | p.R385W | [54,64] | Mildly symptomatic |
| 54 | c.1923G>C | p.L641P | [54] | Mildly symptomatic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; McKenzie, M. The Pathogenesis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. Biomolecules 2025, 15, 416. https://doi.org/10.3390/biom15030416
Sharma S, McKenzie M. The Pathogenesis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. Biomolecules. 2025; 15(3):416. https://doi.org/10.3390/biom15030416
Chicago/Turabian StyleSharma, Shashwat, and Matthew McKenzie. 2025. "The Pathogenesis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency" Biomolecules 15, no. 3: 416. https://doi.org/10.3390/biom15030416
APA StyleSharma, S., & McKenzie, M. (2025). The Pathogenesis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. Biomolecules, 15(3), 416. https://doi.org/10.3390/biom15030416