

The Role of Post-Translational Modifications in Necroptosis

 , , , and
, , , and
Abstract
1. Background
2. Current Knowledge of Necroptosis
2.1. TNFR1
2.2. Fas or TRAILR1/2
2.3. DR3 and DR6
2.4. TLR3/4 and ZBP1
2.5. ROS
2.6. IFN
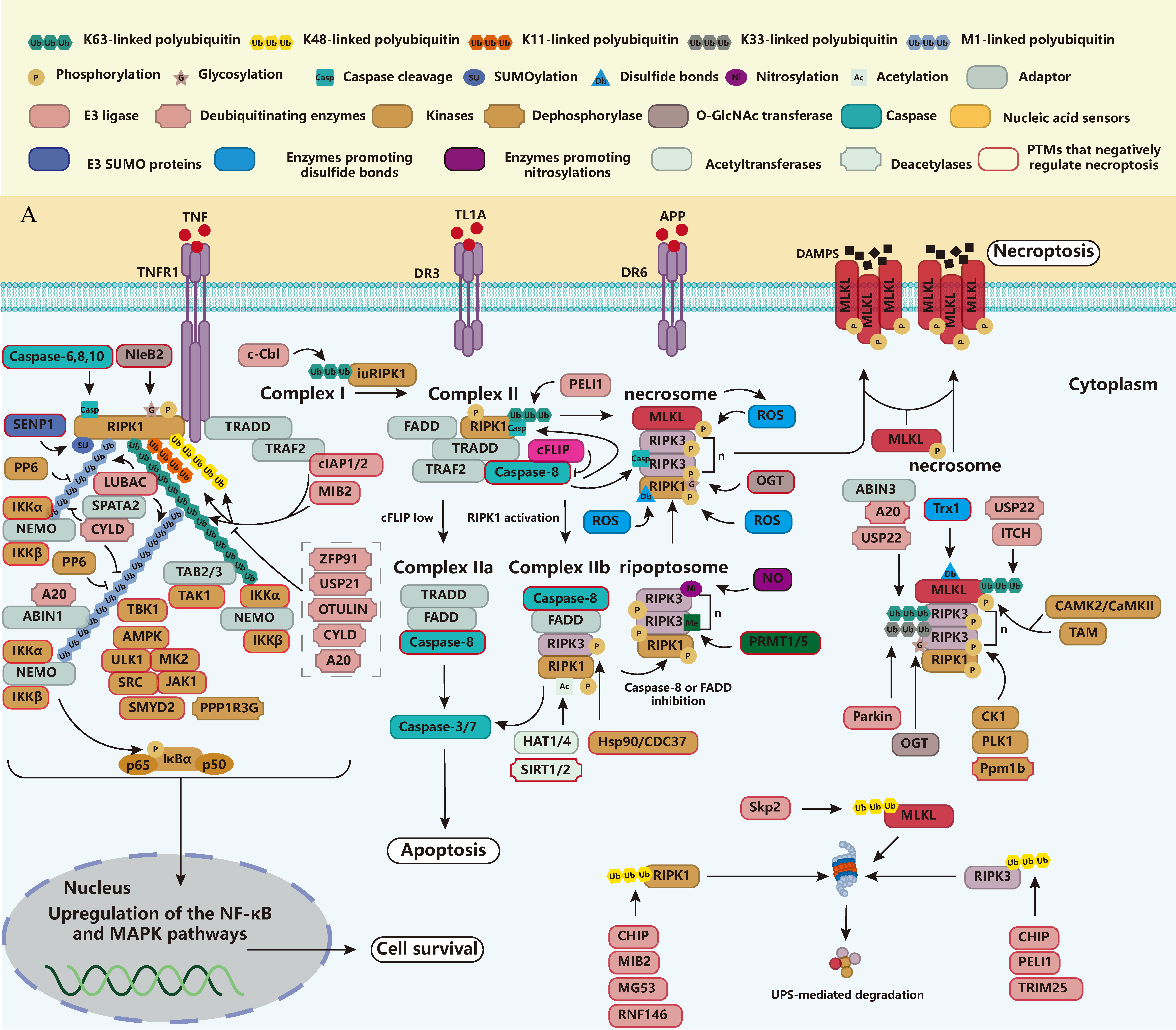
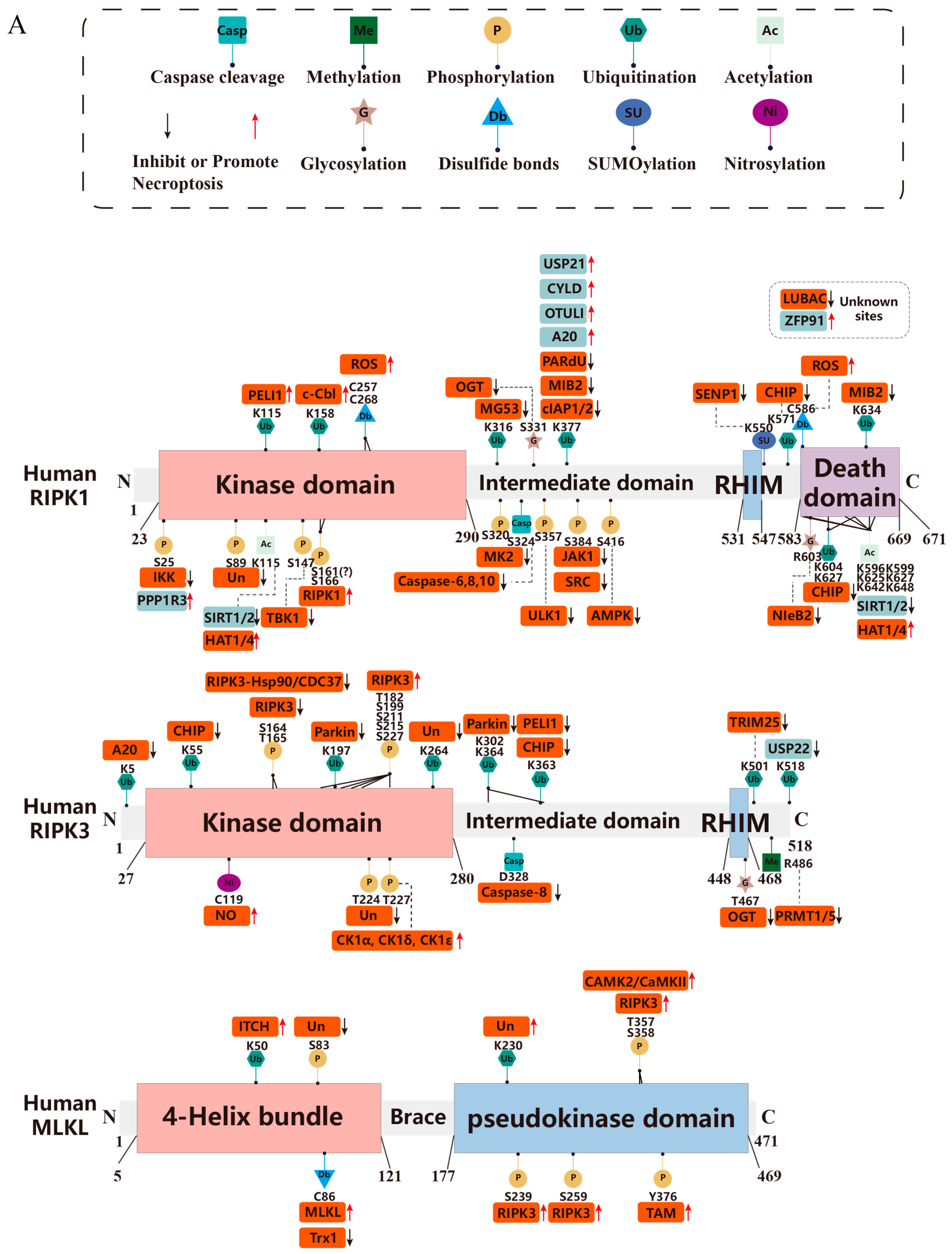
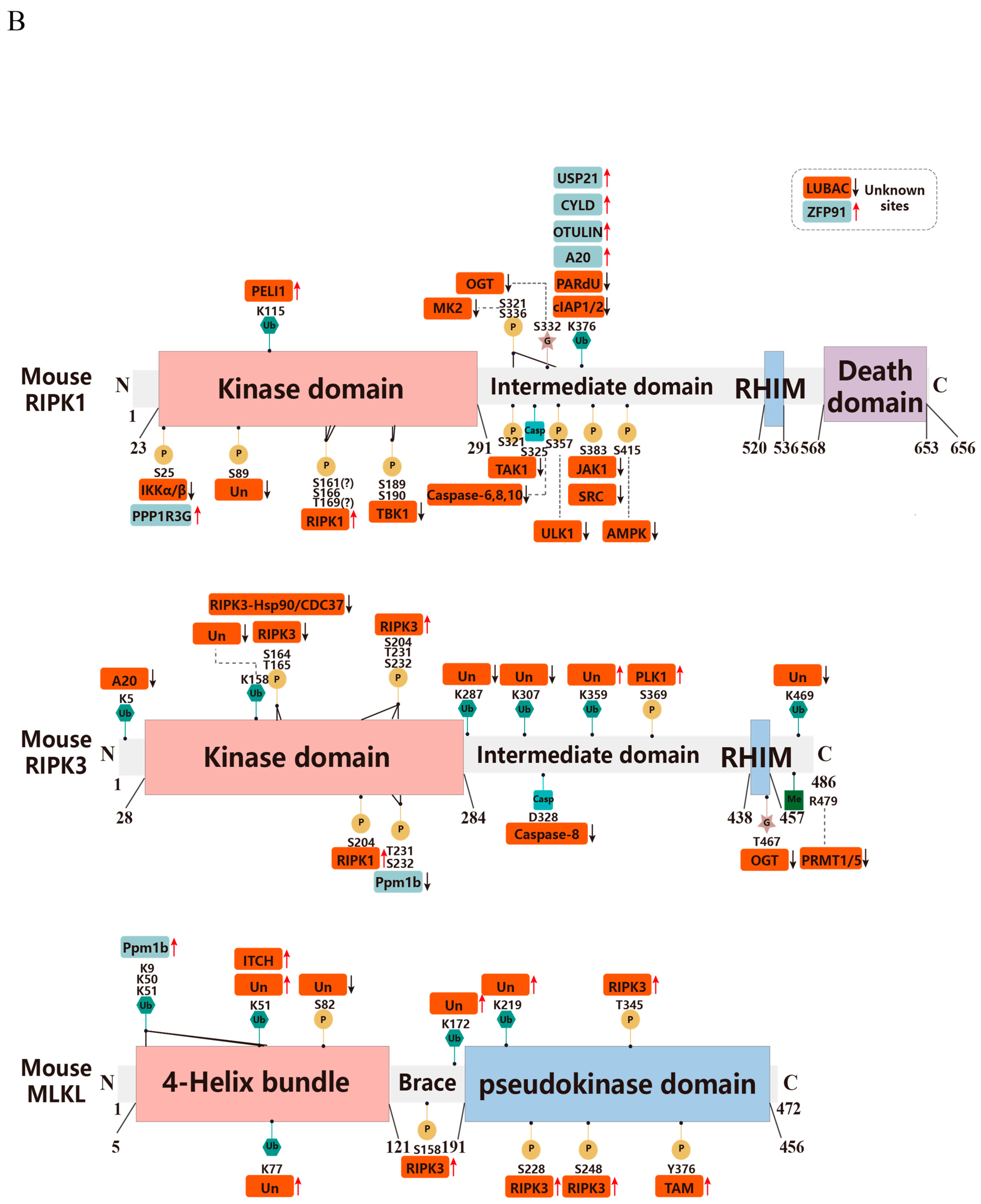
3. Post-Translational Modifications in Necroptosis
3.1. Ubiquitination
3.1.1. RIPK1
3.1.2. RIPK3
3.1.3. MLKL
3.1.4. FADD
3.1.5. Caspase-8
3.1.6. cFLIP
3.1.7. TNFR1
3.2. Phosphorylation
3.2.1. RIPK1
3.2.2. RIPK3
3.2.3. MLKL
3.2.4. FADD
3.2.5. Caspase-8
3.2.6. cFLIP
3.3. Glycosylation
3.4. Methylation
3.5. Acetylation
3.6. Disulfide Bonds
3.7. Caspase Cleavage
3.8. Nitrosylation
3.9. SUMOylation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 4HBD | 4-Helical bundle domain |
| ABIN | A20 binding and inhibitor of NF-kB |
| ABIN3 | A20 binding and inhibitor of NF-kB 3 |
| ACD | Accidental cell death |
| Acetyl-CoA | Acetyl coenzyme A |
| AD | Alzheimer’s disease |
| Akt1 | AKT serine/threonine kinase 1 |
| AMPK | Adenylate-activated protein kinase |
| AMPK | AMP-activated protein kinase |
| APC11 | Anaphase-promoting complex subunit 11 |
| APP | Amyloid precursor protein |
| Aur-A | Aurora-A |
| CAMK2/CaMKII | Calcium/calmodulin-dependent protein kinase II |
| CB2R | Cannabinoid receptor 2 |
| CHIP | Hsp70-interacting protein |
| cIAP1/2 | Cellular inhibitor of apoptosis 1 and 2 |
| CK1 | Casein kinase 1 |
| CKIα | Casein kinase I alpha |
| CSNK1G2 | Casein kinase 1G2 |
| CTSB | Cathepsin B |
| CUL3 | Cullin 3 |
| CYLD | Cylindromatosis |
| DAI | DNA-dependent activator of IFN regulatory factors |
| DAMPs | Danger-associated molecular patterns |
| DD | Death domain |
| DED | Death effector domain |
| DFG | Asp-Phe-Gly |
| DISC | Death-inducing signaling complex |
| DR | Death receptor |
| DR6 | Death Receptor 6 |
| DRP1 | Dynamin-related protein 1 |
| DSB | Double-strand break |
| DUB | Deubiquitinating enzyme |
| EPEC | Enteropathogenic Escherichia coli |
| ER | Endoplasmic reticulum |
| FADD | FAS associated via death domain |
| FHA | Forkhead-associated |
| FKBP12 | FK506-binding protein 12 |
| O-GlcNAc | O-Conjugated β-N-acetylglucosamine |
| HATs | Histone acetyltransferases |
| HBP | Hexosamine biosynthetic pathway |
| HDACs | Histone deacetylases |
| HECTD3 | Homologous to the E6-AP Carboxyl Terminus 3 |
| HOIL-1L | Heme-oxidized IRP2 ubiquitin ligase-1 |
| HOIP | HOIL-1-interacting protein |
| HOSCN | Hypothiocyanic acid |
| HRD | His-Arg-Asp |
| Hsp90 | Heat shock protein 90 |
| IAV | Influenza A virus |
| IFN-I | Interferon type I |
| IKK | IkB kinase |
| IRF1 | TNF-IFN regulatory factor 1 |
| iuRIPK1 | Insoluble RIPK1 complex |
| K | Lysine |
| KATs | Lysine acetyltransferases |
| KMT | Lysine methyltransferase |
| LPS | Lipopolysaccharide |
| LRRK2 | Leucine-rich repeat kinase 2 |
| LUBAC | Linear ubiquitin chain assembly complex |
| M1 | Methionine |
| MAPK | Mitogen-activated protein kinase |
| MAPKAPK2, MK2 | MAPK activated protein kinase 2 |
| MG53 | Mitsugumin 53 |
| MIB2 | Mind Bomb-2 |
| MIRI | Myocardial ischemia–reperfusion injury |
| MKRN1 | Makorin ring finger protein 1 |
| MLKL | Mixed lineage kinase domain-like protein |
| mtROS | Mitochondria ROS |
| MyD88 | Myeloid differentiation primary response 88 |
| NATs | Nα-acetyltransferases |
| NEMO | NF-κB essential modulator |
| OASL | Oligoadenylate synthetase-like |
| OGA | O-GlcNAcase |
| OGT | O-GlcNAc transferase |
| OTULIN | OTU deubiquitinase with linear linkage specificity |
| PARdU | PARylation-dependent ubiquitination |
| PARP5A | Poly(ADP-ribose) polymerase 5A |
| PARylation | Poly ADP-ribosylation |
| PBC | Primary biliary cholangitis |
| PDK 1 | 3-Phosphatidylinositol-dependent protein kinase 1 |
| PELI1 | Pellino E3 ubiquitin protein ligase 1 |
| PGAM5 | phosphoglycerate mutase family member 5 |
| PIPs | Phosphatidylinositol phosphates |
| PKC | Protein kinase C |
| PKR | Protein kinase R |
| PLK1 | Polo-like kinase 1 |
| poly(I:C) | Polyinosine–polycytidylic acid |
| PP1γ | Protein phosphatase 1 γ |
| Ppm1b | Protein phosphatase 1B |
| PPP1R3G | Protein phosphatase 1 regulatory subunit 3G |
| PRMT | Protein arginine methyltransferase |
| PRR | Pattern recognition receptor |
| PTMs | Post-translational modifications |
| RCD | Regulated cell death |
| RDA | RIPK1 kinase activity-dependent apoptosis |
| cFLIPL | Long isoform of cellular FADD-like interleukin (IL)-1β-converting enzyme (FLICE)-inhibitory protein |
| RHIMs | RIP homotypic interaction motifs |
| RIPK1 | Receptor-interacting protein kinase 1 |
| RIPK3 | Receptor-interacting protein kinase 3 |
| RNF146 | RING finger protein 146 |
| ROS | Reactive oxygen species |
| RSK | Ribosomal protein S6 kinase A1 |
| SCF | Skp1-Cullin-1-F-box |
| SCFSkp2 | SCF Cullin-Ring E3 ubiquitin ligase complex |
| SENP1 | SUMO-specific protease 1 |
| SEVO | Sevoflurane |
| SHARPIN | SHANK-associated RH domain-interacting protein |
| SIRTs | NAD+-dependent sirtuins |
| Skp2 | S-phase kinase-associated protein 2 |
| SMYD2 | Protein 2 containing SET and MYND structural domains |
| SPATA2 | Spermatogenesis associated 2 |
| T3SS | Type III secretion system |
| TAB2/3 | TAK1 Binding Protein 2/3 |
| TAK1 | TGF activating kinase 1 |
| TAM | Tyro3, Axl, and Mer |
| TAX1BP1 | Tax1-binding protein 1 |
| TBK1 | TANK binding kinase 1 |
| TMG | Thiamet-G |
| TNF | Tumor necrosis factor |
| TNFRSF | TNF receptor superfamily |
| TRADD | TNFRSF1A associated via death domain |
| TRAF2 | TNF receptor associated factor 2 |
| TRIF | Toll/IL-1 receptor domain-containing adaptor inducing interferon-beta |
| Trx1 | Thioredoxin-1 |
| S. typhimurium | Salmonella typhimurium |
| Ub | Ubiquitin |
| UBA | Ubiquitin-associated |
| UPS | Ubiquitin proteasome system |
| USP22 | Ubiquitin-specific peptidase 22 |
| ZBP1 | Z-DNA binding protein 1 |
| ZFP91 | Zinc finger protein 91 |
References
- Steller, H. Mechanisms and genes of cellular suicide. Science 1995, 267, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Ai, Y.; Meng, Y.; Yan, B.; Zhou, Q.; Wang, X. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol. Cell 2024, 84, 170–179. [Google Scholar] [CrossRef]
- Newton, K.; Strasser, A.; Kayagaki, N.; Dixit, V.M. Cell death. Cell 2024, 187, 235–256. [Google Scholar] [CrossRef]
- Yuan, J.; Ofengeim, D. A guide to cell death pathways. Nat. Rev. Mol. Cell. Biol. 2024, 25, 379–395. [Google Scholar] [CrossRef]
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 196. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Z.; Ren, J.; Morgan, S.; Assa, C.; Liu, B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ. Res. 2015, 116, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 2011, 6, 147–163. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Seo, J.; Kim, M.W.; Bae, K.H.; Lee, S.C.; Song, J.; Lee, E.W. The roles of ubiquitination in extrinsic cell death pathways and its implications for therapeutics. Biochem. Pharmacol. 2019, 162, 21–40. [Google Scholar] [CrossRef]
- Li, W.; Li, F.; Zhang, X.; Lin, H.K.; Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct. Target. Ther. 2021, 6, 422. [Google Scholar] [CrossRef]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell. Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Meier, P.; Legrand, A.J.; Adam, D.; Silke, J. Immunogenic cell death in cancer: Targeting necroptosis to induce antitumour immunity. Nat. Rev. Cancer 2024, 24, 299–315. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.S.; Dixit, V.; Ashkenazi, A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol. 2009, 10, 348–355. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Meng, Y.; Garnish, S.E.; Davies, K.A.; Black, K.A.; Leis, A.P.; Horne, C.R.; Hildebrand, J.M.; Hoblos, H.; Fitzgibbon, C.; Young, S.N.; et al. Phosphorylation-dependent pseudokinase domain dimerization drives full-length MLKL oligomerization. Nat. Commun. 2023, 14, 6804. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. MLKL regulates necrotic plasma membrane permeabilization. Cell Res. 2014, 24, 139–140. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Dovey, C.M.; Diep, J.; Clarke, B.P.; Hale, A.T.; McNamara, D.E.; Guo, H.; Brown, N.W., Jr.; Cao, J.Y.; Grace, C.R.; Gough, P.J.; et al. MLKL Requires the Inositol Phosphate Code to Execute Necroptosis. Mol. Cell 2018, 70, 936–948.e7. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef]
- Zhou, Z.; Han, V.; Han, J. New components of the necroptotic pathway. Protein Cell 2012, 3, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Cho, N.J.; Lee, S.W.; Lee, J.G.; Lee, J.H.; Yi, J.; Choi, M.S.; Park, S.; Gil, H.W.; Oh, J.C.; et al. RIPK3 causes mitochondrial dysfunction and albuminuria in diabetic podocytopathy through PGAM5-Drp1 signaling. Metabolism 2024, 159, 155982. [Google Scholar] [CrossRef]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007, 14, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, H.; Yang, Q.; Wan, L.; Zhao, J.; Wu, Y.; Wang, J.; Yang, Y.; Niu, M.; Liu, H.; et al. Increased Drp1 promotes autophagy and ESCC progression by mtDNA stress mediated cGAS-STING pathway. J. Exp. Clin. Cancer Res. 2022, 41, 76. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300.e16. [Google Scholar] [CrossRef]
- Rothe, J.; Lesslauer, W.; Lötscher, H.; Lang, Y.; Koebel, P.; Köntgen, F.; Althage, A.; Zinkernagel, R.; Steinmetz, M.; Bluethmann, H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364, 798–802. [Google Scholar] [CrossRef]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef]
- Xanthoulea, S.; Pasparakis, M.; Kousteni, S.; Brakebusch, C.; Wallach, D.; Bauer, J.; Lassmann, H.; Kollias, G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 2004, 200, 367–376. [Google Scholar] [CrossRef]
- Seo, J.; Nam, Y.W.; Kim, S.; Oh, D.B.; Song, J. Necroptosis molecular mechanisms: Recent findings regarding novel necroptosis regulators. Exp. Mol. Med. 2021, 53, 1007–1017. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, N.; Darding, M.; Walczak, H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016, 26, 445–461. [Google Scholar] [CrossRef]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Deribe, Y.L.; Skånland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef]
- Silke, J.; Brink, R. Regulation of TNFRSF and innate immune signalling complexes by TRAFs and cIAPs. Cell Death Differ. 2010, 17, 35–45. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Delanghe, T.; Rojas-Rivera, D.; Priem, D.; Delvaeye, T.; Bruggeman, I.; Van Herreweghe, F.; Vandenabeele, P.; Bertrand, M.J.M. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 2017, 19, 1237–1247. [Google Scholar] [CrossRef]
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; Wicky John, S.; Liccardi, G.; et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol. Cell 2017, 66, 698–710.e5. [Google Scholar] [CrossRef]
- Menon, M.B.; Gropengießer, J.; Fischer, J.; Novikova, L.; Deuretzbacher, A.; Lafera, J.; Schimmeck, H.; Czymmeck, N.; Ronkina, N.; Kotlyarov, A.; et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 2017, 19, 1248–1259. [Google Scholar] [CrossRef]
- Tu, H.; Xiong, W.; Zhang, J.; Zhao, X.; Lin, X. Tyrosine phosphorylation regulates RIPK1 activity to limit cell death and inflammation. Nat. Commun. 2022, 13, 6603. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, X.; Berleth, N.; Deitersen, J.; Wallot-Hieke, N.; Böhler, P.; Schlütermann, D.; Stuhldreier, F.; Cox, J.; Schmitz, K.; et al. The Autophagy-Initiating Kinase ULK1 Controls RIPK1-Mediated Cell Death. Cell Rep. 2020, 31, 107547. [Google Scholar] [CrossRef]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491.e19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, D.; Trefts, E.; Lv, M.; Inuzuka, H.; Song, G.; Liu, M.; Lu, J.; Liu, J.; Chu, C.; et al. Metabolic orchestration of cell death by AMPK-mediated phosphorylation of RIPK1. Science 2023, 380, 1372–1380. [Google Scholar] [CrossRef]
- Meng, H.; Liu, Z.; Li, X.; Wang, H.; Jin, T.; Wu, G.; Shan, B.; Christofferson, D.E.; Qi, C.; Yu, Q.; et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E2001–E2009. [Google Scholar] [CrossRef]
- Petersen, S.L.; Chen, T.T.; Lawrence, D.A.; Marsters, S.A.; Gonzalvez, F.; Ashkenazi, A. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ. 2015, 22, 1846–1857. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Newton, K. Multitasking Kinase RIPK1 Regulates Cell Death and Inflammation. Cold Spring Harb. Perspect. Biol. 2020, 12, a036368. [Google Scholar] [CrossRef]
- Priem, D.; Devos, M.; Druwé, S.; Martens, A.; Slowicka, K.; Ting, A.T.; Pasparakis, M.; Declercq, W.; Vandenabeele, P.; van Loo, G.; et al. A20 protects cells from TNF-induced apoptosis through linear ubiquitin-dependent and -independent mechanisms. Cell Death Dis. 2019, 10, 692. [Google Scholar] [CrossRef]
- Dziedzic, S.A.; Su, Z.; Jean Barrett, V.; Najafov, A.; Mookhtiar, A.K.; Amin, P.; Pan, H.; Sun, L.; Zhu, H.; Ma, A.; et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat. Cell Biol. 2018, 20, 58–68. [Google Scholar] [CrossRef]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Dou, Z.; Bonacci, T.R.; Shou, P.; Landoni, E.; Woodcock, M.G.; Sun, C.; Savoldo, B.; Herring, L.E.; Emanuele, M.J.; Song, F.; et al. 4-1BB-encoding CAR causes cell death via sequestration of the ubiquitin-modifying enzyme A20. Cell. Mol. Immunol. 2024, 21, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Y.; Xu, C.; Zhao, Q.; Liu, J.; Xing, M.; Li, X.; Zhang, H.; Wu, X.; Wang, L.; et al. Ubiquitin-binding domain in ABIN1 is critical for regulating cell death and inflammation during development. Cell Death Differ. 2022, 29, 2034–2045. [Google Scholar] [CrossRef] [PubMed]
- Enesa, K.; Zakkar, M.; Chaudhury, H.; Luong, L.A.; Rawlinson, L.; Mason, J.C.; Haskard, D.O.; Dean, J.L.; Evans, P.C. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: A novel negative feedback loop in pro-inflammatory signaling. J. Biol. Chem. 2008, 283, 7036–7045. [Google Scholar] [CrossRef]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef] [PubMed]
- Xinyu, W.; Qian, W.; Yanjun, W.; Jingwen, K.; Keying, X.; Jiazheng, J.; Haibing, Z.; Kai, W.; Xiao, X.; Lixing, Z. Polarity protein AF6 functions as a modulator of necroptosis by regulating ubiquitination of RIPK1 in liver diseases. Cell Death Dis. 2023, 14, 673. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, Z.H.; Wang, J.Y.; Xing, Y.; Ri, M.H.; Jin, H.L.; Zuo, H.X.; Li, M.Y.; Ma, J.; Jin, X. Zinc finger protein 91 mediates necroptosis by initiating RIPK1-RIPK3-MLKL signal transduction in response to TNF receptor 1 ligation. Toxicol. Lett. 2022, 356, 75–88. [Google Scholar] [CrossRef]
- He, S.; Wang, X. RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 2018, 19, 912–922. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Fu, T.M.; Li, Y.; Lu, A.; Li, Z.; Vajjhala, P.R.; Cruz, A.C.; Srivastava, D.B.; DiMaio, F.; Penczek, P.A.; Siegel, R.M.; et al. Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol. Cell 2016, 64, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.L.; Hughes, M.A.; Meng, X.; Sarnowska, N.A.; Powley, I.R.; Jukes-Jones, R.; Dinsdale, D.; Ragan, T.J.; Fairall, L.; Schwabe, J.W.R.; et al. Cryo-EM structural analysis of FADD:Caspase-8 complexes defines the catalytic dimer architecture for co-ordinated control of cell fate. Nat. Commun. 2021, 12, 819. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Maecker, H.; Sharp, D.; Lawrence, D.; Renz, M.; Vucic, D.; Ashkenazi, A. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 2005, 280, 40599–40608. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729.e5. [Google Scholar] [CrossRef]
- Migone, T.S.; Zhang, J.; Luo, X.; Zhuang, L.; Chen, C.; Hu, B.; Hong, J.S.; Perry, J.W.; Chen, S.F.; Zhou, J.X.; et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 2002, 16, 479–492. [Google Scholar] [CrossRef]
- Bittner, S.; Knoll, G.; Ehrenschwender, M. Death receptor 3 mediates necroptotic cell death. Cell. Mol. Life Sci. 2017, 74, 543–554. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, J.; Zhang, D.; Pan, Y.; Zeng, R.; Xu, C.; Shi, S.; Xu, J.; Qi, Q.; Dong, X.; et al. Necroptosis plays a role in TL1A-induced airway inflammation and barrier damage in asthma. Respir. Res. 2024, 25, 271. [Google Scholar] [CrossRef]
- Hassan-Zahraee, M.; Ye, Z.; Xi, L.; Baniecki, M.L.; Li, X.; Hyde, C.L.; Zhang, J.; Raha, N.; Karlsson, F.; Quan, J.; et al. Antitumor Necrosis Factor-like Ligand 1A Therapy Targets Tissue Inflammation and Fibrosis Pathways and Reduces Gut Pathobionts in Ulcerative Colitis. Inflamm. Bowel Dis. 2022, 28, 434–446. [Google Scholar] [CrossRef]
- Pan, G.; Bauer, J.H.; Haridas, V.; Wang, S.; Liu, D.; Yu, G.; Vincenz, C.; Aggarwal, B.B.; Ni, J.; Dixit, V.M. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett. 1998, 431, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shen, Q.; Liao, H.; Fu, H.; Wang, Q.; Yu, J.; Zhang, W.; Chen, C.; Dong, Y.; Yang, X.; et al. Multi-Arm PEG/Peptidomimetic Conjugate Inhibitors of DR6/APP Interaction Block Hematogenous Tumor Cell Extravasation. Adv. Sci. 2021, 8, e2003558. [Google Scholar] [CrossRef]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Polykratis, A.; Hermance, N.; Zelic, M.; Roderick, J.; Kim, C.; Van, T.M.; Lee, T.H.; Chan, F.K.M.; Pasparakis, M.; Kelliher, M.A. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 2014, 193, 1539–1543. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis that Is Targeted by Murine Cytomegalovirus vIRA. Cell Host Microbe 2019, 26, 564. [Google Scholar] [CrossRef]
- Koerner, L.; Wachsmuth, L.; Kumari, S.; Schwarzer, R.; Wagner, T.; Jiao, H.; Pasparakis, M. ZBP1 causes inflammation by inducing RIPK3-mediated necroptosis and RIPK1 kinase activity-independent apoptosis. Cell Death Differ. 2024, 31, 938–953. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Lin, J.; Kaya, G.G.; Ju, E.; Kondylis, V.; Kelepouras, K.; Liccardi, G.; Kim, C.; Pasparakis, M. The RIPK1 death domain restrains ZBP1- and TRIF-mediated cell death and inflammation. Immunity 2024, 57, 1497–1513.e6. [Google Scholar] [CrossRef]
- Byun, H.S.; Ju, E.; Park, K.A.; Sohn, K.C.; Jung, C.S.; Hong, J.H.; Ro, H.; Lee, H.Y.; Quan, K.T.; Park, I.; et al. Rubiarbonol B induces RIPK1-dependent necroptosis via NOX1-derived ROS production. Cell Biol. Toxicol. 2023, 39, 1677–1696. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Lim, J.W.; Kim, H. Astaxanthin induces NADPH oxidase activation and receptor-interacting protein kinase 1-mediated necroptosis in gastric cancer AGS cells. Mol. Med. Rep. 2021, 24, 837. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tan, H.; Zou, Z.; Gong, J.; Zhou, J.; Peng, N.; Su, L.; Maegele, M.; Cai, D.; Gu, Z. Preventing necroptosis by scavenging ROS production alleviates heat stress-induced intestinal injury. Int. J. Hyperth. 2020, 37, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Feng, H.; Sun, W.; Liu, K.; Lu, J.J.; Chen, X. Tert-butyl hydroperoxide (t-BHP) induced apoptosis and necroptosis in endothelial cells: Roles of NOX4 and mitochondrion. Redox Biol. 2017, 11, 524–534. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, J.; Yu, Z.; Qu, X.; Guan, S. 1,3-Dichloro-2-propanol-Induced Renal Tubular Cell Necroptosis through the ROS/RIPK3/MLKL Pathway. J. Agric. Food Chem. 2022, 70, 10847–10857. [Google Scholar] [CrossRef]
- Huang, Y.T.; Liang, Q.Q.; Zhang, H.R.; Chen, S.Y.; Xu, L.H.; Zeng, B.; Xu, R.; Shi, F.L.; Ouyang, D.Y.; Zha, Q.B.; et al. Baicalin inhibits necroptosis by decreasing oligomerization of phosphorylated MLKL and mitigates caerulein-induced acute pancreatitis in mice. Int. Immunopharmacol. 2022, 108, 108885. [Google Scholar] [CrossRef]
- Du, J.; Zhang, X.; Zhang, J.; Huo, S.; Li, B.; Wang, Q.; Song, M.; Shao, B.; Li, Y. Necroptosis and NLPR3 inflammasome activation mediated by ROS/JNK pathway participate in AlCl(3)-induced kidney damage. Food Chem. Toxicol. 2023, 178, 113915. [Google Scholar] [CrossRef]
- Wang, K.J.; Meng, X.Y.; Chen, J.F.; Wang, K.Y.; Zhou, C.; Yu, R.; Ma, Q. Emodin Induced Necroptosis and Inhibited Glycolysis in the Renal Cancer Cells by Enhancing ROS. Oxidative Med. Cell. Longev. 2021, 2021, 8840590. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lu, B.; Wang, C.; Wang, Z.; Luo, T.; Piao, M.; Meng, F.; Chi, G.; Luo, Y.; Ge, P. RIP1 and RIP3 contribute to shikonin-induced DNA double-strand breaks in glioma cells via increase of intracellular reactive oxygen species. Cancer Lett. 2017, 390, 77–90. [Google Scholar] [CrossRef]
- Jia, Y.; Wang, F.; Guo, Q.; Li, M.; Wang, L.; Zhang, Z.; Jiang, S.; Jin, H.; Chen, A.; Tan, S.; et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 2018, 19, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, X.; Feng, J.; Yu, J.; Chen, G. TRADD Mediates RIPK1-Independent Necroptosis Induced by Tumor Necrosis Factor. Front. Cell Dev. Biol. 2019, 7, 393. [Google Scholar] [CrossRef] [PubMed]
- Weindel, C.G.; Martinez, E.L.; Zhao, X.; Mabry, C.J.; Bell, S.L.; Vail, K.J.; Coleman, A.K.; VanPortfliet, J.J.; Zhao, B.; Wagner, A.R.; et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 2022, 185, 3214–3231.e23. [Google Scholar] [CrossRef]
- Thapa, R.J.; Nogusa, S.; Chen, P.; Maki, J.L.; Lerro, A.; Andrake, M.; Rall, G.F.; Degterev, A.; Balachandran, S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. USA 2013, 110, E3109–E3118. [Google Scholar] [CrossRef]
- Schock, S.N.; Chandra, N.V.; Sun, Y.; Irie, T.; Kitagawa, Y.; Gotoh, B.; Coscoy, L.; Winoto, A. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. 2017, 24, 615–625. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, M.; Cao, D.; Yang, C.; Jin, J.; Wu, L.; Hong, X.; Li, W.; Lu, L.; Li, J.; et al. ZBP1-MLKL necroptotic signaling potentiates radiation-induced antitumor immunity via intratumoral STING pathway activation. Sci. Adv. 2021, 7, eabf6290. [Google Scholar] [CrossRef]
- McComb, S.; Cessford, E.; Alturki, N.A.; Joseph, J.; Shutinoski, B.; Startek, J.B.; Gamero, A.M.; Mossman, K.L.; Sad, S. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc. Natl. Acad. Sci. USA 2014, 111, E3206–E3213. [Google Scholar] [CrossRef]
- Hershko, A. Lessons from the discovery of the ubiquitin system. Trends Biochem. Sci. 1996, 21, 445–449. [Google Scholar] [CrossRef]
- Datta, A.B.; Hura, G.L.; Wolberger, C. The structure and conformation of Lys63-linked tetraubiquitin. J. Mol. Biol. 2009, 392, 1117–1124. [Google Scholar] [CrossRef]
- Emmerich, C.H.; Bakshi, S.; Kelsall, I.R.; Ortiz-Guerrero, J.; Shpiro, N.; Cohen, P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem. Biophys. Res. Commun. 2016, 474, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Annibaldi, A.; Wicky John, S.; Vanden Berghe, T.; Swatek, K.N.; Ruan, J.; Liccardi, G.; Bianchi, K.; Elliott, P.R.; Choi, S.M.; Van Coillie, S.; et al. Ubiquitin-Mediated Regulation of RIPK1 Kinase Activity Independent of IKK and MK2. Mol. Cell 2018, 69, 566–580.e5. [Google Scholar] [CrossRef]
- Kist, M.; Kőműves, L.G.; Goncharov, T.; Dugger, D.L.; Yu, C.; Roose-Girma, M.; Newton, K.; Webster, J.D.; Vucic, D. Impaired RIPK1 ubiquitination sensitizes mice to TNF toxicity and inflammatory cell death. Cell Death Differ. 2021, 28, 985–1000. [Google Scholar] [CrossRef] [PubMed]
- Taraborrelli, L.; Peltzer, N.; Montinaro, A.; Kupka, S.; Rieser, E.; Hartwig, T.; Sarr, A.; Darding, M.; Draber, P.; Haas, T.L.; et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 2018, 9, 3910. [Google Scholar] [CrossRef]
- Peltzer, N.; Darding, M.; Montinaro, A.; Draber, P.; Draberova, H.; Kupka, S.; Rieser, E.; Fisher, A.; Hutchinson, C.; Taraborrelli, L.; et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 2018, 557, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Li, D.; Liang, W.; Sun, W.; Xie, X.; Tong, Y.; Shan, B.; Zhang, M.; Lu, X.; Yuan, J.; et al. PP6 negatively modulates LUBAC-mediated M1-ubiquitination of RIPK1 and c-FLIP(L) to promote TNFα-mediated cell death. Cell Death Dis. 2022, 13, 773. [Google Scholar] [CrossRef]
- Feltham, R.; Jamal, K.; Tenev, T.; Liccardi, G.; Jaco, I.; Domingues, C.M.; Morris, O.; John, S.W.; Annibaldi, A.; Widya, M.; et al. Mind Bomb Regulates Cell Death during TNF Signaling by Suppressing RIPK1’s Cytotoxic Potential. Cell Rep. 2018, 23, 470–484. [Google Scholar] [CrossRef]
- Hou, S.; Zhang, J.; Jiang, X.; Yang, Y.; Shan, B.; Zhang, M.; Liu, C.; Yuan, J.; Xu, D. PARP5A and RNF146 phase separation restrains RIPK1-dependent necroptosis. Mol. Cell 2024, 84, 938–954.e8. [Google Scholar] [CrossRef]
- Wang, Q.; Park, K.H.; Geng, B.; Chen, P.; Yang, C.; Jiang, Q.; Yi, F.; Tan, T.; Zhou, X.; Bian, Z.; et al. MG53 Inhibits Necroptosis Through Ubiquitination-Dependent RIPK1 Degradation for Cardiac Protection Following Ischemia/Reperfusion Injury. Front. Cardiovasc. Med. 2022, 9, 868632. [Google Scholar] [CrossRef]
- Wang, H.; Meng, H.; Li, X.; Zhu, K.; Dong, K.; Mookhtiar, A.K.; Wei, H.; Li, Y.; Sun, S.C.; Yuan, J. PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP. Proc. Natl. Acad. Sci. USA 2017, 114, 11944–11949. [Google Scholar] [CrossRef] [PubMed]
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y.; et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5944–E5953. [Google Scholar] [CrossRef]
- Seo, J.; Lee, E.W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.K.; Kim, J.H.; Han, S.Y.; et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302. [Google Scholar] [CrossRef]
- Li, X.; Zhang, M.; Huang, X.; Liang, W.; Li, G.; Lu, X.; Li, Y.; Pan, H.; Shi, L.; Zhu, H.; et al. Ubiquitination of RIPK1 regulates its activation mediated by TNFR1 and TLRs signaling in distinct manners. Nat. Commun. 2020, 11, 6364. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Laurien, L.; Nagata, M.; Schünke, H.; Delanghe, T.; Wiederstein, J.L.; Kumari, S.; Schwarzer, R.; Corona, T.; Krüger, M.; Bertrand, M.J.M.; et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 2020, 11, 1747. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Delanghe, T.; Priem, D.; Wynosky-Dolfi, M.A.; Sorobetea, D.; Rojas-Rivera, D.; Giansanti, P.; Roelandt, R.; Gropengiesser, J.; Ruckdeschel, K.; et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 2019, 10, 1729. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef]
- McQuade, T.; Cho, Y.; Chan, F.K. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 2013, 456, 409–415. [Google Scholar] [CrossRef]
- Du, J.; Xiang, Y.; Liu, H.; Liu, S.; Kumar, A.; Xing, C.; Wang, Z. RIPK1 dephosphorylation and kinase activation by PPP1R3G/PP1γ promote apoptosis and necroptosis. Nat. Commun. 2021, 12, 7067. [Google Scholar] [CrossRef]
- Yu, Y.Q.; Thonn, V.; Patankar, J.V.; Thoma, O.M.; Waldner, M.; Zielinska, M.; Bao, L.L.; Gonzalez-Acera, M.; Wallmüller, S.; Engel, F.B.; et al. SMYD2 targets RIPK1 and restricts TNF-induced apoptosis and necroptosis to support colon tumor growth. Cell Death Dis. 2022, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, Y.; Ji, S.; Kim, H.B.; Jung, H.; Yi, E.C.; Lee, Y.H.; Shin, I.; Yang, W.H.; Cho, J.W. O-GlcNAcylation of RIPK1 rescues red blood cells from necroptosis. Front. Immunol. 2023, 14, 1160490. [Google Scholar] [CrossRef] [PubMed]
- Giogha, C.; Scott, N.E.; Wong Fok Lung, T.; Pollock, G.L.; Harper, M.; Goddard-Borger, E.D.; Pearson, J.S.; Hartland, E.L. NleB2 from enteropathogenic Escherichia coli is a novel arginine-glucose transferase effector. PLoS Pathog. 2021, 17, e1009658. [Google Scholar] [CrossRef]
- Carafa, V.; Nebbioso, A.; Cuomo, F.; Rotili, D.; Cobellis, G.; Bontempo, P.; Baldi, A.; Spugnini, E.P.; Citro, G.; Chambery, A.; et al. RIP1-HAT1-SIRT Complex Identification and Targeting in Treatment and Prevention of Cancer. Clin. Cancer Res.. 2018, 24, 2886–2900. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Boyden, S.E.; Oda, H.; Wood, G.M.; Stone, D.L.; Chau, D.; Liu, L.; Stoffels, M.; Kratina, T.; Lawlor, K.E.; et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 2020, 577, 103–108. [Google Scholar] [CrossRef]
- van Raam, B.J.; Ehrnhoefer, D.E.; Hayden, M.R.; Salvesen, G.S. Intrinsic cleavage of receptor-interacting protein kinase-1 by caspase-6. Cell Death Differ. 2013, 20, 86–96. [Google Scholar] [CrossRef]
- Cho, M.; Dho, S.H.; Shin, S.; Lee, Y.; Kim, Y.; Lee, J.; Yu, S.J.; Park, S.H.; Lee, K.A.; Kim, L.K. Caspase-10 affects the pathogenesis of primary biliary cholangitis by regulating inflammatory cell death. J. Autoimmun. 2022, 133, 102940. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, T.; Wang, K.; Chen, Z.; Yang, Y.; Shan, B.; Sun, Q.; Zhang, M.; Zhang, Y.; Zhong, Y.; et al. SENP1 prevents steatohepatitis by suppressing RIPK1-driven apoptosis and inflammation. Nat. Commun. 2022, 13, 7153. [Google Scholar] [CrossRef]
- Onizawa, M.; Oshima, S.; Schulze-Topphoff, U.; Oses-Prieto, J.A.; Lu, T.; Tavares, R.; Prodhomme, T.; Duong, B.; Whang, M.I.; Advincula, R.; et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 2015, 16, 618–627. [Google Scholar] [CrossRef]
- Zhou, M.; He, J.; Shi, Y.; Liu, X.; Luo, S.; Cheng, C.; Ge, W.; Qu, C.; Du, P.; Chen, Y. ABIN3 Negatively Regulates Necroptosis-induced Intestinal Inflammation Through Recruiting A20 and Restricting the Ubiquitination of RIPK3 in Inflammatory Bowel Disease. J. Crohns Colitis 2021, 15, 99–114. [Google Scholar] [CrossRef]
- Frank, D.; Garnish, S.E.; Sandow, J.J.; Weir, A.; Liu, L.; Clayer, E.; Meza, L.; Rashidi, M.; Cobbold, S.A.; Scutts, S.R.; et al. Ubiquitylation of RIPK3 beyond-the-RHIM can limit RIPK3 activity and cell death. iScience 2022, 25, 104632. [Google Scholar] [CrossRef] [PubMed]
- Roedig, J.; Kowald, L.; Juretschke, T.; Karlowitz, R.; Ahangarian Abhari, B.; Roedig, H.; Fulda, S.; Beli, P.; van Wijk, S.J. USP22 controls necroptosis by regulating receptor-interacting protein kinase 3 ubiquitination. EMBO Rep. 2021, 22, e50163. [Google Scholar] [CrossRef]
- Choi, S.W.; Park, H.H.; Kim, S.; Chung, J.M.; Noh, H.J.; Kim, S.K.; Song, H.K.; Lee, C.W.; Morgan, M.J.; Kang, H.C.; et al. PELI1 Selectively Targets Kinase-Active RIP3 for Ubiquitylation-Dependent Proteasomal Degradation. Mol. Cell 2018, 70, 920–935.e7. [Google Scholar] [CrossRef] [PubMed]
- Mei, P.; Xie, F.; Pan, J.; Wang, S.; Gao, W.; Ge, R.; Gao, B.; Gao, S.; Chen, X.; Wang, Y.; et al. E3 ligase TRIM25 ubiquitinates RIP3 to inhibit TNF induced cell necrosis. Cell Death Differ. 2021, 28, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K. Regulation of RIPK3- and RHIM-dependent Necroptosis by the Proteasome. J. Biol. Chem. 2016, 291, 5948–5959. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Han, S.A.; Fan, Y.; Guo, L.S.; Aziz, K.; Nowsheen, S.; Kim, S.S.; Park, S.Y.; Luo, Q.; et al. The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat. Cell Biol. 2019, 21, 940–951. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, Z.; Li, L.; Zhong, C.Q.; Zheng, X.; Wu, X.; Zhang, Y.; Ma, H.; Huang, D.; Li, W.; et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 2013, 288, 16247–16261. [Google Scholar] [CrossRef]
- Meng, Y.; Davies, K.A.; Fitzgibbon, C.; Young, S.N.; Garnish, S.E.; Horne, C.R.; Luo, C.; Garnier, J.M.; Liang, L.Y.; Cowan, A.D.; et al. Human RIPK3 maintains MLKL in an inactive conformation prior to cell death by necroptosis. Nat. Commun. 2021, 12, 6783. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Meng, Y.; Horne, C.R.; Samson, A.L.; Dagley, L.F.; Young, S.N.; Sandow, J.J.; Czabotar, P.E.; Murphy, J.M. Human RIPK3 C-lobe phosphorylation is essential for necroptotic signaling. Cell Death Dis. 2022, 13, 565. [Google Scholar] [CrossRef]
- Li, D.; Ai, Y.; Guo, J.; Dong, B.; Li, L.; Cai, G.; Chen, S.; Xu, D.; Wang, F.; Wang, X. Casein kinase 1G2 suppresses necroptosis-promoted testis aging by inhibiting receptor-interacting kinase 3. Elife 2020, 9, e61564. [Google Scholar] [CrossRef]
- Li, D.; Chen, J.; Guo, J.; Li, L.; Cai, G.; Chen, S.; Huang, J.; Yang, H.; Zhuang, Y.; Wang, F.; et al. A phosphorylation of RIPK3 kinase initiates an intracellular apoptotic pathway that promotes prostaglandin(2α)-induced corpus luteum regression. Elife 2021, 10, e67409. [Google Scholar] [CrossRef]
- Hanna-Addams, S.; Liu, S.; Liu, H.; Chen, S.; Wang, Z. CK1α, CK1δ, and CK1ε are necrosome components which phosphorylate serine 227 of human RIPK3 to activate necroptosis. Proc. Natl. Acad. Sci. USA 2020, 117, 1962–1970. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, J.; Li, L.; Zhang, Z.; Ren, J.; Liang, Y.; Chen, F.; Yang, C.; Zhou, Z.; Su, S.S.; et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat. Cell Biol. 2015, 17, 434–444. [Google Scholar] [CrossRef]
- Li, X.; Gong, W.; Wang, H.; Li, T.; Attri, K.S.; Lewis, R.E.; Kalil, A.C.; Bhinderwala, F.; Powers, R.; Yin, G.; et al. O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity 2019, 50, 576–590.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, P.; Hua, F.; Hu, Y.; Xiao, F.; Liu, Q.; Huang, D.; Deng, F.; Wei, G.; Deng, W.; et al. Sevoflurane postconditioning reduces myocardial ischemia reperfusion injury-induced necroptosis by up-regulation of OGT-mediated O-GlcNAcylated RIPK3. Aging 2020, 12, 25452–25468. [Google Scholar] [CrossRef]
- Zhang, B.; Li, M.D.; Yin, R.; Liu, Y.; Yang, Y.; Mitchell-Richards, K.A.; Nam, J.H.; Li, R.; Wang, L.; Iwakiri, Y.; et al. O-GlcNAc transferase suppresses necroptosis and liver fibrosis. JCI Insight 2019, 4, e127709. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ha, H.J.; Chung, E.S.; Baek, S.H.; Cho, Y.; Kim, H.K.; Han, J.; Sul, J.H.; Lee, J.; Kim, E.; et al. O-GlcNAcylation ameliorates the pathological manifestations of Alzheimer’s disease by inhibiting necroptosis. Sci. Adv. 2021, 7, eabd3207. [Google Scholar] [CrossRef]
- Wu, F.; Shao, Q.; Cheng, Z.; Xiong, X.; Fang, K.; Zhao, Y.; Dong, R.; Xu, L.; Lu, F.; Chen, G. Traditional herbal formula Wu-Mei-Wan alleviates TNBS-induced colitis in mice by inhibiting necroptosis through increasing RIPK3 O-GlcNAcylation. Chin. Med. 2021, 16, 78. [Google Scholar] [CrossRef]
- Zhang, L.; He, Y.; Jiang, Y.; Wu, Q.; Liu, Y.; Xie, Q.; Zou, Y.; Wu, J.; Zhang, C.; Zhou, Z.; et al. PRMT1 reverts the immune escape of necroptotic colon cancer through RIP3 methylation. Cell Death Dis. 2023, 14, 233. [Google Scholar] [CrossRef]
- Chauhan, C.; Martinez-Val, A.; Niedenthal, R.; Olsen, J.V.; Kotlyarov, A.; Bekker-Jensen, S.; Gaestel, M.; Menon, M.B. PRMT5-mediated regulatory arginine methylation of RIPK3. Cell Death Discov. 2023, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Qu, Z.; Shi, K.; Zhang, D.; Zong, Y.; Zhang, G.; Zhang, G.; Hu, S. RIP3 S-nitrosylation contributes to cerebral ischemic neuronal injury. Brain Res. 2015, 1627, 165–176. [Google Scholar] [CrossRef]
- Zhong, Y.; Peng, P.; Zhang, M.; Han, D.; Yang, H.; Yan, X.; Hu, S. Effect of S-Nitrosylation of RIP3 Induced by Cerebral Ischemia on its Downstream Signaling Pathway. J. Stroke Cerebrovasc. Dis. 2022, 31, 106516. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Garcia, L.R.; Tenev, T.; Newman, R.; Haich, R.O.; Liccardi, G.; John, S.W.; Annibaldi, A.; Yu, L.; Pardo, M.; Young, S.N.; et al. Ubiquitylation of MLKL at lysine 219 positively regulates necroptosis-induced tissue injury and pathogen clearance. Nat. Commun. 2021, 12, 3364. [Google Scholar] [CrossRef]
- Liu, Z.; Dagley, L.F.; Shield-Artin, K.; Young, S.N.; Bankovacki, A.; Wang, X.; Tang, M.; Howitt, J.; Stafford, C.A.; Nachbur, U.; et al. Oligomerization-driven MLKL ubiquitylation antagonizes necroptosis. EMBO J. 2021, 40, e103718. [Google Scholar] [CrossRef]
- Yoon, S.; Bogdanov, K.; Wallach, D. Site-specific ubiquitination of MLKL targets it to endosomes and targets Listeria and Yersinia to the lysosomes. Cell Death Differ. 2022, 29, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, L.; Guan, Q.; Hou, X.; Wang, C.; Liu, L.; Wang, J.; Yu, X.; Li, W.; Liu, H. Skp2-mediated MLKL degradation confers cisplatin-resistant in non-small cell lung cancer cells. Commun. Biol. 2023, 6, 805. [Google Scholar] [CrossRef]
- Weinelt, N.; Wächtershäuser, K.N.; Celik, G.; Jeiler, B.; Gollin, I.; Zein, L.; Smith, S.; Andrieux, G.; Das, T.; Roedig, J.; et al. LUBAC-mediated M1 Ub regulates necroptosis by segregating the cellular distribution of active MLKL. Cell Death Dis. 2024, 15, 77. [Google Scholar] [CrossRef]
- Davies, K.A.; Fitzgibbon, C.; Young, S.N.; Garnish, S.E.; Yeung, W.; Coursier, D.; Birkinshaw, R.W.; Sandow, J.J.; Lehmann, W.I.L.; Liang, L.Y.; et al. Distinct pseudokinase domain conformations underlie divergent activation mechanisms among vertebrate MLKL orthologues. Nat. Commun. 2020, 11, 3060. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Zhan, Q.; Jeon, J.; Li, Y.; Huang, Y.; Xiong, J.; Wang, Q.; Xu, T.L.; Li, Y.; Ji, F.H.; Du, G.; et al. CAMK2/CaMKII activates MLKL in short-term starvation to facilitate autophagic flux. Autophagy 2022, 18, 726–744. [Google Scholar] [CrossRef]
- Rodriguez, D.A.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef]
- Tanzer, M.C.; Tripaydonis, A.; Webb, A.I.; Young, S.N.; Varghese, L.N.; Hall, C.; Alexander, W.S.; Hildebrand, J.M.; Silke, J.; Murphy, J.M. Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem. J. 2015, 471, 255–265. [Google Scholar] [CrossRef]
- Ying, Z.; Pan, C.; Shao, T.; Liu, L.; Li, L.; Guo, D.; Zhang, S.; Yuan, T.; Cao, R.; Jiang, Z.; et al. Mixed Lineage Kinase Domain-like Protein MLKL Breaks Down Myelin following Nerve Injury. Mol. Cell 2018, 72, 457–468.e5. [Google Scholar] [CrossRef] [PubMed]
- Najafov, A.; Mookhtiar, A.K.; Luu, H.S.; Ordureau, A.; Pan, H.; Amin, P.P.; Li, Y.; Lu, Q.; Yuan, J. TAM Kinases Promote Necroptosis by Regulating Oligomerization of MLKL. Mol. Cell 2019, 75, 457–468.e4. [Google Scholar] [CrossRef]
- Zhu, X.; Yang, N.; Yang, Y.; Yuan, F.; Yu, D.; Zhang, Y.; Shu, Z.; Nan, N.; Hu, H.; Liu, X.; et al. Spontaneous necroptosis and autoinflammation are blocked by an inhibitory phosphorylation on MLKL during neonatal development. Cell Res. 2022, 32, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Johnston, A.; Hanna-Addams, S.; Reynoso, E.; Xiang, Y.; Wang, Z. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E7450–E7459. [Google Scholar] [CrossRef]
- Reynoso, E.; Liu, H.; Li, L.; Yuan, A.L.; Chen, S.; Wang, Z. Thioredoxin-1 actively maintains the pseudokinase MLKL in a reduced state to suppress disulfide bond-dependent MLKL polymer formation and necroptosis. J. Biol. Chem. 2017, 292, 17514–17524. [Google Scholar] [CrossRef]
- Seo, J.; Lee, E.W.; Shin, J.; Seong, D.; Nam, Y.W.; Jeong, M.; Lee, S.H.; Lee, C.; Song, J. K6 linked polyubiquitylation of FADD by CHIP prevents death inducing signaling complex formation suppressing cell death. Oncogene 2018, 37, 4994–5006. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.W.; Kim, J.H.; Ahn, Y.H.; Seo, J.; Ko, A.; Jeong, M.; Kim, S.J.; Ro, J.Y.; Park, K.M.; Lee, H.W.; et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat. Commun. 2012, 3, 978. [Google Scholar] [CrossRef] [PubMed]
- Newson, J.P.M.; Scott, N.E.; Yeuk Wah Chung, I.; Wong Fok Lung, T.; Giogha, C.; Gan, J.; Wang, N.; Strugnell, R.A.; Brown, N.F.; Cygler, M.; et al. Salmonella Effectors SseK1 and SseK3 Target Death Domain Proteins in the TNF and TRAIL Signaling Pathways. Mol. Cell. Proteom. 2019, 18, 1138–1156. [Google Scholar] [CrossRef]
- Xue, J.; Hu, S.; Huang, Y.; Zhang, Q.; Yi, X.; Pan, X.; Li, S. Arg-GlcNAcylation on TRADD by NleB and SseK1 Is Crucial for Bacterial Pathogenesis. Front. Cell Dev. Biol. 2020, 8, 641. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, L.; Yao, Q.; Li, L.; Dong, N.; Rong, J.; Gao, W.; Ding, X.; Sun, L.; Chen, X.; et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 2013, 501, 242–246. [Google Scholar] [CrossRef]
- Fritsch, J.; Stephan, M.; Tchikov, V.; Winoto-Morbach, S.; Gubkina, S.; Kabelitz, D.; Schütze, S. Cell fate decisions regulated by K63 ubiquitination of tumor necrosis factor receptor 1. Mol. Cell. Biol. 2014, 34, 3214–3228. [Google Scholar] [CrossRef]
- Marín-Rubio, J.L.; Pérez-Gómez, E.; Fernández-Piqueras, J.; Villa-Morales, M. S194-P-FADD as a marker of aggressiveness and poor prognosis in human T-cell lymphoblastic lymphoma. Carcinogenesis 2019, 40, 1260–1268. [Google Scholar] [CrossRef]
- Alappat, E.C.; Feig, C.; Boyerinas, B.; Volkland, J.; Samuels, M.; Murmann, A.E.; Thorburn, A.; Kidd, V.J.; Slaughter, C.A.; Osborn, S.L.; et al. Phosphorylation of FADD at serine 194 by CKIalpha regulates its nonapoptotic activities. Mol. Cell 2005, 19, 321–332. [Google Scholar] [CrossRef]
- Vilmont, V.; Filhol, O.; Hesse, A.M.; Couté, Y.; Hue, C.; Rémy-Tourneur, L.; Mistou, S.; Cochet, C.; Chiocchia, G. Modulatory role of the anti-apoptotic protein kinase CK2 in the sub-cellular localization of Fas associated death domain protein (FADD). Biochim. Biophys. Acta 2015, 1853, 2885–2896. [Google Scholar] [CrossRef]
- Jang, M.S.; Lee, S.J.; Kang, N.S.; Kim, E. Cooperative phosphorylation of FADD by Aur-A and Plk1 in response to taxol triggers both apoptotic and necrotic cell death. Cancer Res. 2011, 71, 7207–7215. [Google Scholar] [CrossRef]
- Choi, S.G.; Kim, H.; Jeong, E.I.; Lee, H.J.; Park, S.; Lee, S.Y.; Lee, H.J.; Lee, S.W.; Chung, C.H.; Jung, Y.K. SUMO-Modified FADD Recruits Cytosolic Drp1 and Caspase-10 to Mitochondria for Regulated Necrosis. Mol. Cell. Biol. 2017, 37, e00254-16. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef]
- Tomar, D.; Prajapati, P.; Sripada, L.; Singh, K.; Singh, R.; Singh, A.K.; Singh, R. TRIM13 regulates caspase-8 ubiquitination, translocation to autophagosomes and activation during ER stress induced cell death. Biochim. Biophys. Acta 2013, 1833, 3134–3144. [Google Scholar] [CrossRef]
- Li, Y.; Kong, Y.; Zhou, Z.; Chen, H.; Wang, Z.; Hsieh, Y.C.; Zhao, D.; Zhi, X.; Huang, J.; Zhang, J.; et al. The HECTD3 E3 ubiquitin ligase facilitates cancer cell survival by promoting K63-linked polyubiquitination of caspase-8. Cell Death Dis. 2013, 4, e935. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Lawrence, D.; Yang, B.; Yee, S.; Pitti, R.; Marsters, S.; Pham, V.C.; Stephan, J.P.; Lill, J.; Ashkenazi, A. TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol. Cell 2012, 48, 888–899. [Google Scholar] [CrossRef]
- Yang, Z.H.; Wu, X.N.; He, P.; Wang, X.; Wu, J.; Ai, T.; Zhong, C.Q.; Wu, X.; Cong, Y.; Zhu, R.; et al. A Non-canonical PDK1-RSK Signal Diminishes Pro-caspase-8-Mediated Necroptosis Blockade. Mol. Cell 2020, 80, 296–310.e6. [Google Scholar] [CrossRef]
- Peng, C.; Cho, Y.Y.; Zhu, F.; Zhang, J.; Wen, W.; Xu, Y.; Yao, K.; Ma, W.Y.; Bode, A.M.; Dong, Z. Phosphorylation of caspase-8 (Thr-263) by ribosomal S6 kinase 2 (RSK2) mediates caspase-8 ubiquitination and stability. J. Biol. Chem. 2011, 286, 6946–6954. [Google Scholar] [CrossRef] [PubMed]
- Helmke, C.; Raab, M.; Rödel, F.; Matthess, Y.; Oellerich, T.; Mandal, R.; Sanhaji, M.; Urlaub, H.; Rödel, C.; Becker, S.; et al. Ligand stimulation of CD95 induces activation of Plk3 followed by phosphorylation of caspase-8. Cell Res. 2016, 26, 914–934. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Kristensson, M.; Melander, F.; Leandersson, K.; Rönnstrand, L.; Wernstedt, C.; Andersson, T. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J. Exp. Med. 2004, 199, 449–458. [Google Scholar] [CrossRef]
- Cursi, S.; Rufini, A.; Stagni, V.; Condò, I.; Matafora, V.; Bachi, A.; Bonifazi, A.P.; Coppola, L.; Superti-Furga, G.; Testi, R.; et al. Src kinase phosphorylates Caspase-8 on Tyr380: A novel mechanism of apoptosis suppression. EMBO J. 2006, 25, 1895–1905. [Google Scholar] [CrossRef]
- Matthess, Y.; Raab, M.; Sanhaji, M.; Lavrik, I.N.; Strebhardt, K. Cdk1/cyclin B1 controls Fas-mediated apoptosis by regulating caspase-8 activity. Mol. Cell. Biol. 2010, 30, 5726–5740. [Google Scholar] [CrossRef] [PubMed]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kim, T.H.; Chung, H.T.; Talanian, R.V.; Yin, X.M.; Billiar, T.R. Nitric oxide prevents tumor necrosis factor alpha-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology 2000, 32, 770–778. [Google Scholar] [CrossRef]
- Besnault-Mascard, L.; Leprince, C.; Auffredou, M.T.; Meunier, B.; Bourgeade, M.F.; Camonis, J.; Lorenzo, H.K.; Vazquez, A. Caspase-8 sumoylation is associated with nuclear localization. Oncogene 2005, 24, 3268–3273. [Google Scholar] [CrossRef]
- Tang, Y.; Joo, D.; Liu, G.; Tu, H.; You, J.; Jin, J.; Zhao, X.; Hung, M.C.; Lin, X. Linear ubiquitination of cFLIP induced by LUBAC contributes to TNFα-induced apoptosis. J. Biol. Chem. 2018, 293, 20062–20072. [Google Scholar] [CrossRef]
- Nakabayashi, O.; Takahashi, H.; Moriwaki, K.; Komazawa-Sakon, S.; Ohtake, F.; Murai, S.; Tsuchiya, Y.; Koyahara, Y.; Saeki, Y.; Yoshida, Y.; et al. MIND bomb 2 prevents RIPK1 kinase activity-dependent and -independent apoptosis through ubiquitylation of cFLIP(L). Commun. Biol. 2021, 4, 80. [Google Scholar] [CrossRef] [PubMed]
- Poukkula, M.; Kaunisto, A.; Hietakangas, V.; Denessiouk, K.; Katajamäki, T.; Johnson, M.S.; Sistonen, L.; Eriksson, J.E. Rapid turnover of c-FLIPshort is determined by its unique C-terminal tail. J. Biol. Chem. 2005, 280, 27345–27355. [Google Scholar] [CrossRef]
- Wilkie-Grantham, R.P.; Matsuzawa, S.; Reed, J.C. Novel phosphorylation and ubiquitination sites regulate reactive oxygen species-dependent degradation of anti-apoptotic c-FLIP protein. J. Biol. Chem. 2013, 288, 12777–12790. [Google Scholar] [CrossRef]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef]
- Roberts, J.Z.; Holohan, C.; Sessler, T.; Fox, J.; Crawford, N.; Riley, J.S.; Khawaja, H.; Majkut, J.; Evergren, E.; Humphreys, L.M.; et al. The SCF(Skp2) ubiquitin ligase complex modulates TRAIL-R2-induced apoptosis by regulating FLIP(L). Cell Death Differ. 2020, 27, 2726–2741. [Google Scholar] [CrossRef]
- Dold, M.N.; Ng, X.; Alber, C.; Gentle, I.E.; Häcker, G.; Weber, A. The deubiquitinase Usp27x as a novel regulator of cFLIP(L) protein expression and sensitizer to death-receptor-induced apoptosis. Apoptosis 2022, 27, 112–132. [Google Scholar] [CrossRef]
- Shi, B.; Tran, T.; Sobkoviak, R.; Pope, R.M. Activation-induced degradation of FLIP(L) is mediated via the phosphatidylinositol 3-kinase/Akt signaling pathway in macrophages. J. Biol. Chem. 2009, 284, 14513–14523. [Google Scholar] [CrossRef] [PubMed]
- Chanvorachote, P.; Nimmannit, U.; Wang, L.; Stehlik, C.; Lu, B.; Azad, N.; Rojanasakul, Y. Nitric oxide negatively regulates Fas CD95-induced apoptosis through inhibition of ubiquitin-proteasome-mediated degradation of FLICE inhibitory protein. J. Biol. Chem. 2005, 280, 42044–42050. [Google Scholar] [CrossRef]
- Scaffidi, C.; Medema, J.P.; Krammer, P.H.; Peter, M.E. FLICE is predominantly expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. J. Biol. Chem. 1997, 272, 26953–26958. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Qu, X.; Che, X.; Guo, T.; Li, C.; Ma, R.; Fan, Y.; Ma, Y.; Hou, K.; et al. DR5-Cbl-b/c-Cbl-TRAF2 complex inhibits TRAIL-induced apoptosis by promoting TRAF2-mediated polyubiquitination of caspase-8 in gastric cancer cells. Mol. Oncol. 2017, 11, 1733–1751. [Google Scholar] [CrossRef] [PubMed]
- Oztürk, S.; Schleich, K.; Lavrik, I.N. Cellular FLICE-like inhibitory proteins (c-FLIPs): Fine-tuners of life and death decisions. Exp Cell Res. 2012, 318, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Jeffrey, P.D.; Shi, Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc. Natl. Acad. Sci. USA 2009, 106, 8169–8174. [Google Scholar] [CrossRef]
- Li, X.; Wilmanns, M.; Thornton, J.; Köhn, M. Elucidating human phosphatase-substrate networks. Sci. Signal. 2013, 6, rs10. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. NF-κB-Independent Role of IKKα/IKKβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef]
- Blanchett, S.; Dondelinger, Y.; Barbarulo, A.; Bertrand, M.J.M.; Seddon, B. Phosphorylation of RIPK1 serine 25 mediates IKK dependent control of extrinsic cell death in T cells. Front. Immunol. 2022, 13, 1067164. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fu, J.; Chai, Y.; Liu, Y.; Chen, Y.; Yin, J.; Pu, Y.; Chen, C.; Wang, F.; Liu, Z.; et al. Accumulation of RIPK1 into mitochondria is requisite for oxidative stress-mediated necroptosis and proliferation in Rat Schwann cells. Int. J. Med. Sci. 2022, 19, 1965–1976. [Google Scholar] [CrossRef]
- Wu, X.N.; Yang, Z.H.; Wang, X.K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Chan, W.; Zhang, J.; Wang, J.; Wang, Z.; Liu, J.; Li, J.; Liu, X.; Wang, M.; Hao, L.; et al. Regulation of RIP1-Mediated necroptosis via necrostatin-1 in periodontitis. J. Periodontal Res. 2023, 58, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Liu, B. PLK1-mediated S369 phosphorylation of RIPK3 during G2 and M phases enables its ripoptosome incorporation and activity. iScience 2021, 24, 102320. [Google Scholar] [CrossRef]
- Wu, X.; Fan, X.; McMullen, M.R.; Miyata, T.; Kim, A.; Pathak, V.; Wu, J.; Day, L.Z.; Hardesty, J.E.; Welch, N.; et al. Macrophage-derived MLKL in alcohol-associated liver disease: Regulation of phagocytosis. Hepatology 2023, 77, 902–919. [Google Scholar] [CrossRef]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell 1996, 85, 817–827. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef]
- Powley, I.R.; Hughes, M.A.; Cain, K.; MacFarlane, M. Caspase-8 tyrosine-380 phosphorylation inhibits CD95 DISC function by preventing procaspase-8 maturation and cycling within the complex. Oncogene 2016, 35, 5629–5640. [Google Scholar] [CrossRef]
- Barbero, S.; Barilà, D.; Mielgo, A.; Stagni, V.; Clair, K.; Stupack, D. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. J. Biol. Chem. 2008, 283, 13031–13034. [Google Scholar] [CrossRef]
- Senft, J.; Helfer, B.; Frisch, S.M. Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res. 2007, 67, 11505–11509. [Google Scholar] [CrossRef]
- Jia, S.H.; Parodo, J.; Kapus, A.; Rotstein, O.D.; Marshall, J.C. Dynamic regulation of neutrophil survival through tyrosine phosphorylation or dephosphorylation of caspase-8. J. Biol. Chem. 2008, 283, 5402–5413. [Google Scholar] [CrossRef] [PubMed]
- Günster, R.A.; Matthews, S.A.; Holden, D.W.; Thurston, T.L.M. SseK1 and SseK3 Type III Secretion System Effectors Inhibit NF-κB Signaling and Necroptotic Cell Death in Salmonella-Infected Macrophages. Infect. Immun. 2017, 85, e00010-17. [Google Scholar] [CrossRef]
- Pan, X.; Luo, J.; Li, S. Bacteria-Catalyzed Arginine Glycosylation in Pathogens and Host. Front. Cell. Infect. Microbiol. 2020, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Pan, X.; Du, L.; Yao, Q.; Xue, J.; Yao, H.; Wang, D.C.; Li, S.; Shao, F. Structural and Functional Insights into Host Death Domains Inactivation by the Bacterial Arginine GlcNAcyltransferase Effector. Mol. Cell 2019, 74, 922–935.e6. [Google Scholar] [CrossRef]
- Lake, A.N.; Bedford, M.T. Protein methylation and DNA repair. Mutat. Res. 2007, 618, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Denu, J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta 2009, 1789, 45–57. [Google Scholar] [CrossRef]
- Wu, W.; Wang, J.; Xiao, C.; Su, Z.; Su, H.; Zhong, W.; Mao, J.; Liu, X.; Zhu, Y.Z. SMYD2-mediated TRAF2 methylation promotes the NF-κB signaling pathways in inflammatory diseases. Clin. Transl. Med. 2021, 11, e591. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Sedmaki, K.; Karnam, K.; Sharma, P.; Mahale, A.; Routholla, G.; Ghosh, B.; Prakash Kulkarni, O. HDAC6 inhibition attenuates renal injury by reducing IL-1β secretion and RIP kinase mediated necroptosis in acute oxalate nephropathy. Int. Immunopharmacol. 2022, 110, 108919. [Google Scholar] [CrossRef]
- Yang, L.; Chen, S.; Xia, J.; Zhou, Y.; Peng, L.; Fan, H.; Han, Y.; Duan, L.; Cheng, G.; Yang, H.; et al. Histone deacetylase 3 facilitates TNFα-mediated NF-κB activation through suppressing CTSB induced RIP1 degradation and is required for host defense against bacterial infection. Cell Biosci. 2022, 12, 81. [Google Scholar] [CrossRef]
- Bozonet, S.M.; Magon, N.J.; Schwartfeger, A.J.; Konigstorfer, A.; Heath, S.G.; Vissers, M.C.M.; Morris, V.K.; Göbl, C.; Murphy, J.M.; Salvesen, G.S.; et al. Oxidation of caspase-8 by hypothiocyanous acid enables TNF-mediated necroptosis. J. Biol. Chem. 2023, 299, 104792. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687.e13. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, J.; Yu, J.; Chen, G. FKBP12 mediates necroptosis by initiating RIPK1-RIPK3-MLKL signal transduction in response to TNF receptor 1 ligation. J. Cell Sci. 2019, 132, jcs227777. [Google Scholar] [CrossRef]
- Gao, P.; Cao, M.; Jiang, X.; Wang, X.; Zhang, G.; Tang, X.; Yang, C.; Komuro, I.; Ge, J.; Li, L.; et al. Cannabinoid Receptor 2-Centric Molecular Feedback Loop Drives Necroptosis in Diabetic Heart Injuries. Circulation 2023, 147, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Chang, L.C.; Jung, W.; Bowman, J.W.; Kim, D.; Chen, W.; Foo, S.S.; Choi, Y.J.; Choi, U.Y.; Bowling, A.; et al. OASL phase condensation induces amyloid-like fibrillation of RIPK3 to promote virus-induced necroptosis. Nat. Cell Biol. 2023, 25, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Xia, X.; Lei, L.; Wang, S.; Hu, J.; Zhang, G. Necroptosis and its role in infectious diseases. Apoptosis 2020, 25, 169–178. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Thadathil, N.; Selvarani, R.; Nicklas, E.H.; Wang, D.; Miller, B.F.; Richardson, A.; Deepa, S.S. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 2021, 20, e13512. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Zhao, H.; Ge, D.; Liao, J.; Shao, L.; Mo, A.; Hu, L.; Xu, K.; Wu, J.; Mu, M.; et al. Necroptosis in pulmonary macrophages promotes silica-induced inflammation and interstitial fibrosis in mice. Toxicol. Lett. 2022, 355, 150–159. [Google Scholar] [CrossRef]
- Saeed, W.K.; Jun, D.W.; Jang, K.; Koh, D.H. Necroptosis signaling in liver diseases: An update. Pharmacol. Res. 2019, 148, 104439. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef]
- Della Torre, L.; Nebbioso, A.; Stunnenberg, H.G.; Martens, J.H.A.; Carafa, V.; Altucci, L. The Role of Necroptosis: Biological Relevance and Its Involvement in Cancer. Cancers 2021, 13, 684. [Google Scholar] [CrossRef]
- Lalaoui, N.; Brumatti, G. Relevance of necroptosis in cancer. Immunol. Cell Biol. 2017, 95, 137–145. [Google Scholar] [CrossRef]
- Liu, X.; Yao, Y.; Zhu, Y.; Lu, F.; Chen, X. Inhibition of Adipocyte Necroptosis Alleviates Fat Necrosis and Fibrosis After Grafting in a Murine Model. Aesthetic Surg. J. 2024, 44, NP585–NP605. [Google Scholar] [CrossRef]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef]
- Preston, S.P.; Stutz, M.D.; Allison, C.C.; Nachbur, U.; Gouil, Q.; Tran, B.M.; Duvivier, V.; Arandjelovic, P.; Cooney, J.P.; Mackiewicz, L.; et al. Epigenetic Silencing of RIPK3 in Hepatocytes Prevents MLKL-mediated Necroptosis From Contributing to Liver Pathologies. Gastroenterology 2022, 163, 1643–1657.e14. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, J.; Hou, M.; Tan, Z.; Chen, F.; Zhang, J.; Liu, Y.; Leng, Y. Ursolic acid improves necroptosis via STAT3 signaling in intestinal ischemia/reperfusion injury. Int. Immunopharmacol. 2024, 138, 112463. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Ohwada, W.; Yano, T.; Kouzu, H.; Sato, T.; Ogawa, T.; Osanami, A.; Toda, Y.; Nagahama, H.; Tanno, M.; et al. Contribution of MLKL to the development of doxorubicin-induced cardiomyopathy and its amelioration by rapamycin. J. Pharmacol. Sci. 2024, 156, 9–18. [Google Scholar] [CrossRef]
- Liang, J.; Tian, X.; Zhou, M.; Yan, F.; Fan, J.; Qin, Y.; Chen, B.; Huo, X.; Yu, Z.; Tian, Y.; et al. Shikonin and chitosan-silver nanoparticles synergize against triple-negative breast cancer through RIPK3-triggered necroptotic immunogenic cell death. Biomaterials 2024, 309, 122608. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, L.; Xu, M.F.; Huang, D.; Sun, X.L.; Zhang, Y.X.; Li, H.M.; Wu, C.Z. Novel isobavachalcone derivatives induce apoptosis and necroptosis in human non-small cell lung cancer H1975 cells. J. Enzym. Inhib. Med. Chem. 2024, 39, 2292006. [Google Scholar] [CrossRef]
- Persenaire, C.; Babbs, B.; Yamamoto, T.M.; Nebbia, M.; Jordan, K.R.; Adams, S.; Lambert, J.R.; Bitler, B.G. VDX-111, a novel small molecule, induces necroptosis to inhibit ovarian cancer progression. Mol. Carcinog. 2024, 63, 1248–1259. [Google Scholar] [CrossRef]
- Vissers, M.; Heuberger, J.; Groeneveld, G.J.; Oude Nijhuis, J.; De Deyn, P.P.; Hadi, S.; Harris, J.; Tsai, R.M.; Cruz-Herranz, A.; Huang, F.; et al. Safety, pharmacokinetics and target engagement of novel RIPK1 inhibitor SAR443060 (DNL747) for neurodegenerative disorders: Randomized, placebo-controlled, double-blind phase I/Ib studies in healthy subjects and patients. Clin. Transl. Sci. 2022, 15, 2010–2023. [Google Scholar] [CrossRef] [PubMed]
- Majdi, A.; Aoudjehane, L.; Ratziu, V.; Islam, T.; Afonso, M.B.; Conti, F.; Mestiri, T.; Lagouge, M.; Foufelle, F.; Ballenghien, F.; et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J. Hepatol. 2020, 72, 627–635. [Google Scholar] [CrossRef]
- Cao, L.; Mu, W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol. Res. 2021, 163, 105297. [Google Scholar] [CrossRef]
- Hofmans, S.; Devisscher, L.; Martens, S.; Van Rompaey, D.; Goossens, K.; Divert, T.; Nerinckx, W.; Takahashi, N.; De Winter, H.; Van Der Veken, P.; et al. Tozasertib Analogues as Inhibitors of Necroptotic Cell Death. J. Med. Chem. 2018, 61, 1895–1920. [Google Scholar] [CrossRef]
- Harris, P.A.; Marinis, J.M.; Lich, J.D.; Berger, S.B.; Chirala, A.; Cox, J.A.; Eidam, P.M.; Finger, J.N.; Gough, P.J.; Jeong, J.U.; et al. Identification of a RIP1 Kinase Inhibitor Clinical Candidate (GSK3145095) for the Treatment of Pancreatic Cancer. ACS Med. Chem. Lett. 2019, 10, 857–862. [Google Scholar] [CrossRef]
- Duan, X.; Liu, X.; Liu, N.; Huang, Y.; Jin, Z.; Zhang, S.; Ming, Z.; Chen, H. Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis. 2020, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Zhu, F.; Yang, C.; Wu, S.; Lin, Y.; Ma, H.; Yu, X.; Zhao, C.; Ji, Y.; Ge, W.; et al. Discovery of a Potent RIPK3 Inhibitor for the Amelioration of Necroptosis-Associated Inflammatory Injury. Front. Cell Dev. Biol. 2020, 8, 606119. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.; Li, C.M.; Kang, J.; Najafov, A.; Jung, M.; Kang, S.; Wang, S.; Yuan, J.; Jung, Y.K. Casein kinase-1γ1 and 3 stimulate tumor necrosis factor-induced necroptosis through RIPK3. Cell Death Dis. 2019, 10, 923. [Google Scholar] [CrossRef]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef]
- Syed, R.U.; Afsar, S.; Aboshouk, N.A.M.; Salem Alanzi, S.; Abdalla, R.A.H.; Khalifa, A.A.S.; Enrera, J.A.; Elafandy, N.M.; Abdalla, R.A.H.; Ali, O.H.H.; et al. LncRNAs in necroptosis: Deciphering their role in cancer pathogenesis and therapy. Pathol. Res. Pract. 2024, 256, 155252. [Google Scholar] [CrossRef] [PubMed]
- Balusu, S.; Horré, K.; Thrupp, N.; Craessaerts, K.; Snellinx, A.; Serneels, L.; T’Syen, D.; Chrysidou, I.; Arranz, A.M.; Sierksma, A.; et al. MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer’s disease. Science 2023, 381, 1176–1182. [Google Scholar] [CrossRef]
- Lu, H.; Xu, L.; Steriopoulos, J.; McLeod, P.; Huang, X.; Min, J.; Peng, T.; Jevnikar, A.M.; Zhang, Z.X. An acidic pH environment converts necroptosis to apoptosis. Biochem. Biophys. Res. Commun. 2024, 725, 150215. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Site | Domain | Protein | Necroptosis | References |
|---|---|---|---|---|---|
| Ubiquitination | |||||
| Human | K377 | Intermediate | cIAP1, cIAP2 | ↓ | [42] [114] |
| Mouse | K376 | Intermediate | cIAP1, cIAP2 | ↓ | [42] [114] |
| Human and Mouse | Unknown | Unknown | cIAP1(UBA domain) | ↓ | [113] |
| Human and Mouse | Unknown | Unknown | LUBAC | ↓ | [44] [45] [46] [47] |
| Human | K377 | Intermediate | MIB2 | ↓ | [118] |
| Human | K634 | Death domain | MIB2 | ↓ | [118] |
| Mouse | K376 | Intermediate | PARdU | ↓ | [119] |
| Human | K377 | Intermediate | PARdU | ↓ | [119] |
| Human | K316 | Intermediate | MG53 | ↓ | [120] |
| Human | K604, K627 | Death domain | MG53 | ↓ | [120] |
| Human | K377 | Intermediate | A20, OTULIN CYLD, USP21, | ↑ | [62] [63] [64] [65] [66] [67] |
| Mouse | K376 | Intermediate | A20, OTULIN CYLD, USP21, | ↑ | [62] [63] [64] [65] [66] [67] |
| Human and Mouse | Unknown | Unknown | ZFP91 | ↑ | [68] |
| Human | K115 | Kinase domain | PELI1 | ↑ | [121] |
| Mouse | K115 | Kinase domain | PELI1 | ↑ | [121] |
| Human | K158 | Kinase domain | c-Cbl | ↑ | [122] |
| Human | K571 | Intermediate | CHIP | ↓ | [123] |
| Human | K604, K627 | Death domain | CHIP | ↓ | [123] |
| Mouse | K612 | Death domain | Unknown | ↓ | [124] |
| Human | K13 | N-terminal (prior to Kinase domain) | Unknown | Unknown | [124] |
| Human | K302, K305, K306, K396, K530, K565, K585 | Intermediate | Unknown | Unknown | [124] |
| Human | K30, K45, K49, K65, K77, K97, K105, K132, K137, K140, K153, K163, K167, K184, K185, K204, K265, K284 | Kinase domain | Unknown | Unknown | [124] |
| Human | K596, K599, K642, K648 | Death domain | Unknown | Unknown | [124] |
| Mouse | K13, K20 | N-terminal (prior to Kinase domain) | Unknown | Unknown | [124] |
| Mouse | K306, K307, K392, K395, K429, K519, K550, K584 | Intermediate | Unknown | Unknown | [124] |
| Mouse | K30, K45, K46, K65, K77, K105, K137, K140, K153, K163, K167, K205 | Kinase domain | Unknown | Unknown | [124] |
| Mouse | K589, K619, K627, K633 | Death domain | Unknown | Unknown | [124] |
| Phosphorylation | |||||
| Human | S166 | Kinase domain | RIPK1 | ↑ | [125] [126] |
| Human | S161 | Kinase domain | RIPK1 | ↑ | [94] |
| Human | S14, S15, S20 | N-terminal (prior to Kinase domain) | RIPK1 | No effect | [127] |
| Mouse | S14, S15 | N-terminal (prior to Kinase domain) | RIPK1 | No effect | [127] |
| Mouse | T169 | Kinase domain | RIPK1 | Unknown | [127] |
| Human | S25 | Kinase domain | IKKα/β | ↓ | [127] |
| Mouse | S25 | Kinase domain | IKKα/β | ↓ | [127] |
| Human | S320 | Intermediate | MK2 | ↓ | [49] [50] [51] |
| Mouse | S321, S336 | Intermediate | MK2 | ↓ | [49] [50] [51] |
| Mouse | S321 | Intermediate | TAK1 | ↓ | [128] |
| Human | Y384 | Intermediate | JAK1, SRC | ↓ | [52] |
| Mouse | Y383 | Intermediate | JAK1, SRC | ↓ | [52] |
| Human | S357 | Intermediate | ULK1 | ↓ | [53] |
| Human | T147 | Kinase domain | TBK1 | ↓ | [54] |
| Mouse | S189, S190 | Kinase domain | TBK1 | ↓ | [54] |
| Human | S416 | Intermediate | AMPK | ↓ | [55] |
| Mouse | S415 | Intermediate | AMPK | ↓ | [55] |
| Human | S89 | Kinase domain | Unknown | ↓ | [129] |
| Mouse, rat, xenopus, zebrafish | S89 | Kinase domain | Unknown | ↓ | [129] |
| Human | S25 | Kinase domain | PPP1R3G | ↑ | [130] |
| Mouse | S25 | Kinase domain | PPP1R3G | ↑ | [130] |
| Human | Unknown | Unknown | SMYD2 | ↓ | [131] |
| Glycosylation | |||||
| Human | S331 | Intermediate | OGT | ↓ | [132] |
| Mouse | S332 | Intermediate | OGT | ↓ | [132] |
| Human | R603 | Death domain | NIeB2 | ↓ | [133] |
| Acetylation | |||||
| Human | K115 | Kinase domain | SIRT1, SIRT2 | ↓ | [134] |
| Human | K115 | Kinase domain | HAT1, HAT4 | ↑ | [134] |
| Human | K625, K627, K642, K648 | Death domain | SIRT1, SIRT2 | ↓ | [134] |
| Human | K625, K627, K642, K648 | Death domain | HAT1, HAT4 | ↑ | [134] |
| Human | K596, K599 | Intermediate | SIRT1, SIRT2 | ↓ | [134] |
| Human | K596, K599 | Intermediate | HAT1, HAT4 | ↑ | [134] |
| Disulfide bonds | |||||
| Human | C257, C268 | Kinase domain | ROS | ↑ | [94] |
| Human | C586 | Intermediate | ROS | ↑ | [94] |
| Caspase cleavage | |||||
| Human | D324 | Intermediate | Caspase-6, -8, -10 | ↓ | [135] [136] [137] |
| Mouse | D325 | Intermediate | Caspase-6, -8, -10 | ↓ | [135] [136] [137] |
| SUMOylation | |||||
| Human | K550 | Intermediate | SENP1 | ↓ | [138] |
| Species | Site | Domain | Protein | Necroptosis | References |
|---|---|---|---|---|---|
| Ubiquitination | |||||
| Human and Mouse | K5 | N-terminal (prior to Kinase domain) | A20 | ↓ | [139] [140] |
| Mouse | K158, K287 | Kinase domain | Unknown | ↓ | [139] |
| Mouse | K307 | Intermediate | Unknown | ↓ | [139] |
| Mouse | K469 | C-terminal (after RHIM domain) | Unknown | ↓ | [139] |
| Mouse | K359 | Intermediate | Unknown | ↑ | [139] |
| Human | K42 | Kinase domain | USP22 | No effect | [142] |
| Human | K351 | Intermediate | USP22 | No effect | [142] |
| Human | K518 | C-terminal (after RHIM domain) | USP22 | ↑ | [142] |
| Human | K55 | Kinase domain | CHIP | ↓ | [123] |
| Human | K363 | Intermediate | CHIP | ↓ | [123] |
| Human | K363 | Intermediate | PELI1 | ↓ | [143] |
| Human | K501 | C-terminal (after RHIM domain) | TRIM25 | ↓ | [144] |
| Human | K264 | Kinase domain | Unknown | ↓ | [145] |
| Human | K197 | Kinase domain | Parkin | ↓ | [146] |
| Human | K302, K364 | Intermediate | Parkin | ↓ | [146] |
| Phosphorylation | |||||
| Human | S199, S211, S215, S227, T182, T215 | Kinase domain | RIPK3 | ↑ | [147] [148] [149] [150] [151] [150] |
| Mouse | S204, S232, T231 | Kinase domain | RIPK3 | ↑ | [147] [148] [149] [150] |
| Human and Mouse | S164, T165 | Kinase domain | RIPK3 | ↓ | [152] |
| Human | T224 | Kinase domain | Unknown | ↑ | [150] |
| Human | S227 | Kinase domain | CK1α, CK1δ, CK1ε | ↑ | [153] |
| Mouse | S204 | Kinase domain | RIPK1 | ↑ | [129] |
| Human and Mouse | S164, T165 | Kinase domain | RIPK3-Hsp90/CDC37 | ↓ | [152] |
| Mouse | S369 | Intermediate | PLK1 | ↑ | [127] |
| Mouse | S232, T231 | Kinase domain | Ppm1b | ↓ | [154] |
| Glycosylation | |||||
| Human | T467 | RHIM domain | OGT | ↓ | [155] [156] [157] [158] [159] |
| Methylation | |||||
| Human | R486 | C-terminal (after RHIM domain) | PRMT1, PRMT5 | ↓ | [160] [161] |
| Mouse | R479 | C-terminal (after RHIM domain) | PRMT1, PRMT5 | ↓ | [160] [161] |
| Caspase cleavage | |||||
| Human | D328 | Intermediate | Caspase-8 | ↓ | [162] |
| Mouse | D328 | Intermediate | Caspase-8 | ↓ | [162] |
| Rat | D328 | Intermediate | Caspase-8 | ↓ | [162] |
| Nitrosylation | |||||
| Human | C119 | Kinase domain | NO | ↑ | [163] [164] |
| Species | Site | Domain | Protein | Necroptosis | References |
|---|---|---|---|---|---|
| Ubiquitination | |||||
| Mouse | K51, K77 | 4HB | Unknown | ↑ | [166] |
| Mouse | K172 | Brace | Unknown | ↑ | [166] |
| Mouse | K219 | Pseudokinase | Unknown | ↑ | [166] |
| Human | K230 | Pseudokinase | Unknown | ↑ | [166] |
| Mouse | K9, K51, K69 | 4HB | USP21 | ↑ | [167] |
| Human | K50 | 4HB | ITCH | ↑ | [168] |
| Mouse | K50, K51 | 4HB | ITCH | ↑ | [168] |
| Human and Mouse | Unknown | Unknown | Skp2 | ↓ | [169] |
| Phosphorylation | |||||
| Human | T357, S358 | Pseudokinase | RIPK3 | ↑ | [165] [171] [172] |
| Human | T357, S358 | Pseudokinase | CAMK2/CaMKII | ↑ | [173] |
| Mouse | S345 | Pseudokinase | RIPK3 | ↑ | [165] [171] [172] |
| Mouse | S347 | Pseudokinase | RIPK3 | No effect | [174] [175] |
| Mouse | S158 | Brace | RIPK3 | ↑ | [174] [175] |
| Mouse | S228, S248 | Pseudokinase | RIPK3 | ↑ | [174] [175] |
| Mouse | T349 | Pseudokinase | RIPK3 | Unknown | [174] [175] |
| Mouse | S441 | Pseudokinase | Unknown | Unknown | [176] |
| Human and Mouse | Y376 | Pseudokinase | TAM | ↑ | [177] |
| Human | S83 | 4HB | Unknown | ↓ | [178] |
| Mouse | S82 | 4HB | Unknown | ↓ | [178] |
| Disulfide bonds | |||||
| Human | C86 | 4HB | MLKL | ↑ | [179] |
| Human | C86 | 4HB | Trx1 | ↓ | [180] |
| Species | Site | Domain | Protein | Necroptosis | References |
|---|---|---|---|---|---|
| TRADD | |||||
| Glycosylation | |||||
| Human | R235, R245 | Death domain | SseK1 | ↓ | [183] [184] |
| Mouse | R233 | Death domain | SseK1 | ↓ | [183] |
| Human | R235 | Death domain | NIeB | ↓ | [185] |
| TRAILR | |||||
| Glycosylation | |||||
| Human | R359 | Death domain | SseK3 | ↓ | [183] |
| Mouse | R239 | Death domain | SseK3 | ↓ | [183] |
| TNFR1 | |||||
| Glycosylation | |||||
| Human | R376 | Death domain | SseK3 | ↓ | [183] |
| Mouse | R376 | Death domain | SseK3 | ↓ | [183] |
| Ubiquitination | |||||
| Human and Mouse | Unknown | Unknown | RNF8 | ↑ | [186] |
| FADD | |||||
| Ubiquitination | |||||
| Human | K149, K153 | Death domain | CHIP | ↓ | [181] |
| Human and Mouse | Unknown | Unknown | MKRN1 | ↓ | [182] |
| Phosphorylation | |||||
| Human | S194 | Death domain | CKIα | No effect | [187] [188] |
| Mouse | S191 | Death domain | CKIα | No effect | [187] [188] |
| Human | S200 | Death domain | CK2 | No effect | [189] |
| Human | S203 | Death domain | Aur-A, Plk1 | No effect | [190] |
| SUMOylation | |||||
| Human | K120, K125, K149 | Death domain | PIAS3 | Unknown | [191] |
| Caspase-8 | |||||
| Ubiquitination | |||||
| Human | K461 | C-terminal domain (P10 subunit) | CUL3 | ↓ | [192] |
| Human and Mouse | Unknown | Unknown | TRIM13 | ↓ | [193] |
| Human | K215 | Intermediate | HECTD3 | ↓ | [194] |
| Human | K224, K229, K231 | C-terminal domain (P18 subunit) | TRAF2 | Unknown | [195] |
| Phosphorylation | |||||
| Mouse | T265 | C-terminal domain (P18 subunit) | p90 RSK | ↑ | [196] |
| Human | T263 | C-terminal domain (P18 subunit) | P90 RSK (RSK2) | Unknown | [197] |
| Human | T273 | C-terminal domain (P18 subunit) | Plk3 | Unknown | [198] |
| Human | S347 | C-terminal domain (P18 subunit) | P38-MAPK | Unknown | [199] |
| Human | Y380 | C-terminal domain (Intermediate) | Src | Unknown | [200] |
| Human | S387 | C-terminal domain (Intermediate) | CDK1 | Unknown | [201] |
| Human | Y448 | C-terminal domain (P10 subunit) | Lyn | Unknown | [202] |
| Nitrosylation | |||||
| Human and Mouse | Unknown | Unknown | NO | Unknown | [203] |
| SUMOylation | |||||
| Human | K156 | DED domain | Unknown | Unknown | [204] |
| cFLIP | |||||
| Ubiquitination | |||||
| Human | K351, K353 | C-terminal domain (P20 subunit) | LUBAC | ↓ | [205] |
| Human | K351, K353, K381, K386, K389, K460, K462, K473, K474 | C-terminal domain | MIB2 | ↓ | [206] |
| Human | K167, K192, K195 | Intermediate | Unknown | Unknown | [207] [208] [209] |
| Human and Mouse | Unknown | Unknown | SCFSkp2 | Unknown | [210] |
| Human and Mouse | Unknown | Unknown | ITCH | Unknown | [209] |
| Human and Mouse | Unknown | Unknown | Usp27x | Unknown | [211] |
| Human and Mouse | Unknown | Unknown | TRIM28 | Unknown | [211] |
| Phosphorylation | |||||
| Human | T166 | Intermediate | Unknown | Unknown | [207] [208] [209] |
| Human | S193 | Intermediate | PKC | Unknown | [207] [208] [209] |
| Human | S273 | C-terminal domain (P20 subunit) | Akt1 | Unknown | [212] |
| Nitrosylation | |||||
| Human | C254, C259 | C-terminal domain (P20 subunit) | NO | ↑ | [213] |
| Drug Name | Target | Development Stage | Disease/Indication | References |
|---|---|---|---|---|
| GSK2982772 | RIPK1 inhibitor | Phase II Clinical Trial | Psoriasis, Ulcerative Colitis, Rheumatoid Arthritis | [258] |
| SAR443060 (DNL758) | RIPK1 inhibitor | Phase I Clinical Trial | Neurodegenerative Disorders | [265] |
| RIPA-56 | RIPK1 inhibitor | Preclinical | Non-Alcoholic Fatty Liver Disease | [266] |
| Necrostatin-1s (Nec-1s) | RIPK1 inhibitor | Preclinical | Neurodegenerative Disorders, Ischemia–Reperfusion Injury, Cardiovascular Diseases, Renal Diseases, Liver Diseases, etc. | [267] |
| Tozasertib | RIPK1 inhibitor | Phase II Clinical Trial | Mouse Model of TNF-α-induced systemic inflammatory response syndrome | [268] |
| GSK3145095 | RIPK1 inhibitor | Discontinued (Phase I) | Pancreatic Cancer, Colorectal Cancer | [269] |
| Necrosulfonamide (NSA) | MLKL inhibitor | Preclinical | Cardiac Arrest, Spinal Cord Injury, Intracerebral Hemorrhage | [270] |
| GSK’872 | RIPK3 inhibitor | Preclinical | Ischemia–Reperfusion Injury, Sepsis, Neurodegenerative Diseases, Psoriasis, Ulcerative Colitis, Rheumatoid Arthritis | [271] |
| VDX-111 | RIPK1 agonist | Preclinical | Ovarian Cancer | [264] |
| SHK+Chi-Ag NPs | RIPK3 agonist | Preclinical | Triple-Negative Breast Cancer | [262] |
| Seventeen isobavachalcone (IBC) derivatives (1-17) | RIPK3, MLKL agonist | Preclinical | Non-Small Cell Lung Cancer | [263] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, H.; Han, Z.; Xu, M.; Gao, X.; Qiu, S.; Ren, N.; Yi, Y.; Zhou, C. The Role of Post-Translational Modifications in Necroptosis. Biomolecules 2025, 15, 549. https://doi.org/10.3390/biom15040549
Xiao H, Han Z, Xu M, Gao X, Qiu S, Ren N, Yi Y, Zhou C. The Role of Post-Translational Modifications in Necroptosis. Biomolecules. 2025; 15(4):549. https://doi.org/10.3390/biom15040549
Chicago/Turabian StyleXiao, Hao, Zeping Han, Min Xu, Xukang Gao, Shuangjian Qiu, Ning Ren, Yong Yi, and Chenhao Zhou. 2025. "The Role of Post-Translational Modifications in Necroptosis" Biomolecules 15, no. 4: 549. https://doi.org/10.3390/biom15040549
APA StyleXiao, H., Han, Z., Xu, M., Gao, X., Qiu, S., Ren, N., Yi, Y., & Zhou, C. (2025). The Role of Post-Translational Modifications in Necroptosis. Biomolecules, 15(4), 549. https://doi.org/10.3390/biom15040549