
Biogenic Amine Metabolism and Its Genetic Variations in Autism Spectrum Disorder: A Comprehensive Overview

 , , ,
, , ,
Abstract
1. Introduction
2. Classification of Biogenic Amines
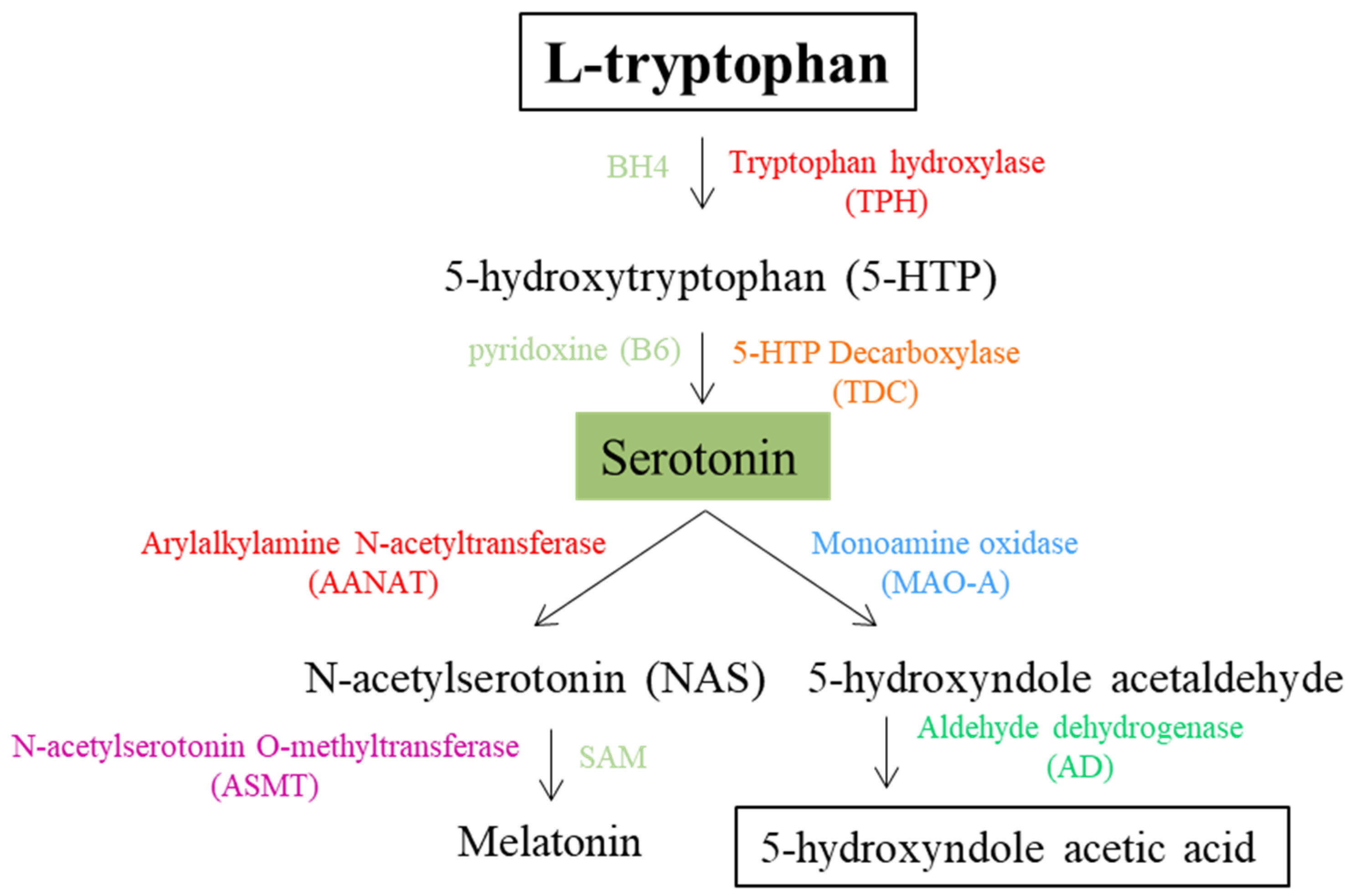
3. Serotonin
4. Histamine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Symbol | Name | Protein ID | E.C. Number | Gene Name | Gene ID | OMIM Gene ID | Disease Association (MIM Phenotype) | Gene Association with ASD (SFARI and Pubmed) | ASD or ASD Associated NDD SNV or CNV | Type of Study | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HDC | L-histidine decarboxylase | P19113 | 4.1.1.22 | HDC | 3067 | *142704 | Gilles de la Tourette syndrome (#137580) | Not listed in SFARI database; found on PubMed in association with autism | CNV, del15q21.2 | Array-CGH | [110] |
| DAO | Diamine oxidase; amine oxidase copper-containing 1 | P19801 | 1.4.3.22 | AOC1 | 26 | *104610 | Not correlated with a MIM phenototype | Not listed in SFARI database; found on PubMed in association with autism | CNV, del7q35q36.1 | Array-CGH | [115] |
| HMT | Histamine N-methyltransferase | P50135 | 2.1.1.8 | HNMT | 3176 | *605238 | Susceptibility to asthma (#600807) and intellectual developmental disorder, autosomal recessive 51—MRT51 (#616739) | Not listed in SFARI database; found on PubMed in association with autism | c.88 C > T (p.Glu30Ter) | WES | [113] |
| MAO (MAO-B) | Monoamine oxidase (2 isoforms) | P27338 | 1.4.3.4 | MAOB | 4129 | *309860 | Not correlated with a MIM phenototype | Strong candidate gene | rs1799836 correlated with serotonin levels in autism | Genotyping | [82] |
| rs6324 correlated with ASD symptom severity | Genotyping | [82] | |||||||||
| CNV, delchrXq11.3q11.3 | Array-CGH | [62] | |||||||||
| CNV, delchrXp11.3-p11.4 | Array-CGH | [63] | |||||||||
| c.392G > T (p.Ser131Ile) | WES | [117] |
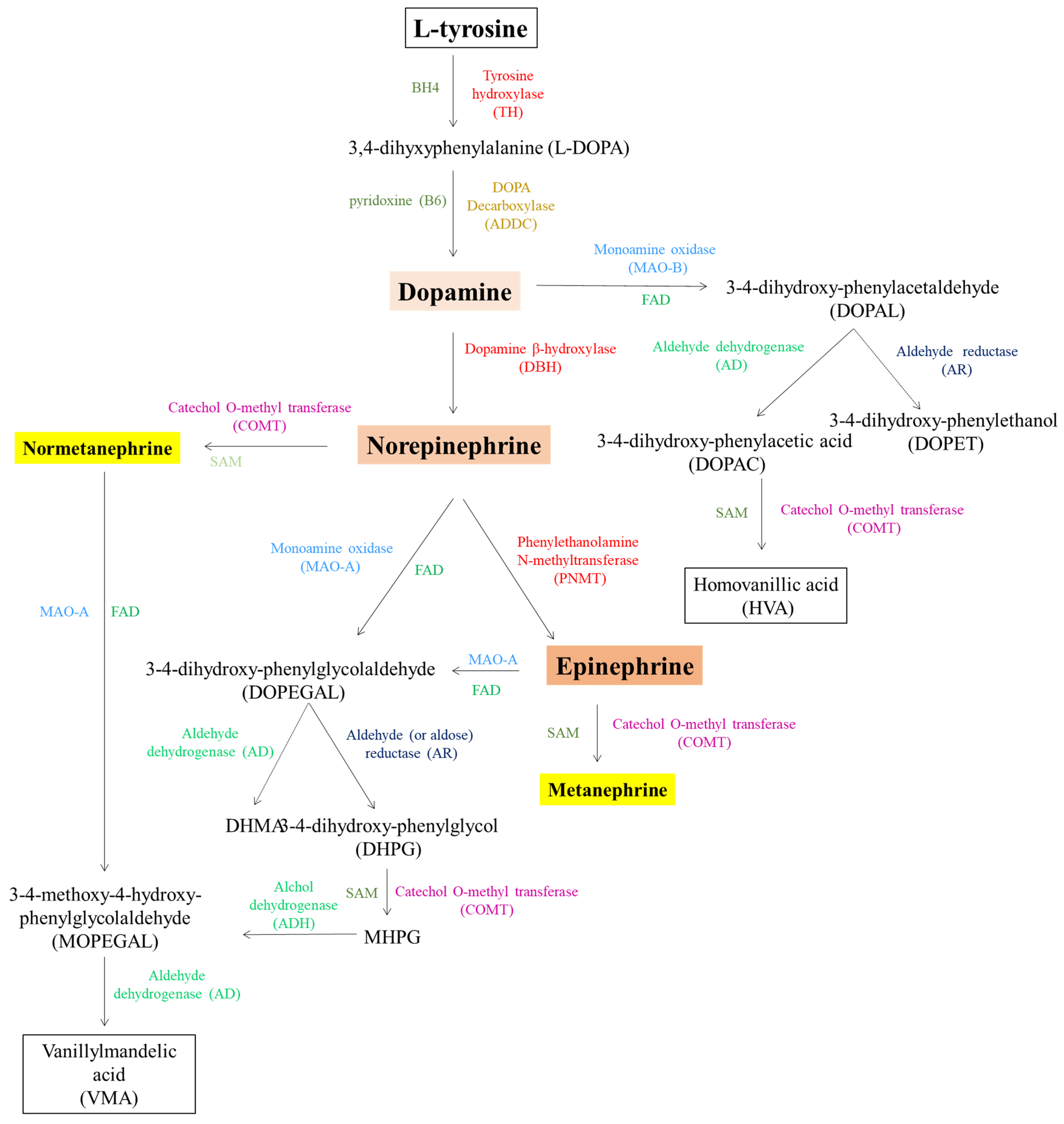
5. Catecholamines
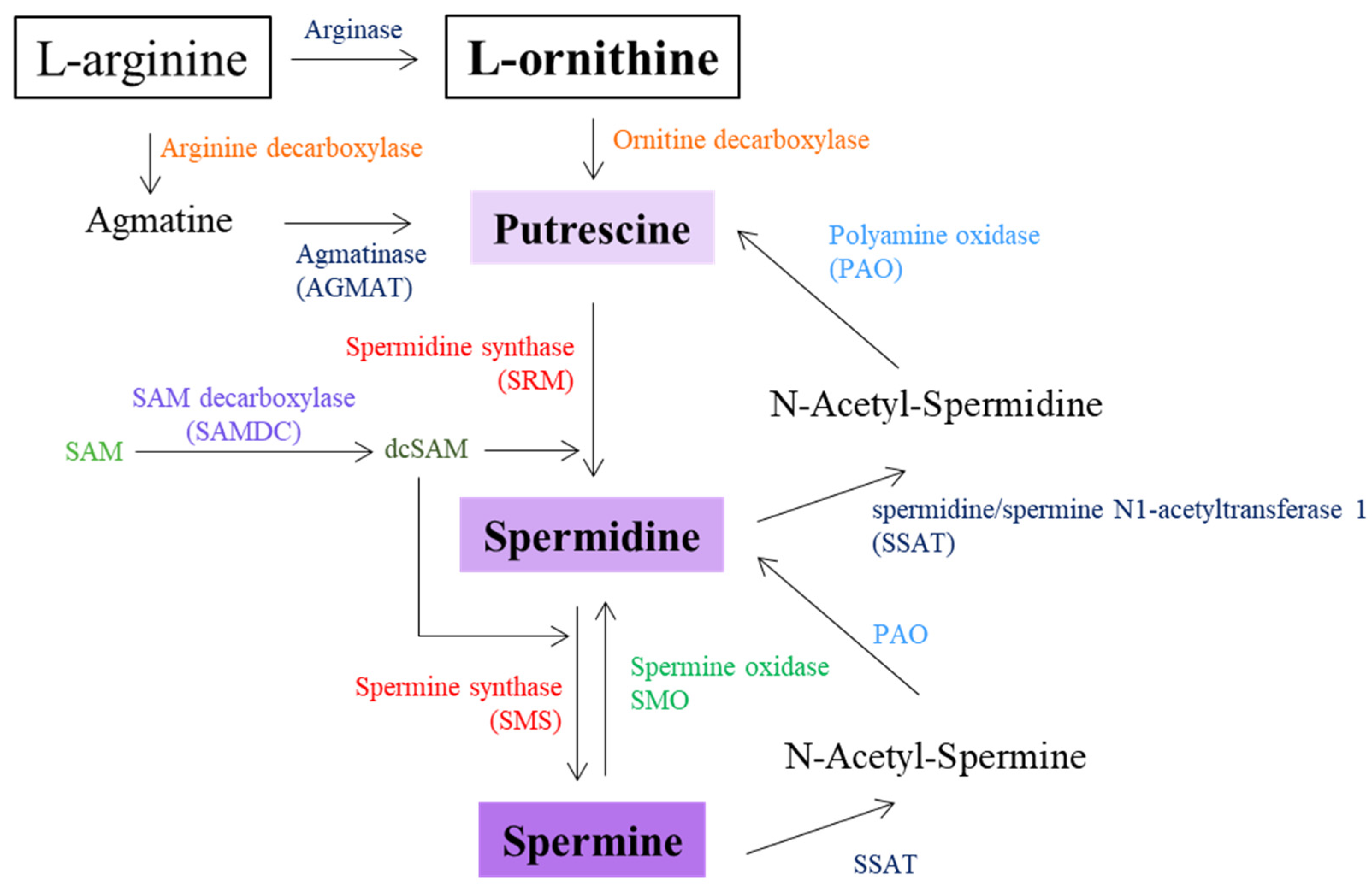
6. Diamines and Polyamines
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2022. [Google Scholar]
- Lai, M.C.; Kassee, C.; Besney, R.; Bonato, S.; Hull, L.; Mandy, W.; Szatmari, P.; Ameis, S.H. Prevalence of co-occurring mental health diagnoses in the autism population: A systematic review and meta-analysis. Lancet Psychiatry 2019, 6, 819–829. [Google Scholar] [CrossRef]
- Micai, M.; Fatta, L.M.; Gila, L.; Caruso, A.; Salvitti, T.; Fulceri, F.; Ciaramella, A.; D’Amico, R.; Del Giovane, C.; Bertelli, M.; et al. Prevalence of co-occurring conditions in children and adults with autism spectrum disorder: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2023, 155, 105436. [Google Scholar] [CrossRef]
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.J.H.; Jones, R.M.; Pickles, A.; State, M.W.; et al. Autism spectrum disorder. Nat. Rev. Dis. Primers 2020, 6, 5. [Google Scholar] [CrossRef]
- Maenner, M.J.; Warren, Z.; Williams, A.R.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Fitzgerald, R.T.; Furnier, S.M.; Hughes, M.M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020. MMWR Surveill. Summ. 2023, 72, 1–14. [Google Scholar] [CrossRef]
- Qiu, Z.; Du, A. Revisiting the genetic architecture of autism spectrum disorders in the genomic era: Insights from East Asian studies. Curr. Opin. Neurobiol. 2025, 90, 102936. [Google Scholar] [CrossRef]
- Ruggeri, B.; Sarkans, U.; Schumann, G.; Persico, A.M. Biomarkers in autism spectrum disorder: The old and the new. Psychopharmacology 2014, 231, 1201–1216. [Google Scholar] [CrossRef]
- Parellada, M.; Andreu-Bernabeu, Á.; Burdeus, M.; San José Cáceres, A.; Urbiola, E.; Carpenter, L.L.; Kraguljac, N.V.; McDonald, W.M.; Nemeroff, C.B.; Rodriguez, C.I.; et al. In Search of Biomarkers to Guide Interventions in Autism Spectrum Disorder: A Systematic Review. Am. J. Psychiatry 2023, 180, 23–40. [Google Scholar] [CrossRef]
- Loth, E.; Charman, T.; Mason, L.; Tillmann, J.; Jones, E.J.H.; Wooldridge, C.; Ahmad, J.; Auyeung, B.; Brogna, C.; Ambrosino, S.; et al. The EU-AIMS Longitudinal European Autism Project (LEAP): Design and methodologies to identify and validate stratification biomarkers for autism spectrum disorders. Mol. Autism 2017, 8, 24. [Google Scholar] [CrossRef]
- Butler, M.G.; Moreno-De-Luca, D.; Persico, A.M. Actionable Genomics in Clinical Practice: Paradigmatic Case Reports of Clinical and Therapeutic Strategies Based upon Genetic Testing. Genes 2022, 13, 323. [Google Scholar] [CrossRef]
- Cortese, S.; Bellato, A.; Gabellone, A.; Marzulli, L.; Matera, E.; Parlatini, V.; Petruzzelli, M.G.; Persico, A.M.; Delorme, R.; Fusar-Poli, P.; et al. Latest clinical frontiers related to autism diagnostic strategies. Cell Rep. Med. 2025, 6, 101916. [Google Scholar] [CrossRef]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Marotta, R.; Risoleo, M.C.; Messina, G.; Parisi, L.; Carotenuto, M.; Vetri, L.; Roccella, M. The Neurochemistry of Autism. Brain Sci. 2020, 10, 163. [Google Scholar] [CrossRef]
- Fetit, R.; Hillary, R.F.; Price, D.J.; Lawrie, S.M. The neuropathology of autism: A systematic review of post-mortem studies of autism and related disorders. Neurosci. Biobehav. Rev. 2021, 129, 35–62. [Google Scholar] [CrossRef]
- Erdag, D.; Merhan, O.; Yildiz, B. Biochemical and pharmacological properties of biogenic amines. In Biogenic Amines; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Lintas, C.; Sacco, R.; Azzarà, A.; Cassano, I.; Gurrieri, F. Genotype-Phenotype Correlations in Relation to Newly Emerging Monogenic Forms of Autism Spectrum Disorder and Associated Neurodevelopmental Disorders: The Importance of Phenotype Reevaluation after Pangenomic Results. J. Clin. Med. 2021, 10, 5060. [Google Scholar] [CrossRef]
- Silla Santos, M.H. Biogenic amines: Their importance in foods. Int. J. Food Microbiol. 1996, 29, 213–231. [Google Scholar] [CrossRef]
- Jairath, G.; Singh, P.K.; Dabur, R.S.; Rani, M.; Chaudhari, M. Biogenic amines in meat and meat products and its public health significance: A review. J. Food Sci. Technol. 2015, 52, 6835–6846. [Google Scholar] [CrossRef]
- Yoon, S.; Kim, M.; Moon, B. Various biogenic amines in Doenjang and changes in concentration depending on boiling and roasting. Appl. Biol. Chem. 2017, 60, 273–279. [Google Scholar] [CrossRef]
- Ly, D.; Mayrhofer, S.; Schmidt, J.M.; Zitz, U.; Domig, K.J. Biogenic Amine Contents and Microbial Characteristics of Cambodian Fermented Foods. Foods 2020, 9, 198. [Google Scholar] [CrossRef]
- Bardócz, S.; Grant, G.; Brown, D.S.; Ralph, A.; Pusztai, A. Polyamines in food—Implications for growth and health. J. Nutr. Biochem. 1993, 4, 66–71. [Google Scholar] [CrossRef]
- Ekici, K.; Omer, A.K. Biogenic amines formation and their importance in fermented foods. BIO Web Conf. 2020, 17, 00232. [Google Scholar] [CrossRef]
- David, J.C.; Coulon, J.F. Octopamine in invertebrates and vertebrates. A review. Prog. Neurobiol. 1985, 24, 141–185. [Google Scholar] [CrossRef]
- Omer, A.K.; Mohammed, R.R.; Ameen, P.S.M.; Abas, Z.A.; Ekici, K. Presence of Biogenic Amines in Food and Their Public Health Implications: A Review. J. Food Prot. 2021, 84, 1539–1548. [Google Scholar] [CrossRef]
- Mistry, S.K.; Burwell, T.J.; Chambers, R.M.; Rudolph-Owen, L.; Spaltmann, F.; Cook, W.J.; Morris, S.M., Jr. Cloning of human agmatinase. An alternate path for polyamine synthesis induced in liver by hepatitis B virus. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G375–G381. [Google Scholar] [CrossRef]
- Ramani, D.; De Bandt, J.P.; Cynober, L. Aliphatic polyamines in physiology and diseases. Clin. Nutr. 2014, 33, 14–22. [Google Scholar] [CrossRef]
- Mehta, P.K.; Christen, P. The molecular evolution of pyridoxal-5′-phosphate-dependent enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 2000, 74, 129–184. [Google Scholar] [CrossRef]
- Jalkanen, S.; Salmi, M. Cell surface monoamine oxidases: Enzymes in search of a function. EMBO J. 2001, 20, 3893–3901. [Google Scholar] [CrossRef]
- Seiler, N.; Raul, F. Polyamines and the intestinal tract. Crit. Rev. Clin. Lab. Sci. 2007, 44, 365–411. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Mattevi, A.; Edmondson, D.E. Structure-function relationships in flavoenzyme-dependent amine oxidations: A comparison of polyamine oxidase and monoamine oxidase. J. Biol. Chem. 2002, 277, 23973–23976. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef]
- Shinka, T.; Onodera, D.; Tanaka, T.; Shoji, N.; Miyazaki, T.; Moriuchi, T.; Fukumoto, T. Serotonin synthesis and metabolism-related molecules in a human prostate cancer cell line. Oncol. Lett. 2011, 2, 211–215. [Google Scholar] [CrossRef]
- Hoyer, D.; Schoeffter, P.; Waeber, C.; Palacios, J.M. Serotonin 5-HT1D receptors. Ann. N. Y. Acad. Sci. 1990, 600, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Pytliak, M.; Vargová, V.; Mechírová, V.; Felšöci, M. Serotonin receptors—From molecular biology to clinical applications. Physiol. Res. 2011, 60, 15–25. [Google Scholar] [CrossRef]
- Mohammad-Zadeh, L.F.; Moses, L.; Gwaltney-Brant, S.M. Serotonin: A review. J. Vet. Pharmacol. Ther. 2008, 31, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, H.G.; Grozdanovic, Z. Psychopharmacology of central serotonergic systems. Pharmacopsychiatry 1995, 28, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Höglund, E.; Øverli, Ø.; Winberg, S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef]
- Comai, S.; Bertazzo, A.; Brughera, M.; Crotti, S. Tryptophan in health and disease. Adv. Clin. Chem. 2020, 95, 165–218. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Amino Acids and Developmental Origins of Hypertension. Nutrients 2020, 12, 1763. [Google Scholar] [CrossRef]
- Finberg, J.P.M. Inhibitors of MAO-B and COMT: Their effects on brain dopamine levels and uses in Parkinson’s disease. J. Neural. Transm. 2019, 126, 433–448. [Google Scholar] [CrossRef]
- Gabriele, S.; Sacco, R.; Persico, A.M. Blood serotonin levels in autism spectrum disorder: A systematic review and meta-analysis. Eur. Neuropsychopharmacol. 2014, 24, 919–929. [Google Scholar] [CrossRef]
- Padmakumar, M.; Van Raes, E.; Van Geet, C.; Freson, K. Blood platelet research in autism spectrum disorders: In search of biomarkers. Res. Pract. Thromb. Haemost. 2019, 3, 566–577. [Google Scholar] [CrossRef]
- Pagan, C.; Benabou, M.; Leblond, C.; Cliquet, F.; Mathieu, A.; Lemière, N.; Goubran-Botros, H.; Delorme, R.; Leboyer, M.; Callebert, J.; et al. Decreased phenol sulfotransferase activities associated with hyperserotonemia in autism spectrum disorders. Transl. Psychiatry 2021, 11, 23. [Google Scholar] [CrossRef]
- Esposito, D.; Cruciani, G.; Zaccaro, L.; Di Carlo, E.; Spitoni, G.F.; Manti, F.; Carducci, C.; Fiori, E.; Leuzzi, V.; Pascucci, T. A Systematic Review on Autism and Hyperserotonemia: State-of-the-Art, Limitations, and Future Directions. Brain Sci. 2024, 14, 481. [Google Scholar] [CrossRef]
- Whitaker-Azmitia, P.M. Behavioral and cellular consequences of increasing serotonergic activity during brain development: A role in autism? Int. J. Dev. Neurosci. 2005, 23, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.L.; Anacker, A.M.J.; Veenstra-VanderWeele, J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 2016, 321, 24–41. [Google Scholar] [CrossRef]
- Marazziti, D.; Muratori, F.; Cesari, A.; Masala, I.; Baroni, S.; Giannaccini, G.; Dell’Osso, L.; Cosenza, A.; Pfanner, P.; Cassano, G.B. Increased density of the platelet serotonin transporter in autism. Pharmacopsychiatry 2000, 33, 165–168. [Google Scholar] [CrossRef]
- Jaiswal, P.; Mohanakumar, K.P.; Rajamma, U. Serotonin mediated immunoregulation and neural functions: Complicity in the aetiology of autism spectrum disorders. Neurosci. Biobehav. Rev. 2015, 55, 413–431. [Google Scholar] [CrossRef]
- Ramoz, N.; Cai, G.; Reichert, J.; Corwin, T.; Kryzak, L.; Smith, C.; Silverman, J.; Hollander, E.; Buxbaum, J. Family-based association study of TPH1 and TPH2 polymorphisms in autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Coon, H.; Dunn, D.; Lainhart, J.; Miller, J.; Hamil, C.; Battaglia, A.; Tancredi, R.; Leppert, M.F.; Weiss, R.; McMahon, W. Possible association between autism and variants in the brain-expressed tryptophan hydroxylase gene (TPH2). Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 135B, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Hervás, A.; Balmaña, N.; Salgado, M.; Maristany, M.; Vilella, E.; Aguilera, F.; Orejuela, C.; Cuscó, I.; Gallastegui, F.; et al. Neurotransmitter systems and neurotrophic factors in autism: Association study of 37 genes suggests involvement of DDC. World J. Biol. Psychiatry 2013, 14, 516–527. [Google Scholar] [CrossRef]
- Mitani, T.; Isikay, S.; Gezdirici, A.; Gulec, E.Y.; Punetha, J.; Fatih, J.M.; Herman, I.; Akay, G.; Du, H.; Calame, D.G.; et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am. J. Hum. Genet. 2021, 108, 1981–2005. [Google Scholar] [CrossRef]
- Schmidt, A.; Danyel, M.; Grundmann, K.; Brunet, T.; Klinkhammer, H.; Hsieh, T.C.; Engels, H.; Peters, S.; Knaus, A.; Moosa, S.; et al. Next-generation phenotyping integrated in a national framework for patients with ultrarare disorders improves genetic diagnostics and yields new molecular findings. Nat. Genet. 2024, 56, 1644–1653. [Google Scholar] [CrossRef]
- Alqahtani, A.S.; Alotibi, R.S.; Aloraini, T.; Almsned, F.; Alassali, Y.; Alfares, A.; Alhaddad, B.; Al Eissa, M.M. Prospect of genetic disorders in Saudi Arabia. Front. Genet. 2023, 14, 1243518. [Google Scholar] [CrossRef]
- Alonso-Gonzalez, A.; Calaza, M.; Amigo, J.; González-Peñas, J.; Martínez-Regueiro, R.; Fernández-Prieto, M.; Parellada, M.; Arango, C.; Rodriguez-Fontenla, C.; Carracedo, A. Exploring the biological role of postzygotic and germinal de novo mutations in ASD. Sci. Rep. 2021, 11, 319. [Google Scholar] [CrossRef]
- Cirnigliaro, M.; Chang, T.S.; Arteaga, S.A.; Pérez-Cano, L.; Ruzzo, E.K.; Gordon, A.; Bicks, L.K.; Jung, J.Y.; Lowe, J.K.; Wall, D.P.; et al. The contributions of rare inherited and polygenic risk to ASD in multiplex families. Proc. Natl. Acad. Sci. USA 2023, 120, e2215632120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Y.; Quaife, C.J.; Palmiter, R.D. Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature 1995, 374, 640–643. [Google Scholar] [CrossRef]
- Verma, D.; Chakraborti, B.; Karmakar, A.; Bandyopadhyay, T.; Singh, A.S.; Sinha, S.; Chatterjee, A.; Ghosh, S.; Mohanakumar, K.P.; Mukhopadhyay, K.; et al. Sexual dimorphic effect in the genetic association of monoamine oxidase A (MAOA) markers with autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 50, 11–20. [Google Scholar] [CrossRef]
- Yoo, H.J.; Lee, S.K.; Park, M.; Cho, I.H.; Hyun, S.H.; Lee, J.C.; Yang, S.Y.; Kim, S.A. Family- and population-based association studies of monoamine oxidase A and autism spectrum disorders in Korean. Neurosci. Res. 2009, 63, 172–176. [Google Scholar] [CrossRef]
- Cohen, I.L.; Liu, X.; Schutz, C.; White, B.N.; Jenkins, E.C.; Brown, W.T.; Holden, J.J. Association of autism severity with a monoamine oxidase A functional polymorphism. Clin. Genet. 2003, 64, 190–197. [Google Scholar] [CrossRef]
- Cohen, I.L.; Liu, X.; Lewis, M.E.; Chudley, A.; Forster-Gibson, C.; Gonzalez, M.; Jenkins, E.C.; Brown, W.T.; Holden, J.J. Autism severity is associated with child and maternal MAOA genotypes. Clin. Genet. 2011, 79, 355–362. [Google Scholar] [CrossRef]
- Saito, M.; Yamagata, T.; Matsumoto, A.; Shiba, Y.; Nagashima, M.; Taniguchi, S.; Jimbo, E.; Momoi, M.Y. MAOA/B deletion syndrome in male siblings with severe developmental delay and sudden loss of muscle tonus. Brain Dev. 2014, 36, 64–69. [Google Scholar] [CrossRef]
- Whibley, A.; Urquhart, J.; Dore, J.; Willatt, L.; Parkin, G.; Gaunt, L.; Black, G.; Donnai, D.; Raymond, F.L. Deletion of MAOA and MAOB in a male patient causes severe developmental delay, intermittent hypotonia and stereotypical hand movements. Eur. J. Hum. Genet. 2010, 18, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.E.; Leffler, M.; Rogers, C.; Shaw, M.; Carroll, R.; Earl, J.; Cheung, N.W.; Champion, B.; Hu, H.; Haas, S.A.; et al. New insights into Brunner syndrome and potential for targeted therapy. Clin. Genet. 2016, 89, 120–127. [Google Scholar] [CrossRef]
- Codina-Solà, M.; Rodríguez-Santiago, B.; Homs, A.; Santoyo, J.; Rigau, M.; Aznar-Laín, G.; Del Campo, M.; Gener, B.; Gabau, E.; Botella, M.P.; et al. Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol. Autism 2015, 6, 21. [Google Scholar] [CrossRef]
- Hu, C.; He, L.; Li, H.; Ding, Y.; Zhang, K.; Li, D.; Zhu, G.; Wu, B.; Xu, X.; Xu, Q. Clinical Targeted Panel Sequencing Analysis in Clinical Evaluation of Children with Autism Spectrum Disorder in China. Genes 2022, 13, 1010. [Google Scholar] [CrossRef]
- Ibarluzea, N.; Hoz, A.B.; Villate, O.; Llano, I.; Ocio, I.; Martí, I.; Guitart, M.; Gabau, E.; Andrade, F.; Gener, B.; et al. Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability. Genes 2020, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Tuncay, I.O.; DeVries, D.; Gogate, A.; Kaur, K.; Kumar, A.; Xing, C.; Goodspeed, K.; Seyoum-Tesfa, L.; Chahrour, M.H. The genetics of autism spectrum disorder in an East African familial cohort. Cell Genom. 2023, 3, 100322. [Google Scholar] [CrossRef] [PubMed]
- Popp, B.; Ekici, A.B.; Thiel, C.T.; Hoyer, J.; Wiesener, A.; Kraus, C.; Reis, A.; Zweier, C. Exome Pool-Seq in neurodevelopmental disorders. Eur. J. Hum. Genet. 2017, 25, 1364–1376. [Google Scholar] [CrossRef]
- Piton, A.; Poquet, H.; Redin, C.; Masurel, A.; Lauer, J.; Muller, J.; Thevenon, J.; Herenger, Y.; Chancenotte, S.; Bonnet, M.; et al. 20 ans après: A second mutation in MAOA identified by targeted high-throughput sequencing in a family with altered behavior and cognition. Eur. J. Hum. Genet. 2014, 22, 776–783. [Google Scholar] [CrossRef]
- Brunner, H.G.; Nelen, M.; Breakefield, X.O.; Ropers, H.H.; van Oost, B.A. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 1993, 262, 578–580. [Google Scholar] [CrossRef]
- Melke, J.; Goubran Botros, H.; Chaste, P.; Betancur, C.; Nygren, G.; Anckarsäter, H.; Rastam, M.; Ståhlberg, O.; Gillberg, I.C.; Delorme, R.; et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol. Psychiatry 2008, 13, 90–98. [Google Scholar] [CrossRef]
- Jonsson, L.; Anckarsäter, H.; Zettergren, A.; Westberg, L.; Walum, H.; Lundström, S.; Larsson, H.; Lichtenstein, P.; Melke, J. Association between ASMT and autistic-like traits in children from a Swedish nationwide cohort. Psychiatr. Genet. 2014, 24, 21–27. [Google Scholar] [CrossRef]
- Nava, C.; Keren, B.; Mignot, C.; Rastetter, A.; Chantot-Bastaraud, S.; Faudet, A.; Fonteneau, E.; Amiet, C.; Laurent, C.; Jacquette, A.; et al. Prospective diagnostic analysis of copy number variants using SNP microarrays in individuals with autism spectrum disorders. Eur. J. Hum. Genet. 2014, 22, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Ruan, Y.; Lu, T.; Liu, C.; Jia, M.; Yue, W.; Liu, J.; Bourgeron, T.; Zhang, D. Sequencing ASMT identifies rare mutations in Chinese Han patients with autism. PLoS ONE 2013, 8, e53727. [Google Scholar] [CrossRef] [PubMed]
- Delorme, R.; Durand, C.M.; Betancur, C.; Wagner, M.; Ruhrmann, S.; Grabe, H.J.; Nygren, G.; Gillberg, C.; Leboyer, M.; Bourgeron, T.; et al. No human tryptophan hydroxylase-2 gene R441H mutation in a large cohort of psychiatric patients and control subjects. Biol. Psychiatry 2006, 60, 202–203. [Google Scholar] [CrossRef]
- Sacco, R.; Papaleo, V.; Hager, J.; Rousseau, F.; Moessner, R.; Militerni, R.; Bravaccio, C.; Trillo, S.; Schneider, C.; Melmed, R.; et al. Case-control and family-based association studies of candidate genes in autistic disorder and its endophenotypes: TPH2 and GLO1. BMC Med. Genet. 2007, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Yoo, H.J.; Cho, I.H.; Park, M.; Kim, S.A. Association with tryptophan hydroxylase 2 gene polymorphisms and autism spectrum disorders in Korean families. Neurosci. Res. 2012, 73, 333–336. [Google Scholar] [CrossRef]
- Singh, A.S.; Chandra, R.; Guhathakurta, S.; Sinha, S.; Chatterjee, A.; Ahmed, S.; Ghosh, S.; Rajamma, U. Genetic association and gene-gene interaction analyses suggest likely involvement of ITGB3 and TPH2 with autism spectrum disorder (ASD) in the Indian population. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 45, 131–143. [Google Scholar] [CrossRef]
- Yu, H.; Liu, J.; Yang, A.; Yang, G.; Yang, W.; Lei, H.; Quan, J.; Zhang, Z. Lack of Association Between Polymorphisms in Dopa Decarboxylase and Dopamine Receptor-1 Genes with Childhood Autism in Chinese Han Population. J. Child Neurol. 2016, 31, 560–564. [Google Scholar] [CrossRef]
- Tassone, F.; Qi, L.; Zhang, W.; Hansen, R.L.; Pessah, I.N.; Hertz-Picciotto, I. MAOA, DBH, and SLC6A4 variants in CHARGE: A case-control study of autism spectrum disorders. Autism Res. 2011, 4, 250–261. [Google Scholar] [CrossRef]
- Chakraborti, B.; Verma, D.; Karmakar, A.; Jaiswal, P.; Sanyal, A.; Paul, D.; Sinha, S.; Singh, A.S.; Guhathakurta, S.; Roychowdhury, A.; et al. Genetic variants of MAOB affect serotonin level and specific behavioral attributes to increase autism spectrum disorder (ASD) susceptibility in males. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 71, 123–136. [Google Scholar] [CrossRef]
- Jonsson, L.; Ljunggren, E.; Bremer, A.; Pedersen, C.; Landén, M.; Thuresson, K.; Giacobini, M.; Melke, J. Mutation screening of melatonin-related genes in patients with autism spectrum disorders. BMC Med. Genom. 2010, 3, 10. [Google Scholar] [CrossRef]
- Pagan, C.; Goubran-Botros, H.; Delorme, R.; Benabou, M.; Lemière, N.; Murray, K.; Amsellem, F.; Callebert, J.; Chaste, P.; Jamain, S.; et al. Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci. Rep. 2017, 7, 2096. [Google Scholar] [CrossRef]
- Benabou, M.; Rolland, T.; Leblond, C.S.; Millot, G.A.; Huguet, G.; Delorme, R.; Leboyer, M.; Pagan, C.; Callebert, J.; Maronde, E.; et al. Heritability of the melatonin synthesis variability in autism spectrum disorders. Sci. Rep. 2017, 7, 17746. [Google Scholar] [CrossRef]
- Ballester-Navarro, P.; Martínez-Madrid, M.; Javaloyes-Sanchís, A.; Belda-Cantó, C.; Aguilar, V.; Inda, M.; Richdale, A.; Muriel, J.; Morales, D.; Peiró, A. Interplay of circadian clock and melatonin pathway gene variants in adults with autism, intellectual disability and sleep problems. Res. Autism Spectr. Disord. 2021, 81, 101715. [Google Scholar] [CrossRef]
- Daly, E.; Tricklebank, M.D.; Wichers, R. Chapter Two—Neurodevelopmental roles and the serotonin hypothesis of autism spectrum disorder. In The Serotonin System; Tricklebank, E.D., Daly, E., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 23–44. [Google Scholar] [CrossRef]
- Miyazaki, K.; Narita, N.; Narita, M. Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: Implication for pathogenesis of autism. Int. J. Dev. Neurosci. 2005, 23, 287–297. [Google Scholar] [CrossRef]
- Zengeler, K.E.; Shapiro, D.A.; Bruch, K.R.; Lammert, C.R.; Ennerfelt, H.; Lukens, J.R. SSRI treatment modifies the effects of maternal inflammation on in utero physiology and offspring neurobiology. Brain Behav. Immun. 2023, 108, 80–97. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yubero-Lahoz, S.; Robledo, P.; Farré, M.; de la Torre, R. Platelet SERT as a peripheral biomarker of serotonergic neurotransmission in the central nervous system. Curr. Med. Chem. 2013, 20, 1382–1396. [Google Scholar] [CrossRef]
- Israelyan, N.; Margolis, K.G. Reprint of: Serotonin as a link between the gut-brain-microbiome axis in autism spectrum disorders. Pharmacol. Res. 2019, 140, 115–120. [Google Scholar] [CrossRef]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the nervous system. Physiol. Rev. 2008, 88, 1183–1241. [Google Scholar] [CrossRef]
- Panula, P.; Sundvik, M.; Karlstedt, K. Developmental roles of brain histamine. Trends Neurosci. 2014, 37, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Carthy, E.; Ellender, T. Histamine, Neuroinflammation and Neurodevelopment: A Review. Front. Neurosci. 2021, 15, 680214. [Google Scholar] [CrossRef]
- Zampeli, E.; Tiligada, E. The role of histamine H4 receptor in immune and inflammatory disorders. Br. J. Pharmacol. 2009, 157, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Alonso, N.; Zappia, C.D.; Cabrera, M.; Davio, C.A.; Shayo, C.; Monczor, F.; Fernández, N.C. Physiological implications of biased signaling at histamine H2 receptors. Front. Pharmacol. 2015, 6, 45. [Google Scholar] [CrossRef]
- Schlicker, E.; Kathmann, M. Role of the Histamine H3 Receptor in the Central Nervous System. Handb. Exp. Pharmacol. 2017, 241, 277–299. [Google Scholar] [CrossRef]
- Panula, P. Histamine receptors, agonists, and antagonists in health and disease. Handb. Clin. Neurol. 2021, 180, 377–387. [Google Scholar] [CrossRef]
- Moriguchi, T.; Takai, J. Histamine and histidine decarboxylase: Immunomodulatory functions and regulatory mechanisms. Genes Cells 2020, 25, 443–449. [Google Scholar] [CrossRef]
- Marone, G.; Granata, F.; Spadaro, G.; Genovese, A.; Triggiani, M. The histamine-cytokine network in allergic inflammation. J. Allergy Clin. Immunol. 2003, 112, S83–S88. [Google Scholar] [CrossRef]
- Chua, R.X.Y.; Tay, M.J.Y.; Ooi, D.S.Q.; Siah, K.T.H.; Tham, E.H.; Shek, L.P.; Meaney, M.J.; Broekman, B.F.P.; Loo, E.X.L. Understanding the Link Between Allergy and Neurodevelopmental Disorders: A Current Review of Factors and Mechanisms. Front. Neurol. 2021, 11, 603571. [Google Scholar] [CrossRef]
- Kovacheva, E.; Gevezova, M.; Maes, M.; Sarafian, V. Mast Cells in Autism Spectrum Disorder-The Enigma to Be Solved? Int. J. Mol. Sci. 2024, 25, 2651. [Google Scholar] [CrossRef]
- Maintz, L.; Novak, N. Histamine and histamine intolerance. Am. J. Clin. Nutr. 2007, 85, 1185–1196. [Google Scholar] [CrossRef]
- Schayer, R.W. Catabolism of physiological quantities of histamine in vivo. Physiol. Rev. 1959, 39, 116–126. [Google Scholar] [CrossRef]
- Ercan-Sencicek, A.G.; Stillman, A.A.; Ghosh, A.K.; Bilguvar, K.; O’Roak, B.J.; Mason, C.E.; Abbott, T.; Gupta, A.; King, R.A.; Pauls, D.L.; et al. L-histidine decarboxylase and Tourette’s syndrome. N. Engl. J. Med. 2010, 362, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Shin, J.H.; Rajpurohit, A.; Deep-Soboslay, A.; Collado-Torres, L.; Brandon, N.J.; Hyde, T.M.; Kleinman, J.E.; Jaffe, A.E.; Cross, A.J.; et al. Altered expression of histamine signaling genes in autism spectrum disorder. Transl. Psychiatry 2017, 7, e1126. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Deng, X.; Zhang, J.; Su, L.; Xu, H.; Liang, H.; Huang, X.; Song, Z.; Deng, H. Mutation screening of the HDC gene in Chinese Han patients with Tourette syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Karagiannidis, I.; Dehning, S.; Sandor, P.; Tarnok, Z.; Rizzo, R.; Wolanczyk, T.; Madruga-Garrido, M.; Hebebrand, J.; Nöthen, M.M.; Lehmkuhl, G.; et al. Support of the histaminergic hypothesis in Tourette syndrome: Association of the histamine decarboxylase gene in a large sample of families. J. Med. Genet. 2013, 50, 760–764. [Google Scholar] [CrossRef]
- Lintas, C.; Sacco, R.; Azzarà, A.; Cassano, I.; Laino, L.; Grammatico, P.; Gurrieri, F. Genetic Dysruption of the Histaminergic Pathways: A Novel Deletion at the 15q21.2 locus Associated with Variable Expressivity of Neuropsychiatric Disorders. Genes 2022, 13, 1685. [Google Scholar] [CrossRef]
- Heidarzadeh-Asl, S.; Maurer, M.; Kiani, A.; Atiakshin, D.; Skov, P.S.; Elieh-Ali-Komi, D. Novel Insights on the Biology and Immunological Effects of Histamine: A Road Map for Allergists and Mast Cell Biologists. J. Allergy Clin. Immunol. 2024. [Google Scholar] [CrossRef]
- Mulatinho, M.V.; de Carvalho Serao, C.L.; Scalco, F.; Hardekopf, D.; Pekova, S.; Mrasek, K.; Liehr, T.; Weise, A.; Rao, N.; Llerena, J.C., Jr. Severe intellectual disability, omphalocele, hypospadia and high blood pressure associated to a deletion at 2q22.1q22.3: Case report. Mol. Cytogenet. 2012, 5, 30. [Google Scholar] [CrossRef]
- Verhoeven, W.M.A.; Egger, J.I.M.; Janssen, P.K.C.; van Haeringen, A. Adult male patient with severe intellectual disability caused by a homozygous mutation in the HNMT gene. BMJ Case Rep. 2020, 13, e235972. [Google Scholar] [CrossRef]
- Blasco-Fontecilla, H.; Bella-Fernández, M.; Wang, P.; Martin-Moratinos, M.; Li, C. Prevalence and Clinical Picture of Diamine Oxidase Gene Variants in Children and Adolescents with Attention Deficit Hyperactivity Disorder: A Pilot Study. J. Clin. Med. 2024, 13, 1659. [Google Scholar] [CrossRef] [PubMed]
- Tosca, L.; Drévillon, L.; Mouka, A.; Lecerf, L.; Briand, A.; Ortonne, V.; Benoit, V.; Brisset, S.; Van Maldergem, L.; Laudouar, Q.; et al. Two new cases of interstitial 7q35q36.1 deletion including CNTNAP2 and KMT2C. Mol. Genet. Genom. Med. 2021, 9, e1645. [Google Scholar] [CrossRef]
- Jones, D.N.; Raghanti, M.A. The role of monoamine oxidase enzymes in the pathophysiology of neurological disorders. J. Chem. Neuroanat. 2021, 114, 101957. [Google Scholar] [CrossRef]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; AlDhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R.; et al. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: A trio study from Saudi families. Sci. Rep. 2017, 7, 5679. [Google Scholar] [CrossRef]
- Rashaid, A.H.B.; Alqhazo, M.T.; Nusair, S.D.; Adams, J.B.; Bashtawi, M.A.; Al-Fawares, O. Profiling plasma levels of thiamine and histamine in Jordanian children with autism spectrum disorder (ASD): Potential biomarkers for evaluation of ASD therapies and diet. Nutr. Neurosci. 2023, 26, 842–849. [Google Scholar] [CrossRef]
- Gnegy, M.E. Chapter 14—Catecholamines. In Basic Neurochemistry, 8th ed.; Brady, S.T., Siegel, G.J., Albers, R.W., Price, D.L., Eds.; Academic Press: New York, NY, USA, 2012; pp. 283–299. [Google Scholar]
- Cortez, V.; Santana, M.; Marques, A.P.; Mota, A.; Rosmaninho-Salgado, J.; Cavadas, C. Regulation of catecholamine release in human adrenal chromaffin cells by β-adrenoceptors. Neurochem. Int. 2012, 60, 387–393. [Google Scholar] [CrossRef]
- Erwin, G.V.; Deitrich, R.A. Brain Aldehyde Dehydrogenase: Localization, purification, and properties. J. Biol. Chem. 1966, 241, 3533–3539. [Google Scholar]
- Tank, A.W.; Weiner, H.; Thurman, J.A. Enzymology and subcellular localization of aldehyde oxidation in rat liver. Oxidation of 3,4-dihydroxyphenylacetaldehyde derived from dopamine to 3,4-dihydroxyphenylacetic acid. Biochem. Pharmacol. 1981, 30, 3265–3275. [Google Scholar] [CrossRef]
- Ichinose, H.; Kurosawa, Y.; Titani, K.; Fujita, K.; Nagatsu, T. Isolation and characterization of a cDNA clone encoding human aromatic L-amino acid decarboxylase. Biochem. Biophys. Res. Commun. 1989, 164, 1024–1030. [Google Scholar] [CrossRef]
- Lundström, K.; Salminen, M.; Jalanko, A.; Savolainen, R.; Ulmanen, I. Cloning and characterization of human placental catechol-O-methyltransferase cDNA. DNA Cell Biol. 1991, 10, 181–189. [Google Scholar] [CrossRef]
- Bertocci, B.; Miggiano, V.; Da Prada, M.; Dembic, Z.; Lahm, H.W.; Malherbe, P. Human catechol-O-methyltransferase: Cloning and expression of the membrane-associated form. Proc. Natl. Acad. Sci. USA 1991, 88, 1416–1420. [Google Scholar] [CrossRef]
- Kawamura, M.; Eisenhofer, G.; Kopin, I.J.; Kador, P.F.; Lee, Y.S.; Fujisawa, S.; Sato, S. Aldose reductase: An aldehyde scavenging enzyme in the intraneuronal metabolism of norepinephrine in human sympathetic ganglia. Auton. Neurosci. 2002, 96, 131–139. [Google Scholar] [CrossRef]
- Laatikainen, L.M.; Sharp, T.; Bannerman, D.M.; Harrison, P.J.; Tunbridge, E.M. Modulation of hippocampal dopamine metabolism and hippocampal-dependent cognitive function by catechol-O-methyltransferase inhibition. J. Psychopharmacol. 2012, 26, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.D.; McMillan, A.; Shaw, K.N. 3-Methoxy-4-hydroxy-D-mandelic acid, a urinary metabolite of norepinephrine. Biochim. Biophys. Acta 1957, 25, 422–423. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.D.; McMillan, A. Studies on the formation of 3-methoxy-4-hydroxy-D-mandelic acid, a urinary metabolite of norepinephrine and epinephrine. Pharmacol. Rev. 1959, 11, 394–401. [Google Scholar] [CrossRef]
- Bernstein, A.I.; Stout, K.A.; Miller, G.W. The vesicular monoamine transporter 2: An underexplored pharmacological target. Neurochem. Int. 2014, 73, 89–97. [Google Scholar] [CrossRef]
- Costa, K.M.; Schoenbaum, G. Dopamine. Curr. Biol. 2022, 32, R817–R824. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef]
- Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Xia, Q.P.; Cheng, Z.Y.; He, L. The modulatory role of dopamine receptors in brain neuroinflammation. Int. Immunopharmacol. 2019, 76, 105908. [Google Scholar] [CrossRef]
- Komada, M.; Nishimura, Y. Epigenetics and Neuroinflammation Associated with Neurodevelopmental Disorders: A Microglial Perspective. Front. Cell Dev. Biol. 2022, 10, 852752. [Google Scholar] [CrossRef]
- Cai, Y.; Xing, L.; Yang, T.; Chai, R.; Wang, J.; Bao, J.; Shen, W.; Ding, S.; Chen, G. The neurodevelopmental role of dopaminergic signaling in neurological disorders. Neurosci. Lett. 2021, 741, 135540. [Google Scholar] [CrossRef] [PubMed]
- Ijomone, O.K.; Oria, R.S.; Ijomone, O.M.; Aschner, M.; Bornhorst, J. Dopaminergic Perturbation in the Aetiology of Neurodevelopmental Disorders. Mol. Neurobiol. 2025, 62, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Korn, C.; Akam, T.; Jensen, K.H.R.; Vagnoni, C.; Huber, A.; Tunbridge, E.M.; Walton, M.E. Distinct roles for dopamine clearance mechanisms in regulating behavioral flexibility. Mol. Psychiatry 2021, 26, 7188–7199. [Google Scholar] [CrossRef]
- Ranjbar-Slamloo, Y.; Fazlali, Z. Dopamine and Noradrenaline in the Brain; Overlapping or Dissociate Functions? Front. Mol. Neurosci. 2020, 12, 334. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.-S.; McNamara, J.O.; Williams, S.M. Projections to the Cerebellum. In Neuroscience, 2nd ed.; Purves, D., Augustine, G.J., Fitzpatrick, D., Katz, L.C., LaMantia, A.-S., McNamara, J.O., Williams, S.M., Eds.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- García-Sáinz, J.A.; Romero-Ávila, M.T.; Medina Ldel, C. α(1D)-Adrenergic receptors constitutive activity and reduced expression at the plasma membrane. Methods Enzymol. 2010, 484, 109–125. [Google Scholar] [CrossRef]
- Pytka, K.; Podkowa, K.; Rapacz, A.; Podkowa, A.; Żmudzka, E.; Olczyk, A.; Sapa, J.; Filipek, B. The role of serotonergic, adrenergic and dopaminergic receptors in antidepressant-like effect. Pharmacol. Rep. 2016, 68, 263–274. [Google Scholar] [CrossRef]
- Motiejunaite, J.; Amar, L.; Vidal-Petiot, E. Adrenergic receptors and cardiovascular effects of catecholamines. Ann. Endocrinol 2021, 82, 193–197. [Google Scholar] [CrossRef]
- Cheslack-Postava, K.; Fallin, M.D.; Avramopoulos, D.; Connors, S.L.; Zimmerman, A.W.; Eberhart, C.G.; Newschaffer, C.J. beta2-Adrenergic receptor gene variants and risk for autism in the AGRE cohort. Mol. Psychiatry 2007, 12, 283–291. [Google Scholar] [CrossRef]
- Esmaiel, N.N.; Ashaat, E.A.; Mosaad, R.; Fayez, A.; Ibrahim, M.; Abdallah, Z.Y.; Issa, M.Y.; Salem, S.; Ramadan, A.; El Wakeel, M.A.; et al. The potential impact of COMT gene variants on dopamine regulation and phenotypic traits of ASD patients. Behav. Brain Res. 2020, 378, 112272. [Google Scholar] [CrossRef]
- Bertrán, M.; Tagle, F.P.; Irarrázaval, M. Psychiatric manifestations of 22q11.2 deletion syndrome: A literature review. Neurologia 2018, 33, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Hidding, E.; Swaab, H.; de Sonneville, L.M.; van Engeland, H.; Vorstman, J.A. The role of COMT and plasma proline in the variable penetrance of autistic spectrum symptoms in 22q11.2 deletion syndrome. Clin. Genet. 2016, 90, 420–427. [Google Scholar] [CrossRef]
- Wongpaiboonwattana, W.; Hnoonual, A.; Limprasert, P. Association between 19-bp Insertion/Deletion Polymorphism of Dopamine β-Hydroxylase and Autism Spectrum Disorder in Thai Patients. Medicina 2022, 58, 1228. [Google Scholar] [CrossRef]
- Jones, M.B.; Palmour, R.M.; Zwaigenbaum, L.; Szatmari, P. Modifier effects in autism at the MAO-A and DBH loci. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 126B, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Koevoet, D.; Deschamps, P.K.H.; Kenemans, J.L. Catecholaminergic and cholinergic neuromodulation in autism spectrum disorder: A comparison to attention-deficit hyperactivity disorder. Front. Neurosci. 2023, 16, 1078586. [Google Scholar] [CrossRef]
- Young, J.G.; Cohen, D.J.; Brown, S.L.; Caparulo, B.K. Decreased urinary free catecholamines in childhood autism. J. Am. Acad. Child Psychiatry 1978, 17, 671–678. [Google Scholar] [CrossRef]
- Launay, J.M.; Bursztejn, C.; Ferrari, P.; Dreux, C.; Braconnier, A.; Zarifian, E.; Lancrenon, S.; Fermanian, J. Catecholamines metabolism in infantile autism: A controlled study of 22 autistic children. J. Autism Dev. Disord. 1987, 17, 333–347. [Google Scholar] [CrossRef]
- Barthelemy, C.; Bruneau, N.; Cottet-Eymard, J.M.; Domenech-Jouve, J.; Garreau, B.; Lelord, G.; Muh, J.P.; Peyrin, L. Urinary free and conjugated catecholamines and metabolites in autistic children. J. Autism Dev. Disord. 1988, 18, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Martineau, J.; Hérault, J.; Petit, E.; Guérin, P.; Hameury, L.; Perrot, A.; Mallet, J.; Sauvage, D.; Lelord, G.; Müh, J.P. Catecholaminergic metabolism and autism. Dev. Med. Child Neurol. 1994, 36, 688–697. [Google Scholar] [CrossRef]
- Minderaa, R.B.; Anderson, G.M.; Volkmar, F.R.; Akkerhuis, G.W.; Cohen, D.J. Noradrenergic and adrenergic functioning in autism. Biol. Psychiatry 1994, 36, 237–241. [Google Scholar] [CrossRef]
- Willemsen, M.A.; Verbeek, M.M.; Kamsteeg, E.J.; de Rijk-van Andel, J.F.; Aeby, A.; Blau, N.; Burlina, A.; Donati, M.A.; Geurtz, B.; Grattan-Smith, P.J.; et al. Tyrosine hydroxylase deficiency: A treatable disorder of brain catecholamine biosynthesis. Brain 2010, 133, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Reyes, Z.M.D.; Lynch, E.; Henry, J.; De Simone, L.M.; Sobotka, S.A. Diagnosis of autism in a rare case of tyrosine hydroxylase deficiency: A case report. BMC Med. Genom. 2023, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Bastos, P.; Gomes, T.; Ribeiro, L. Catechol-O-Methyltransferase (COMT): An Update on Its Role in Cancer, Neurological and Cardiovascular Diseases. Rev. Physiol. Biochem. Pharmacol. 2017, 173, 1–39. [Google Scholar] [CrossRef]
- Moskovitz, J.; Walss-Bass, C.; Cruz, D.A.; Thompson, P.M.; Hairston, J.; Bortolato, M. The enzymatic activities of brain catechol-O-methyltransferase (COMT) and methionine sulphoxide reductase are correlated in a COMT Val/Met allele-dependent fashion. Neuropathol. Appl. Neurobiol. 2015, 41, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Fekih-Romdhane, F.; Kerbage, G.; Hachem, N.; El Murr, M.; Haddad, G.; Loch, A.A.; Abou Khalil, R.; El Hayek, E.; Hallit, S. The moderating role of COMT gene rs4680 polymorphism between maladaptive metacognitive beliefs and negative symptoms in patients with schizophrenia. BMC Psychiatry 2024, 24, 831. [Google Scholar] [CrossRef]
- Sun, H.; Yuan, F.; Shen, X.; Xiong, G.; Wu, J. Role of COMT in ADHD: A systematic meta-analysis. Mol. Neurobiol. 2014, 49, 251–261. [Google Scholar] [CrossRef]
- Carmel, M.; Zarchi, O.; Michaelovsky, E.; Frisch, A.; Patya, M.; Green, T.; Gothelf, D.; Weizman, A. Association of COMT and PRODH gene variants with intelligence quotient (IQ) and executive functions in 22q11.2DS subjects. J. Psychiatr. Res. 2014, 56, 28–35. [Google Scholar] [CrossRef]
- Riley, K.N.; Catalano, L.M.; Bernat, J.A.; Adams, S.D.; Martin, D.M.; Lalani, S.R.; Patel, A.; Burnside, R.D.; Innis, J.W.; Rudd, M.K. Recurrent deletions and duplications of chromosome 2q11.2 and 2q13 are associated with variable outcomes. Am. J. Med. Genet. A 2015, 167A, 2664–2673. [Google Scholar] [CrossRef]
- Zeitz, M.J.; Lerner, P.P.; Ay, F.; Van Nostrand, E.; Heidmann, J.D.; Noble, W.S.; Hoffman, A.R. Implications of COMT long-range interactions on the phenotypic variability of 22q11.2 deletion syndrome. Nucleus 2013, 4, 487–493. [Google Scholar] [CrossRef]
- Gothelf, D.; Law, A.J.; Frisch, A.; Chen, J.; Zarchi, O.; Michaelovsky, E.; Ren-Patterson, R.; Lipska, B.K.; Carmel, M.; Kolachana, B.; et al. Biological effects of COMT haplotypes and psychosis risk in 22q11.2 deletion syndrome. Biol. Psychiatry 2014, 75, 406–413. [Google Scholar] [CrossRef]
- Sun, Z.; Bo, Q.; Mao, Z.; Li, F.; He, F.; Pao, C.; Li, W.; He, Y.; Ma, X.; Wang, C. Reduced Plasma Dopamine-β-Hydroxylase Activity Is Associated with the Severity of Bipolar Disorder: A Pilot Study. Front. Psychiatry 2021, 12, 566091. [Google Scholar] [CrossRef]
- Tang, Y.L.; Epstein, M.P.; Anderson, G.M.; Zabetian, C.P.; Cubells, J.F. Genotypic and haplotypic associations of the DBH gene with plasma dopamine beta-hydroxylase activity in African Americans. Eur. J. Hum. Genet. 2007, 15, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Kurata, H.T.; Marton, L.J.; Nichols, C.G. The polyamine binding site in inward rectifier K+ channels. J. Gen. Physiol. 2006, 127, 467–480. [Google Scholar] [CrossRef]
- Soeters, P.B.; Hallemeesch, M.M.; Bruins, M.J.; van Eijk, H.M.; Deutz, N.E. Quantitative in vivo assessment of arginine utilization and nitric oxide production in endotoxemia. Am. J. Surg. 2002, 183, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef]
- Schneider, J.; Wendisch, V.F. Biotechnological production of polyamines by bacteria: Recent achievements and future perspectives. Appl. Microbiol. Biotechnol. 2011, 91, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, C.; Lentini, A.; Provenzano, B.; Beninati, S. Evidences for a role of protein cross-links in transglutaminase-related disease. Amino Acids 2012, 42, 975–986. [Google Scholar] [CrossRef]
- Sagar, N.A.; Tarafdar, S.; Agarwal, S.; Tarafdar, A.; Sharma, S. Polyamines: Functions, Metabolism, and Role in Human Disease Management. Med. Sci. 2021, 9, 44. [Google Scholar] [CrossRef]
- Yamashita, T.; Nishimura, K.; Saiki, R.; Okudaira, H.; Tome, M.; Higashi, K.; Nakamura, M.; Terui, Y.; Fujiwara, K.; Kashiwagi, K.; et al. Role of polyamines at the G1/S boundary and G2/M phase of the cell cycle. Int. J. Biochem. Cell Biol. 2013, 45, 1042–1050. [Google Scholar] [CrossRef]
- Arthur, R.; Jamwal, S.; Kumar, P. A review on polyamines as promising next-generation neuroprotective and anti-aging therapy. Eur. J. Pharmacol. 2024, 978, 176804. [Google Scholar] [CrossRef]
- Cervelli, M.; Angelucci, E.; Germani, F.; Amendola, R.; Mariottini, P. Inflammation, carcinogenesis and neurodegeneration studies in transgenic animal models for polyamine research. Amino Acids 2014, 46, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Schibalski, R.S.; Shulha, A.S.; Tsao, B.P.; Palygin, O.; Ilatovskaya, D.V. The role of polyamine metabolism in cellular function and physiology. Am. J. Physiol. Cell Physiol. 2024, 327, C341–C356. [Google Scholar] [CrossRef]
- Castagnolo, D.; Schenone, S.; Botta, M. Guanylated diamines, triamines, and polyamines: Chemistry and biological properties. Chem. Rev. 2011, 111, 5247–5300. [Google Scholar] [CrossRef] [PubMed]
- Laube, G.; Bernstein, H.G. Agmatine: Multifunctional arginine metabolite and magic bullet in clinical neuroscience? Biochem. J. 2017, 474, 2619–2640. [Google Scholar] [CrossRef]
- Zhu, H.E.; Yin, J.Y.; Chen, D.X.; He, S.; Chen, H. Agmatinase promotes the lung adenocarcinoma tumorigenesis by activating the NO-MAPKs-PI3K/Akt pathway. Cell Death Dis. 2019, 10, 854. [Google Scholar] [CrossRef]
- Shantz, L.M.; Levin, V.A. Regulation of ornithine decarboxylase during oncogenic transformation: Mechanisms and therapeutic potential. Amino Acids 2007, 33, 213–223. [Google Scholar] [CrossRef]
- Pyronnet, S.; Pradayrol, L.; Sonenberg, N. A cell cycle-dependent internal ribosome entry site. Mol. Cell 2000, 5, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.P.; Loughran, G.; Atkins, J.F. uORFs with unusual translational start codons autoregulate expression of eukaryotic ornithine decarboxylase homologs. Proc. Natl. Acad. Sci. USA 2008, 105, 10079–10084. [Google Scholar] [CrossRef]
- Casero, R.A.; Pegg, A.E. Polyamine catabolism and disease. Biochem. J. 2009, 421, 323–338. [Google Scholar] [CrossRef]
- Cervelli, M.; Fratini, E.; Amendola, R.; Bianchi, M.; Signori, E.; Ferraro, E.; Lisi, A.; Federico, R.; Marcocci, L.; Mariottini, P. Increased spermine oxidase (SMO) activity as a novel differentiation marker of myogenic C2C12 cells. Int. J. Biochem. Cell Biol. 2009, 41, 934–944. [Google Scholar] [CrossRef]
- Pegg, A.E. S-Adenosylmethionine decarboxylase. Essays Biochem. 2009, 46, 25–45. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.N.; Fiorucci, C.; Mariottini, P.; Cervelli, M. Unveiling the hidden players: Noncoding RNAs orchestrating polyamine metabolism in disease. Cell Biosci. 2024, 14, 84. [Google Scholar] [CrossRef]
- Ray, R.M.; Viar, M.J.; Johnson, L.R. Amino acids regulate expression of antizyme-1 to modulate ornithine decarboxylase activity. J. Biol. Chem. 2012, 287, 3674–3690. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Harada, Y.; Moriyama, S.; Furuta, K.; Tanaka, S.; Miyaji, T.; Omote, H.; Moriyama, Y.; Hiasa, M. Vesicular Polyamine Transporter Mediates Vesicular Storage and Release of Polyamine from Mast Cells. J. Biol. Chem. 2017, 292, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Prokop, J.W.; Bupp, C.P.; Frisch, A.; Bilinovich, S.M.; Campbell, D.B.; Vogt, D.; Schultz, C.R.; Uhl, K.L.; VanSickle, E.; Rajasekaran, S.; et al. Emerging Role of ODC1 in Neurodevelopmental Disorders and Brain Development. Genes 2021, 12, 470. [Google Scholar] [CrossRef]
- Mäkitie, L.T.; Kanerva, K.; Polvikoski, T.; Paetau, A.; Andersson, L.C. Brain neurons express ornithine decarboxylase-activating antizyme inhibitor 2 with accumulation in Alzheimer’s disease. Brain Pathol. 2010, 20, 571–580. [Google Scholar] [CrossRef]
- Morrison, L.D.; Cao, X.C.; Kish, S.J. Ornithine decarboxylase in human brain: Influence of aging, regional distribution, and Alzheimer’s disease. J. Neurochem. 1998, 71, 288–294. [Google Scholar] [CrossRef]
- Bachmann, A.S.; VanSickle, E.A.; Michael, J.; Vipond, M.; Bupp, C.P. Bachmann-Bupp syndrome and treatment. Dev. Med. Child Neurol. 2024, 66, 445–455. [Google Scholar] [CrossRef]
- Zhou, X.E.; Schultz, C.R.; Suino Powell, K.; Henrickson, A.; Lamp, J.; Brunzelle, J.S.; Demeler, B.; Vega, I.E.; Bachmann, A.S.; Melcher, K. Structure and Enzymatic Activity of an Intellectual Disability-Associated Ornithine Decarboxylase Variant, G84R. ACS Omega 2022, 7, 34665–34675. [Google Scholar] [CrossRef]
- Schwartz, C.E.; Wang, X.; Stevenson, R.E.; Pegg, A.E. Spermine synthase deficiency resulting in X-linked intellectual disability (Snyder-Robinson syndrome). Methods Mol. Biol. 2011, 720, 437–445. [Google Scholar] [CrossRef]
- Akinyele, O.; Munir, A.; Johnson, M.A.; Perez, M.S.; Gao, Y.; Foley, J.R.; Nwafor, A.; Wu, Y.; Murray-Stewart, T.; Casero, R.A., Jr.; et al. Impaired polyamine metabolism causes behavioral and neuroanatomical defects in a mouse model of Snyder-Robinson syndrome. Dis. Model Mech. 2024, 17, dmm050639. [Google Scholar] [CrossRef]
- Alayoubi, A.M.; Iqbal, M.; Aman, H.; Hashmi, J.A.; Alayadhi, L.; Al-Regaiey, K.; Basit, S. Loss-of-function variant in spermidine/spermine N1-acetyl transferase like 1 (SATL1) gene as an underlying cause of autism spectrum disorder. Sci. Rep. 2024, 14, 5765. [Google Scholar] [CrossRef]
- Halaris, A.; Zhu, H.; Feng, Y.; Piletz, J.E. Plasma agmatine and platelet imidazoline receptors in depression. Ann. N. Y. Acad. Sci. 1999, 881, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Uzbay, T.I. The pharmacological importance of agmatine in the brain. Neurosci. Biobehav. Rev. 2012, 36, 502–519. [Google Scholar] [CrossRef]
- Esnafoglu, E.; İrende, İ. Decreased plasma agmatine levels in autistic subjects. J. Neural Transm. 2018, 125, 735–740. [Google Scholar] [CrossRef]
- Plenis, A.; Olędzka, I.; Kowalski, P.; Miękus, N.; Bączek, T. Recent Trends in the Quantification of Biogenic Amines in Biofluids as Biomarkers of Various Disorders: A Review. J. Clin. Med. 2019, 8, 640. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef]
- Vorstman, J.A.S.; Freitag, C.M.; Persico, A.M. From Genes to Therapy in Autism Spectrum Disorder. Genes 2022, 13, 1377. [Google Scholar] [CrossRef]
- Williams, K.; Brignell, A.; Randall, M.; Silove, N.; Hazell, P. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst. Rev. 2013, 20, CD004677. [Google Scholar] [CrossRef]
- Wisner, K.L.; Zarin, D.A.; Holmboe, E.S.; Appelbaum, P.S.; Gelenberg, A.J.; Leonard, H.L.; Frank, E. Risk-benefit decision making for treatment of depression during pregnancy. Am. J. Psychiatry 2000, 157, 1933–1940. [Google Scholar] [CrossRef]
- Kaplan, Y.C.; Keskin-Arslan, E.; Acar, S.; Sozmen, K. Prenatal selective serotonin reuptake inhibitor use and the risk of autism spectrum disorder in children: A systematic review and meta-analysis. Reprod. Toxicol. 2016, 66, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Sominsky, L.; O’Hely, M.; Berk, M.; Vuillermin, P.; Dawson, S.L. Prenatal environmental risk factors for autism spectrum disorder and their potential mechanisms. BMC Med. 2024, 22, 393. [Google Scholar] [CrossRef]
- Barroso, B.; Mendonça, F.; Mazer, P.; Prata, C.; Pinto, J.O. Methylphenidate and P300 in attention deficit hyperactivity disorder: A systematic review and meta-analysis. Int. J. Psychophysiol. 2025, 211, 112553. [Google Scholar] [CrossRef]
- Paulsen, B.; Velasco, S.; Kedaigle, A.J.; Pigoni, M.; Quadrato, G.; Deo, A.J.; Adiconis, X.; Uzquiano, A.; Sartore, R.; Yang, S.M.; et al. Autism genes converge on asynchronous development of shared neuron classes. Nature 2022, 602, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Ormstad, H.; Bryn, V.; Verkerk, R.; Skjeldal, O.H.; Halvorsen, B.; Saugstad, O.D.; Isaksen, J.; Maes, M. Serum Tryptophan, Tryptophan Catabolites and Brain-derived Neurotrophic Factor in Subgroups of Youngsters with Autism Spectrum Disorders. CNS Neurol. Disord. Drug Targets 2018, 17, 626–639. [Google Scholar] [CrossRef]
- Rangel-Huerta, O.D.; Gomez-Fernández, A.; de la Torre-Aguilar, M.J.; Gil, A.; Perez-Navero, J.L.; Flores-Rojas, K.; Martín-Borreguero, P.; Gil-Campos, M. Metabolic profiling in children with autism spectrum disorder with and without mental regression: Preliminary results from a cross-sectional case-control study. Metabolomics 2019, 15, 99. [Google Scholar] [CrossRef]
- Ristori, M.V.; Quagliariello, A.; Reddel, S.; Ianiro, G.; Vicari, S.; Gasbarrini, A.; Putignani, L. Autism, Gastrointestinal Symptoms and Modulation of Gut Microbiota by Nutritional Interventions. Nutrients 2019, 11, 2812. [Google Scholar] [CrossRef]
- Corbett, B.A.; Kantor, A.B.; Schulman, H.; Walker, W.L.; Lit, L.; Ashwood, P.; Rocke, D.M.; Sharp, F.R. A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol. Psychiatry 2007, 12, 292–306. [Google Scholar] [CrossRef]
- Cortelazzo, A.; De Felice, C.; Guerranti, R.; Signorini, C.; Leoncini, S.; Zollo, G.; Leoncini, R.; Timperio, A.M.; Zolla, L.; Ciccoli, L.; et al. Expression and oxidative modifications of plasma proteins in autism spectrum disorders: Interplay between inflammatory response and lipid peroxidation. Proteom. Clin. Appl. 2016, 10, 1103–1112. [Google Scholar] [CrossRef]
- Frederick, A.L.; Stanwood, G.D. Drugs, biogenic amine targets and the developing brain. Dev. Neurosci. 2009, 31, 7–22. [Google Scholar] [CrossRef]


| Name | IUPAC Name | Classification | Chemical Structure |
|---|---|---|---|
| Serotonin | 3-(2-aminoethyl)-1H-indol-5-ol | Monoamine | Heterocyclic |
| Histamine | 4-(2-aminoethyl)benzene-1,2-diol | Monoamine | Heterocyclic |
| Dopamine | 4-(2-aminoethyl)benzene-1,2-diol | Monoamine | Aromatic |
| Epinephrine | 4-[(1R)-1-hydroxy-2-(methylamino)ethyl]benzene-1,2-diol | Monoamine | Aromatic |
| Norepinephrine | 4-[(1R)-2-amino-1-hydroxyethyl]benzene-1,2-diol | Monoamine | Aromatic |
| Putrescine | butane-1,4-diamine | Diamine | Aliphatic |
| Spermidine | (4-aminobutyl)(3-aminopropyl)amine | Polyamine | Aliphatic |
| Spermine | (3-aminopropyl)({4-[(3-aminopropyl)amino]butyl})amine | Polyamine | Aliphatic |
| Enzyme Symbol | Name | Protein ID | E.C. Number | Gene Name | Gene ID | OMIM Gene ID | Disease Association (MIM Phenotype) | Gene Association with ASD (SFARI and Pubmed) | ASD or ASD Associated NDD SNV or CNV | Type of Study | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TPH | Tryptophan 5-monooxygenase; L-tryptophan 5-hydroxyilase (2 isoforme) | P17752 | 1.14.16.4 | TPH1 | 7166 | *191060 | No association | Not listed in SFARI database; found on PubMed in association with autism | No association with autism | Genotyping | [49] |
| Q8IWU9 | 1.14.16.4 | TPH2 | 121278 | *607478 | Susceptibility to attention deficit-hyperactivity disorder-7 (#613003) and major depressive disorders (#608516) | Not listed in SFARI database; found on PubMed in association with autism | No association with autism | Genotyping | [49] | ||
| Weak association of rs4341581 and rs11179000 with autism | Sanger sequencing | [50] | |||||||||
| AADC | Aromatic-L-acid decarboxylase; dopa decarboxylase | P20711 | 4.1.1.28 | DDC | 1644 | *107930 | Aromatic-L-acid decarboxylase deficiency—AADCD (#608643) | Strong candidate gene | Association of rs6592961 with autism | Case-control | [51] |
| c.1040 G > A (p.Arg347Gln) | WES and WGS | [52] | |||||||||
| c.1066_1068del (p.Arg356del) | WES | [53] | |||||||||
| c.1234 C > T (p.Arg412Trp) | WES and WGS | [54] | |||||||||
| c.1331 T > C (p.Phe444Ser) | WES | [55] | |||||||||
| c.480del (p.Thr161ProfsTer3) | WGS | [56] | |||||||||
| c.759 T > A (p.Asn253Lys) | Meta-analysis | [57] | |||||||||
| c.849 G > C (p.Glu283Asp) | WES | [53] | |||||||||
| MAO (MAO-A) | Monoamine oxidase (2 isoforms) | P21397 | 1.4.3.4 | MAOA | 4128 | *309850 | Brunner syndrome (#300615) | Strong candidate gene | Association of rs6323 with autism (males only) | Case-control | [58] |
| MAOA VNTR polymorphism association with autism | Case-control | [59,60,61] | |||||||||
| CNV, del chrXp11.3 (800kb) | Array-CGH | [62] | |||||||||
| CNV, del chrXp11.3-p11.4 (240 Kb) | Array-CGH | [63] | |||||||||
| c.133C > T (p.Arg45Trp) | WES | [64] | |||||||||
| c.1438-2A > G | WES and transcriptome profiling | [65] | |||||||||
| c.208G > A (p.Val70Met) | Targeted gene panel | [66] | |||||||||
| c.617G > A (p.Arg206Gln) | Targeted gene panel | [67] | |||||||||
| c.710A > T (p.Gln237Leu) | WGS | [68] | |||||||||
| c.730G > A (p.Val244Ile) | WES | [69] | |||||||||
| c.749_750insT (p.Ser251LysfsTer2) | WES | [64] | |||||||||
| c.797_798delinsTT (p.Cys266Phe) | Targeted gene panel | [70] | |||||||||
| c.815C > T (p.Ala272Val) | WES | [55] | |||||||||
| c.886C > T (p.Gln296Ter) | Sanger sequencing | [71] | |||||||||
| AANAT (SNAT) | Serotonin N-acetyltransferase; arylalkylamine N-acetyltransferase | Q16613 | 2.3.1.87 | AANAT | 15 | *600950 | Not correlated with a MIM phenototype | Not listed in SFARI database; not found on PubMed in association with autism | / | / | / |
| ASMT (HIOMT) | Acetylserotonin O-methyltransferase; hydroxyindole O-methyltransferase | P46597 | 2.1.1.4 | ASMT | 438 | *402500 | Not correlated with a MIM phenototype | Strong candidate gene (2) | Association of rs4446909 with autism | Genotyping | [72] |
| Association of rs5989681 with autism | Genotyping | [72] | |||||||||
| Association of several SNPs with autism | Genotyping | [73] | |||||||||
| CNV. delchrX | Array-CGH | [74] | |||||||||
| c.855 G > A (p.Trp257Ter) | Sanger sequencing | [75] | |||||||||
| c.-56C > A upstream variant | Sanger sequencing | [75] | |||||||||
| c.241A > G (p.Lys81Glu) | Sanger sequencing | [72] | |||||||||
| c.343C > T (p.Arg115Trp) | Sanger sequencing | [75] | |||||||||
| c.496G > A (p.Val166Ile) | Sanger sequencing | [75] | |||||||||
| c.51C > A (p.Asn17Ile) | Sanger sequencing | [72] | |||||||||
| c.536T > G (p.Val179Gly) | Sanger sequencing | [75] | |||||||||
| c.569G > A (p.Trp190Ter) | WES | [55] | |||||||||
| c.615G > A (p.Gln205=) | Sanger sequencing | [72] | |||||||||
| c.675C > A (p.Cys225Ter) | WGS | [56] | |||||||||
| c.917G > C (Gly306Ala) | Sanger sequencing | [72] | |||||||||
| c.976C > T (p.Leu326Phe) | Sanger sequencing | [72] | |||||||||
| IVS2 + 943T | Sanger sequencing | [75] | |||||||||
| IVS5 + 2T > C | genotyping | [72] | |||||||||
| IVS5 + 43G > C | Sanger sequencing | [75] |
| Enzyme Symbol | Name | Protein ID | E.C. Number | Gene Name | Gene ID | OMIM Gene ID | Disease Association (MIM Phenotype) | Gene Association with ASD (SFARI and Pubmed) | ASD or ASD Associated NDD SNV or CNV | Type of Study | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TH | Tyrosine hydroxylase; tyrosine 3-monooxygenase | P07101 | 1.14.16.2 | TH | 7054 | *191290 | Segawa syndrome, autosomal recessive (#605407) | Not listed in SFARI database; not found on PubMed in association with autism | / | / | / |
| AADC | Aromatic-L-acid decarboxylase; dopa decarboxylase | P20711 | 4.1.1.28 | DDC | 1644 | *107930 | See above | See above | See above | See above | See above |
| MAO (MAO-A) | Monoamine oxidase | P21397 | 1.4.3.4 | MAOA | 4128 | *309850 | See above | See above | See above | See above | See above |
| COMT | Catechol-O-methyltransferase | P21964 | 2.1.1.6 | COMT | 1312 | *116790 | Panic disorder 1—PAND1(#167870) and schizophrenia—SCZD (#181500) | Not listed in SFARI database; found on PubMed in association with autism | rs4680 associated with increasing autism severity | Sanger sequencing | [145] |
| CNV,delchr22q11.2 | Array-CGH | [146,147] | |||||||||
| DBH | Dopamine beta-hydroxylase; dopamine beta-monooxygenase | P09172 | 1.14.17.1 | DBH | 1621 | *609312 | Othostatic hypotension 1—ORTHYP1 (#223360) | Not listed in SFARI database; found on PubMed in association with autism | 19-bp insertion/deletion polymorphism association with autism | PCR and gel electrophoresis | [148] |
| c.910 G > T association with IQ in ASD | PCR and gel electrophoresis | [149] | |||||||||
| PNMT | Phenylethanolamine N-methyltransferase | P11086 | 2.1.1.28 | PNMT | 5409 | *171190 | Not correlated with a MIM phenotype | Not listed in SFARI database; not found on PubMed in association with autism | / | / | / |
| Enzyme Symbol | Name | Protein ID | E.C. Number | Gene Name | Gene ID | OMIM Gene ID | Disease Association (MIM Phenotype) | Gene Association with ASD (SFARI and Pubmed) | ASD or ASD Associated NDD SNV or CNV | Type of Study | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ODC | Ornithine decarboxylase | P11926 | 4.1.1.17 | ODC1 | 4953 | *165640 | Bachman-Bupp syndrome—BABS (#619075) | Not listed in SFARI database; found on PubMed in association with neurodevelopmental disorders | Preliminary evidence of rs138359527 association with neurodevelopment | In silico analysis | [190] |
| SAMDC | Adenosylmethionine decarboxylase 1 | P17707 | 4.1.1.50 | AMD1 | 262 | *180980 | Not correlated with a MIM phenotype | Not listed in SFARI database; not found on PubMed in association with autism | / | / | / |
| SRM | Spermidine synthase | P19623 | 2.5.1.16 | SRM | 6723 | *182891 | Not correlated with a MIM phenotype | Not listed in SFARI database; not found on PubMed in association with autism | / | / | / |
| SMS | Spermine synthase | P52788 | 2.5.1.22 | SMS | 6611 | *300105 | Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type—MRXSSR (#309583) | Not listed in SFARI database; found on PubMed in association with autism | c.424C > T (p.Leu142Phe) | WES | [117] |
| SSAT | Spermidine/spermine N1-acetyltransferase 1 | P21673 | 2.3.1.57 | SAT1 | 6303 | *313020 | Not correlated with a MIM phenotype | Not listed in SFARI database; not found on PubMed in association with autism | / | / | / |
| PAO | Polyamine oxidase; N(1)-acetylpolyamine oxidase | Q6QHF9 | 1.5.3.13 | PAOX | 196743 | *615853 | Not correlated with a MIM phenotype | Not listed in SFARI database; not found on PubMed in association with autism | / | / | / |
| SMO | Spermina oxidase; polyamine oxidase 1 | Q9NWM0 | 1.5.3.16 | SMOX | 54498 | *615854 | Not correlated with a MIM phenotype | Not listed in SFARI database; not found on PubMed in association with autism | / | / | / |
| MAO (MAO-B) | Monoamine oxidase | P27338 | 1.4.3.4 | MAOB | 4129 | *309860 | See above | See above | See above | See above | See above |
| DAO | Diamine oxidase; amine oxidase copper-containing 1 | P19801 | 1.4.3.22 | AOC1 | 26 | *104610 | See above | See above | See above | See above | See above |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabolacci, C.; Caruso, A.; Micai, M.; Galati, G.; Lintas, C.; Pisanu, M.E.; Scattoni, M.L. Biogenic Amine Metabolism and Its Genetic Variations in Autism Spectrum Disorder: A Comprehensive Overview. Biomolecules 2025, 15, 539. https://doi.org/10.3390/biom15040539
Tabolacci C, Caruso A, Micai M, Galati G, Lintas C, Pisanu ME, Scattoni ML. Biogenic Amine Metabolism and Its Genetic Variations in Autism Spectrum Disorder: A Comprehensive Overview. Biomolecules. 2025; 15(4):539. https://doi.org/10.3390/biom15040539
Chicago/Turabian StyleTabolacci, Claudio, Angela Caruso, Martina Micai, Giulia Galati, Carla Lintas, Maria Elena Pisanu, and Maria Luisa Scattoni. 2025. "Biogenic Amine Metabolism and Its Genetic Variations in Autism Spectrum Disorder: A Comprehensive Overview" Biomolecules 15, no. 4: 539. https://doi.org/10.3390/biom15040539
APA StyleTabolacci, C., Caruso, A., Micai, M., Galati, G., Lintas, C., Pisanu, M. E., & Scattoni, M. L. (2025). Biogenic Amine Metabolism and Its Genetic Variations in Autism Spectrum Disorder: A Comprehensive Overview. Biomolecules, 15(4), 539. https://doi.org/10.3390/biom15040539