
Structural Characteristics of Mitochondrial Genomes of Two Species of Mackerel and Phylogenetic Analysis of Scombridae Family


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials and DNA Extraction
2.2. Sequencing, Gene Annotation, and Analysis
2.3. Phylogenetic Analysis
3. Results and Analysis
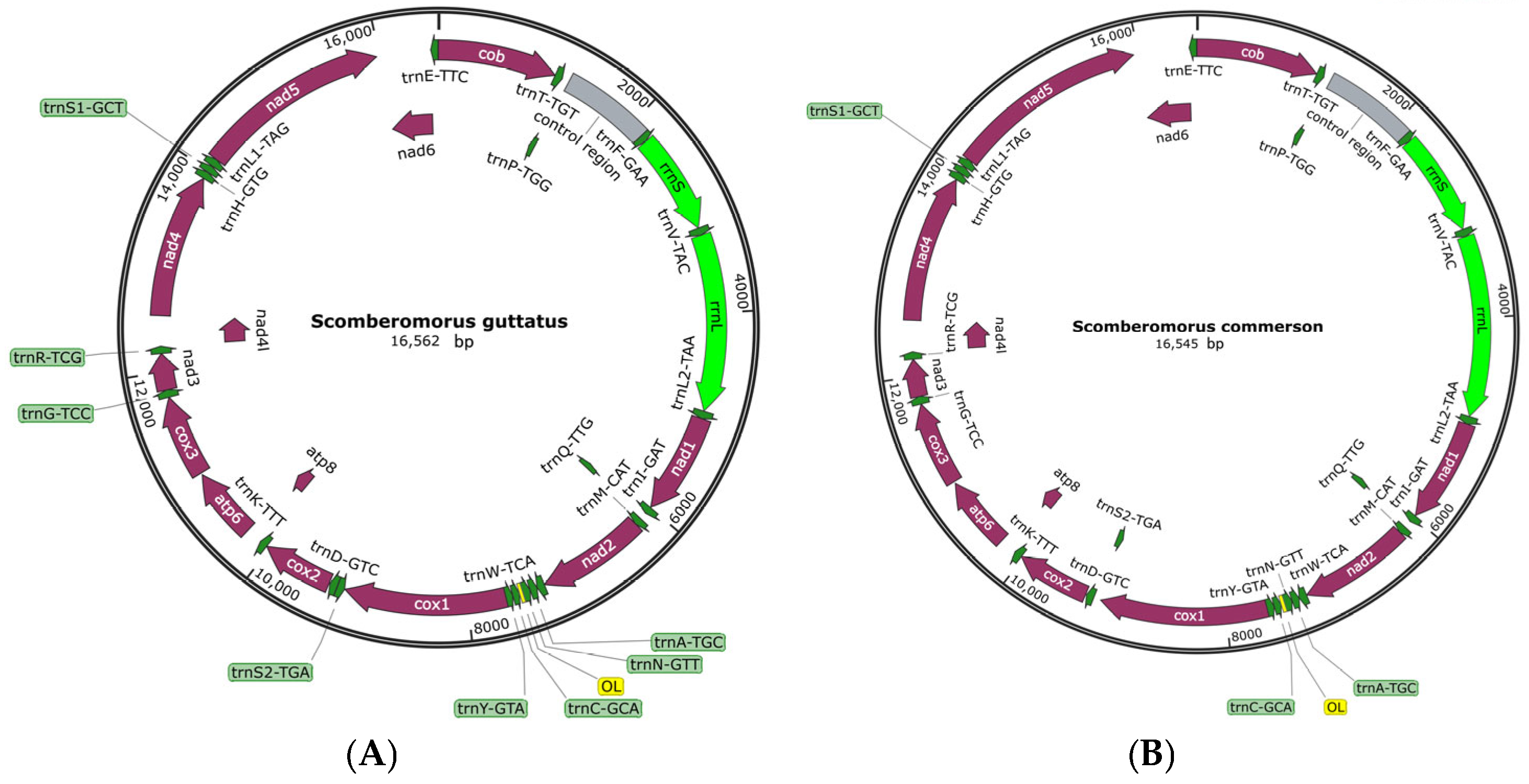
3.1. Composition and Localization of Mitochondrial Genome
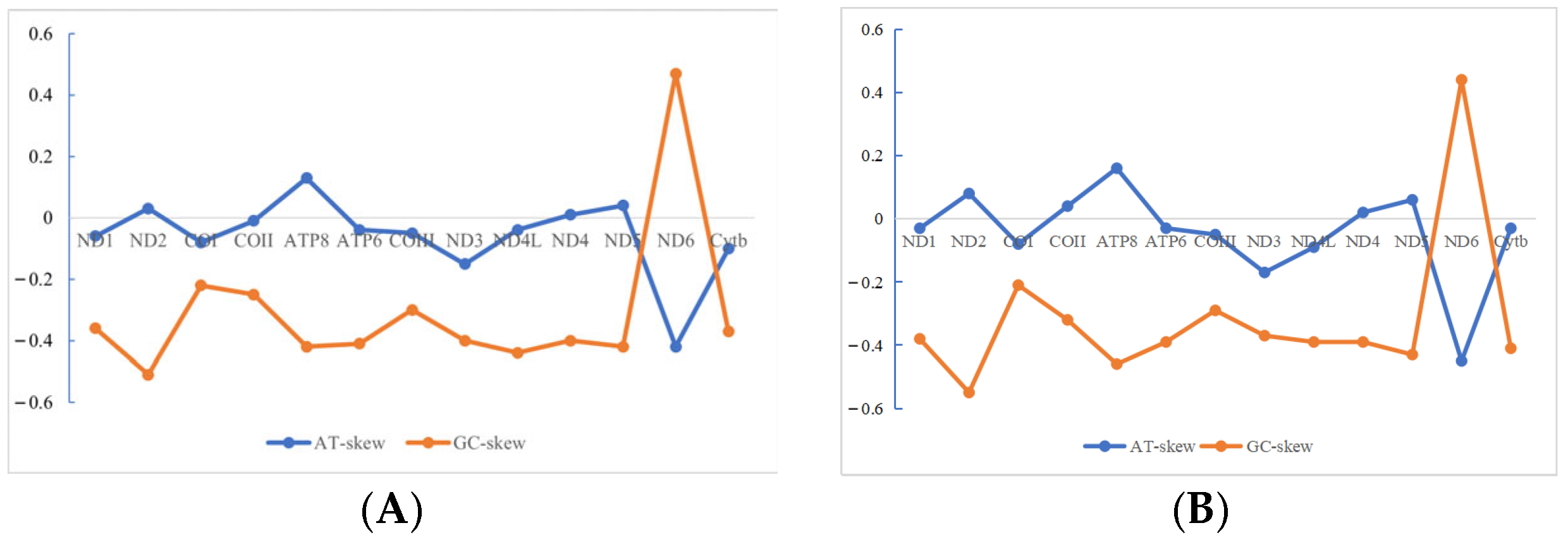
3.2. Analysis of Mitochondrial Genome Base Composition
3.3. Analysis of Gene Segments in Mitochondrial Genome
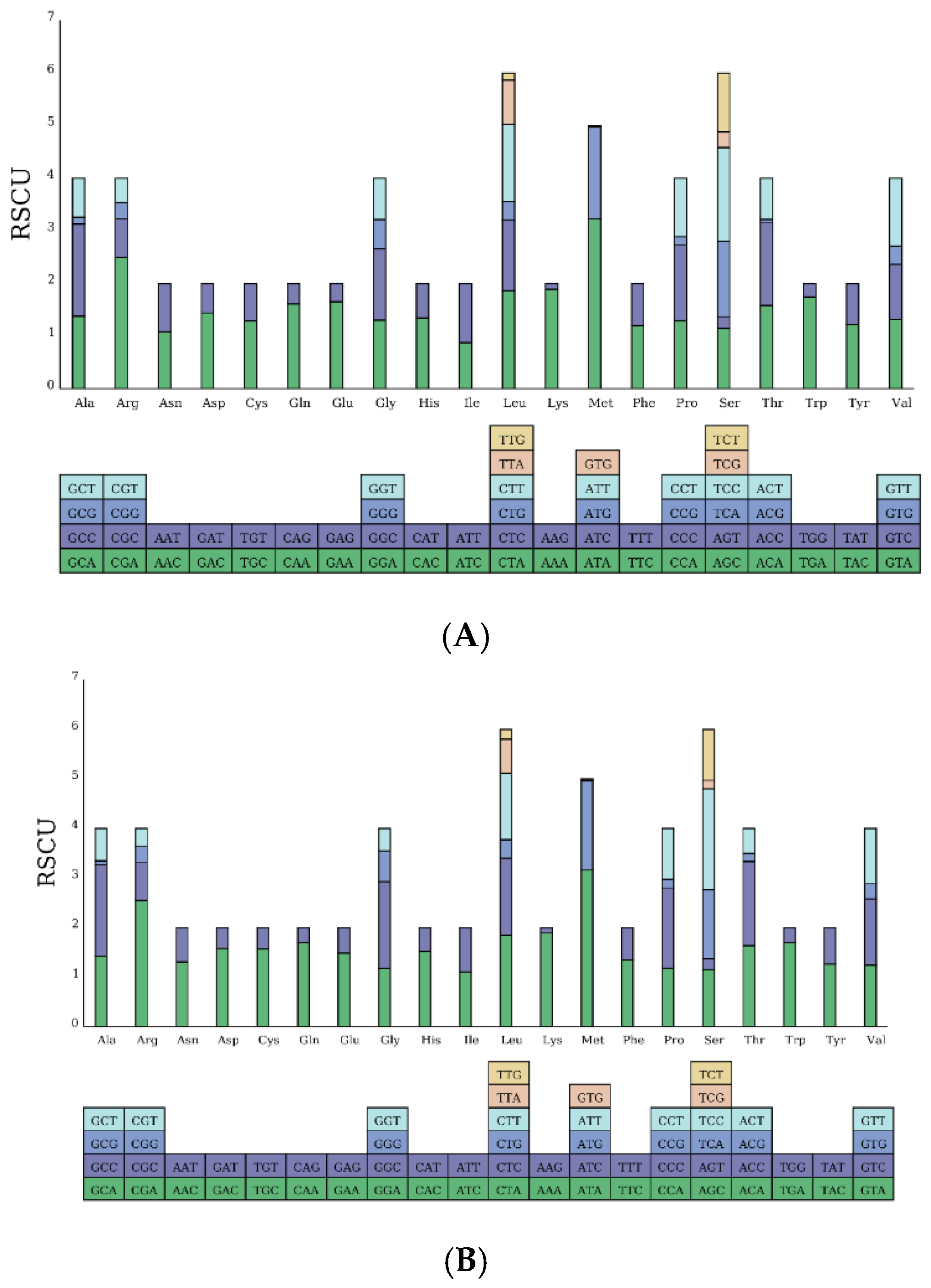
3.3.1. Base Composition and Content of Protein-Coding Genes
3.3.2. rRNA and tRNA Structural Features
3.3.3. Structural Features of the Mitochondrial Genome Control Region
Termination-Associated Sequence Region
Central Conserved Domain Translation
The Conservative Sequence Region
3.4. Results of Phylogenetic Analysis
4. Discussion
4.1. Mitochondrial DNA Sequence Structural Features of S. guttatus and S. commerson
4.2. Molecular Phylogenetic Analysis of Scombridae
5. Conclusions
- (1)
- The mitochondrial DNA sequences of spotted mackerel and king mackerel are 16,562 bp and 16,545 bp, respectively. Both species possess 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes, and 1 non-coding region (D-loop). The base composition consists of A (28.5%, 28.1%), T (26.6%, 25.3%), G (16.0%, 16.2%), and C (28.9%, 30.3%), indicating a high A + T content and a low G + C content, demonstrating a significant AT bias. Protein-coding genes in both species exhibit a relatively uniform distribution of bases at the first codon position, while the second and third positions show a clear anti-G bias. Among the 20 amino acids encoded, Leucine (Leu) is the most frequently used. This study reveals that the absolute value of the free energy of the 12S rRNA gene is lower than that of the 16S rRNA gene. In both species, the tRNA-Ser lacks the DHU arm, preventing the formation of a cloverleaf structure. In control area of S. guttatus and S. commerson, the terminal-associated sequence (TAS) was identified, and a total of three core sequences with repeated “---TACAT---ATGTA---” were found. In the control region of S. guttatus and S. commerson, termination signal regions, central conserved regions. The central conserved region exhibits a typical CSB-E structure and CSB-D structure, but no CSB-F structure was found. The conserved sequence block did not reveal a CSB-1 structure.
- (2)
- In this study, a phylogenetic tree of 60% of the Scombridae fish species was constructed using concatenated sequences of 12 protein-coding genes encoded by the heavy (H) strand of the mitochondrial genome. The tree was built using the NJ and ML methods. The results revealed a close relationship between the Scomber genus and the Acanthocybium genus, while the genus Scomberomorus showed closer affinity with the genera Thunnus, Auxis, Katsuwonus, Acanthocybium, Gymnosarda, Scomberomorus, Grammatorcynus, and Rastrelliger. Furthermore, within the genus Scomberomorus, a monophyletic group was formed, with the spotted mackerel and the king mackerel diverging early. S. niphonius and the S. munroi formed a separate clade, whereas S. concolor, S. sierra, and S. maculatus were sister groups. This study provides a more comprehensive understanding of the phylogenetic relationships within the Scombridae family, further confirming the evolutionary relationships among its members.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, D.; Zhang, M. Chinese Marine Fish; Ocean University of China Press: Qingdao, China, 2015. [Google Scholar]
- Holland, L.Z.; Holland, P.W.H.; Holland, N.D.; Ferraris, J.D.; Palumbi, S.R. Molecular Zoology: Advances, Strategies, and Protocols; Wiley: New York, NY, USA, 1996; pp. 267–282. [Google Scholar]
- Nelson, J.S.; Grande, T.C.; Wilson, M.V.H. Fishes of the World; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Welch, D.J.; Newman, S.J.; Buckworth, R.C.; Ovenden, J.R.; Broderick, D.; Lester, R.J.; Newman, S.J.; Buckworth, R.C.; Ovenden, J.R.; Broderick, D.; et al. Integrating different approaches in the definition of biological stocks: A northern Australian multi-jurisdictional fisheries example using grey mackerel. Scomberomorus Semifasciatus 2015, 55, 73–80. [Google Scholar]
- Broderick, D.; Ovenden, J.R.; Buckworth, R.C.; Newman, S.J.; Lester, R.J.G.; Welch, D.J. Genetic population structure of grey mackerel Scomberomorus semifasciatus in northern Australia. J. Fish Biol. 2011, 79, 633–661. [Google Scholar] [PubMed]
- Da Cunha, D.B.; Rodrigues-Filho LF, S.; de Luna Sales, J.B.; Rêgo, P.; Queiroz, C.; Sampaio, I.; Vallinoto, M. Molecular inferences on Scomberomorus brasiliensis, from the Western South Atlantic, based on two mitochondrial genes. Front. Mar. Sci. 2020, 7, 558902. [Google Scholar]
- López, M.D.; Alcocer, M.U.; Jaimes, P.D. Phylogeography and historical demography of the Pacific Sierra mackerel (Scomberomorus sierra) in the Eastern Pacific. BMC Genet. 2010, 11, 34. [Google Scholar]
- Milá, B.; Van Tassell, J.L.; Calderón, J.A.; Rüber, L.; Zardoya, R. Cryptic Lineage Divergence in Marine Environments: Genetic Differentiation at Multiple Spatial and Temporal Scales in the Widespread Intertidal Goby Gobiosoma Bosc. Ecol. Evol. 2017, 7, 5514–5523. [Google Scholar]
- Santa Brígida, E.L.; Cunha, D.B.D.; Rego, P.S.D.; Sampaio, I.; Schneider, H.; Vallinoto, M. Population Analysis of Scomberomorus Cavalla (Cuvie) (Perciformes, Scombridae) From the Northern and Northeastern Coast of Brazil. Braz. J. Biol. 2007, 67, 919–924. [Google Scholar]
- Abedi, E.; Mohammadi, M.; Qasemi, A. Genetic Stock Structure of Scomberomorus Guttatus Using Microsatellite Markers in the Persian Gulf. J. Oceanogr. 2011, 2, 15–21. [Google Scholar]
- Takeyama, H.; Chow, S.; Tsuzuki, H.; Matunaga, T. Mitochondrial DNA sequence variation within and between tuna Thunnus species and its application to species identification. J. Fish Biol. 2001, 58, 1646–1657. [Google Scholar]
- Chow, S.; Nohara, K.; Tanabe, T.; Itoh, T.; Tsuji, S.; Nishikawa, Y.; Uyeyanagi, S.; Uchikawa, K. Genetic and morphological identification of larval and small juvenile tunas (Pisces: Scombridae) caught by a mid-water trawl in the western Pacific. Bull.-Fish. Res. Agency Jpn. 2003, 8, 1–14. [Google Scholar]
- Huang, H.; Cheng, Q.; Zheng, J. New identification methods for three Scombridae fishes based on morphological and molecular markers. MarineFisheries 2011, 33, 297–303. [Google Scholar]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. A mitogenomic perspective on the basal teleostean phylogeny: Resolving higher-level relationships with longer DNA sequences. Mol. Phylogenetics Evol. 2001, 20, 275–285. [Google Scholar]
- Harrison, R.G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar]
- Liu, Y.; Hu, J.; Ning, Z.; Xiao, P.; Yang, T. Mitochondrial genome sequence characteristics and phylogenetic analysis of Schizothorax argentatus. Chin. J. Biotechnol. 2023, 39, 2965–2985. [Google Scholar]
- Liu, S.F.; Wu, R.X.; Zhu, L.; Zhuang, Z.M. Complete Sequence and Gene Organization of Mitochondrial DNA of The Small Yellow Croaker Larimichthys polyactis. Oceanol. Limnol. Sin. 2010, 41, 885–894. [Google Scholar]
- Liang, Y.; Zhang, G.; Chen, C.; Zhao, X.; Gao, N.; Li, J.; Fang, T.; Yang, K.; Yin, F.; Guo, W. Complete mitochondrial genome sequencing and phylogeny analysis of goby (Taenioides cirratus). J. Dalian Ocean. Unive Rsity 2023, 38, 772–778. [Google Scholar]
- Li, Z.; Li, C.; Kong, L.; Bao, C.; He, C.; Jin, W.; Chen, X. Mitochondrial genome structure and phylogenetic analysis oftriploid rainbow trout. J. Northwest AF Univ. (Nat. Sci. Ed.) 2023, 51, 9–17+28. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar]
- Tomita, K.; Ueda, T.; Watanabe, K. RNA editing in the acceptor stem of squid mitochondrial tRNATyr. Nucleic Acids Res. 1996, 24, 4987–4991. [Google Scholar] [PubMed]
- Liu, H. Structure and Evolution of the Mitochondrial DNA Control Region in Fishes: A Case Study of Rhodeus ocellatus. Adv. Nat. Sci. 2002, 3, 44–48. [Google Scholar]
- Lee, W.J.; Conroy, J.; Howell, W.H.; Kocher, T.D. Structure and evolution of teleost mitochondrial control regions. Mol. Evol. 1995, 41, 54–66. [Google Scholar]
- Zeng, Q.; Liu, H. Study on mitochondrial DNA control region of the Ictiobus cyprinellus. J. Hubei Univ. Nat. Sci. Ed. 2001, 23, 261–264. [Google Scholar]
- Lee, W.J.; Kocher, T.D. Complete sequence of a sea lamprey (Petromyzonmarinus) mitochondrial genome: Early establishment of the vertebrate genome organization. Genetics 1995, 139, 873–887. [Google Scholar]
- Zhong, D.; Zhao, G.; Zhang, Z.S.; Xun, A.L. Advance in the Entire Balance and Local Unbalance of Base Distribution in Genome. Hereditas 2002, 24, 351–355. [Google Scholar]
- Frank, A.C.; Lobry, J.R. Asymmetric substitution patterns: A review of possible underlying mutational or selective mechanisms. Gene 1999, 238, 65–77. [Google Scholar]
- Li, Y. Determination and Molecular Evolution Analysis of Mitochondrial Genomes of Three Species of Schizothoracine Fishes from the Qinghai-Tibetan Plateau. Ph.D. Thesis, Fudan University, Shanghai, China, 2012. [Google Scholar]
- Bao, M. The Staructure Analysis of Complete Mitochondrial Genome and Development of Microsatellite Markers for Schizothorax pseudaksaieusis; Huazhong Agricultural University: Wuhan, China, 2015. [Google Scholar]
- Miya, M.; Sato, Y.; Fukunaga, T.; Sado, T.; Poulsen, J.Y.; Sato, K.; Minamoto, T.; Yamamoto, S.; Yamanaka, H.; Araki, H.; et al. MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: Detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2015, 2, 150088. [Google Scholar]
- Lian, Z.Q.; Gun, S.B.; Li, L.; Zhang, F.; Xiao, W.; Wu, X.D. Sequencing and Analysis of The Complete Mitochondrial Genome of Silurus Lanzhouensis Based on Next Generation Sequencing Technologies. Acta Hydrobiol. Sin. 2017, 41, 334–345. [Google Scholar]
- Lynch, M. Mutation accumulation in nuclear, organelle, and prokaryotic transfer RNA genes. Mol. Biol. Evol. 1997, 14, 914–925. [Google Scholar]
- Xiao, W.; Zhang, Y. Genetics and evolution of mitochondrial DNA in fish. Acta Hydrobiol. Sin. 2000, 24, 384–391. [Google Scholar] [CrossRef]
- Liu, H.; Yu, L.; Zhang, F. Molecular Structure and Application Progress of Fish Mitochondrial DNA Control Region. Water Conserv. Fish. 2008, 28, 4–8. [Google Scholar]
- Qiu, F.; Su, Y.; Fu, M.; Wang, J. Molecular phylogenetic analysis in Scombridae based on Cytb and ITS1 fragment sequences. J. Fish. Sci. China 2010, 17, 201–211. [Google Scholar]
- Qiu, F. Genetic Diversity of Western Pacific Bluefin Tuna and Yellowfin Tuna and Molecular Systematics of Scombridae; Xiamen University: Xiamen, China, 2009. [Google Scholar]
- Miya, M.; Takeshima, H.; Endo, H.; Ishiguro, N.B.; Inoue, J.G.; Mukai, T.; Satoh, T.P.; Yamaguchi, M.; Kawaguchi, A.; Mabuchi, K. Major patterns of higher teleostean phylogenies: A new perspective based on 100 complete mitochondrial DNA sequences. Mol. Phylogenetics Evol. 2003, 26, 121–138. [Google Scholar] [CrossRef]
- Chen, Y. Phylogenetic Study of Scombridae Fishes and Molecular Phylogeography of Katsuwonus Pelamis Inferred from Mitochondrial DNA; Shanghai Ocean University: Shanghai, China, 2014. [Google Scholar]
- Jeena, N.S.; Rahuman, S.; Roul, S.K.; Azeez, P.A.; Vinothkumar, R.; Manas, H.M.; Nesnas, E.A.; Rathinam, A.M.M.; Surya, S.; Rohit, P. Resolved and redeemed: A new fleck to the evolutionary divergence in the genus Scomberomorus Lacepède, 1801 (Scombridae) with cryptic speciation. Front. Mar. Sci. 2022, 9, 888463. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genera | Species | GenBank ID |
|---|---|---|
| Auxis | Auxis rochei | NC_005313 |
| Auxis thazard | NC_005318 | |
| Euthynnus | Euthynnus alletteratus | NC_004530 |
| Euthynnus affinis | AP012946.1 | |
| Gasterochisma | Gasterochisma melampus | HQ425781 |
| Katsuwonus | Katsuwonus pelamis | NC_005316 |
| Rastrelliger | Rastrelliger brachysoma | NC_013485 |
| Rastrelliger kanagurta | JX524134 | |
| Scomber | Scomber australasicus | NC_013725 |
| Scomber colias | NC_013724 | |
| Scomber japonicus | NC_013723 | |
| Scomber scombrus | NC_006398 | |
| Scomberomorus | Scomberomorus cavalla | DQ536428.1 |
| Scomberomorus niphonius | NC_016420 | |
| Scomberomorus concolor | KX925518.1 | |
| Scomberomorus guttatus | this study | |
| Scomberomorus commerson | this study | |
| Scomberomorus maculatus | OP056995.2 | |
| Scomberomorus munroi | NC_021390.1 | |
| Scomberomorus semifasciatus | NC_021391.1 | |
| Scomberomorus sierra | KX925517.1 | |
| Thunnus | Thunnus alalunga | NC_005317 |
| Thunnus albacares | NC_014061 | |
| Thunnus atlanticus | NC_025519.1 | |
| Thunnus maccoyii | NC_014101 | |
| Thunnus obesus | NC_014059 | |
| Thunnus orientalis | NC_008455 | |
| Thunnus thynnus | NC_014052 | |
| Thunnus tonggol | HQ425780 | |
| Acanthocybium | Acanthocybium solandri | NC_067731.1 |
| Sarda | Sarda orientalis | AP012949.1 |
| Gymnosarda | Gymnosarda unicolor | AP012510.1 |
| Sequence | Position | Length | Start Codon | Stop Codon | Spacing Nucleotide Count | Coding Strand | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S.g | S.c | S.g | S.c | S.g | S.c | S.g | S.c | S.g | S.c | S.g | S.c | |
| Cytb | 1–1141 | - | 1141 | - | ATG | T-- | - | 4 | - | H | ||
| tRNA-Thr | 1142–1215 | 1142–1213 | 74 | 72 | 0 | - | H | |||||
| tRNA-Pro | 1215–1284 | 1213–1282 | 70 | - | −1 | - | L | |||||
| D-loop | 1285–2141 | 1283–2137 | 857 | 855 | 0 | - | H | |||||
| tRNA-Phe | 2142–2209 | 2138–2205 | 68 | - | 0 | - | H | |||||
| s-rRNA | 2210–3176 | 2206–3162 | 967 | 957 | 0 | - | H | |||||
| tRNA-Val | 3177–3248 | 3163–3234 | 72 | - | 0 | - | H | |||||
| l-rRNA | 3249–4945 | 3235–4927 | 1697 | 1693 | 0 | - | H | |||||
| tRNA-Leu | 4946–5019 | 4928–5001 | 74 | - | 0 | - | H | |||||
| ND1 | 5020–5994 | 5002–5976 | 975 | - | ATG | TAA | - | 0 | - | H | ||
| tRNA-Ile | 5999–6069 | 5981–6051 | 71 | - | 4 | - | H | |||||
| tRNA-Gln | 6069–6139 | 6051–6121 | 71 | - | −1 | - | L | |||||
| tRNA-Met | 6139–6207 | 6121–6189 | 69 | - | −1 | - | H | |||||
| ND2 | 6208–7253 | 6190–7235 | 1046 | - | ATG | TA- | - | 0 | - | H | ||
| tRNA-Trp | 7254–7326 | 7236–7308 | 73 | - | 0 | - | H | |||||
| tRNA-Ala | 7328–7396 | 7310–7378 | 69 | - | 1 | - | L | |||||
| tRNA-Asn | 7398–7470 | 7380–7452 | 73 | - | 1 | - | L | |||||
| OL(rep_origin) | 7471–7503 | 7453–7486 | 33 | 34 | 0 | - | H | |||||
| tRNA-Cys | 7504–7569 | 7487–7552 | 66 | - | 0 | - | L | |||||
| tRNA-Tyr | 7570–7636 | 7553–7620 | 67 | 68 | 0 | - | L | |||||
| COI | 7638–9185 | 7622–9181 | 1548 | 1560 | GTG | AGA | AGG | 1 | - | H | ||
| tRNA-Ser | 9188–9259 | 9173–9244 | 72 | - | 2 | −9 | L | |||||
| tRNA-Asp | 9263–9335 | 9248–9320 | 73 | - | 3 | - | H | |||||
| COII | 9344–10,034 | 9329–10,019 | 691 | - | ATG | T-- | - | 8 | - | H | ||
| tRNA-Lys | 10,035–10,108 | 10,020–10,093 | 74 | - | 0 | - | H | |||||
| ATP8 | 10,110–10,277 | 10,095–10,262 | 168 | - | ATG | TAA | - | 1 | - | H | ||
| ATP6 | 10,268–10,951 | 10,253–10,936 | 684 | - | ATG | TAA | - | −10 | - | H | ||
| COIII | 10,971–11,755 | 10,955–11,739 | 785 | - | ATG | TA- | - | 19 | 18 | H | ||
| tRNA-Gly | 11,756–11,827 | 11,740–11,811 | 72 | - | 0 | - | H | |||||
| ND3 | 11,828–12,176 | 11,812–12,160 | 349 | - | ATG | T-- | - | 0 | - | H | ||
| tRNA-Arg | 12,177–12,245 | 12,161–12,229 | 69 | - | 0 | - | H | |||||
| ND4L | 12,246–12,542 | 12,230–12,526 | 297 | - | ATG | TAA | - | 0 | - | H | ||
| ND4 | 12,536–13,916 | 12,520–13,900 | 1381 | - | ATG | T-- | - | −7 | - | H | ||
| tRNA-His | 13,917–13,987 | 13,901–13,970 | 71 | 70 | 0 | - | H | |||||
| tRNA-Ser | 13,988–14,055 | 13,971–14,038 | 68 | - | 0 | - | H | |||||
| tRNA-Leu | 14,060–14,132 | 14,043–14,115 | 73 | - | 4 | - | H | |||||
| ND5 | 14,133–15,971 | 14,116–15,954 | 1839 | - | ATG | TAA | - | 0 | - | H | ||
| ND6 | 15,968–16,489 | 15,951–16,472 | 522 | - | ATG | TAA | TAG | −4 | - | L | ||
| tRNA-Glu | 16,490–16,558 | 16,473–16,541 | 69 | - | 0 | - | L | |||||
| Sequence | A% | T% | G% | C% | A + T% | G + C% | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S.g | S.c | S.g | S.c | S.g | S.c | S.g | S.c | S.g | S.c | S.g | S.c | S.g | S.c | |
| mtDNA complete sequence | 28.5 | 28.1 | 26.6 | 25.3 | 16 | 16.2 | 28.9 | 30.3 | 55.1 | 53.5 | 44.9 | 46.5 | 16,562 | 16,545 |
| Protein-coding genes | 26.2 | 25.7 | 28.6 | 27.1 | 15.3 | 15.6 | 29.9 | 31.6 | 54.8 | 52.8 | 45.2 | 47.3 | 11,426 | 11,438 |
| tRNA genes | 27.5 | 27 | 27.2 | 26.9 | 23.7 | 24.4 | 21.6 | 21.7 | 54.7 | 53.9 | 45.3 | 46 | 1558 | 1556 |
| rRNA gene | 31.3 | 32.2 | 22.5 | 21.5 | 21.1 | 21 | 25.1 | 25.3 | 53.8 | 53.7 | 46.2 | 46.3 | 2664 | 2650 |
| Control region | 32.2 | 31.1 | 30.9 | 32.1 | 16 | 14.9 | 20.9 | 22 | 63.1 | 63.1 | 36.9 | 36.8 | 857 | 855 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Li, A.; Liu, S. Structural Characteristics of Mitochondrial Genomes of Two Species of Mackerel and Phylogenetic Analysis of Scombridae Family. Biomolecules 2025, 15, 555. https://doi.org/10.3390/biom15040555
Yang J, Li A, Liu S. Structural Characteristics of Mitochondrial Genomes of Two Species of Mackerel and Phylogenetic Analysis of Scombridae Family. Biomolecules. 2025; 15(4):555. https://doi.org/10.3390/biom15040555
Chicago/Turabian StyleYang, Jianqi, Ang Li, and Shufang Liu. 2025. "Structural Characteristics of Mitochondrial Genomes of Two Species of Mackerel and Phylogenetic Analysis of Scombridae Family" Biomolecules 15, no. 4: 555. https://doi.org/10.3390/biom15040555
APA StyleYang, J., Li, A., & Liu, S. (2025). Structural Characteristics of Mitochondrial Genomes of Two Species of Mackerel and Phylogenetic Analysis of Scombridae Family. Biomolecules, 15(4), 555. https://doi.org/10.3390/biom15040555