
The B22 Dilemma: Structural Basis for Conformational Differences in Proinsulin B-Chain Arg22 Mutants



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
- FVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
WT:
- FVNQHLCGSHLVEALYLVCGEEGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
R(B22)E:
- FVNQHLCGSHLVEALYLVCGEQGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
R(B22)Q:
- FVNQHLCGSHLVEALYLVCGEAGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
R(B22)A:
- FVNQHLCGSHLVEALYLVCGEGGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
R(B22)G:
- FVNQHLCGSHLVEALYLVCGEFGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
R(B22)F:
- FVNQHLCGSHLVEALYLVCGEKGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
R(B22)K:
- FVNQHLCGSHLVEALYLVCGEYGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
R(B22)Y:
- FVNQHLCGSHLVEALYLVCGEDGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
R(B22)D:
3. Results
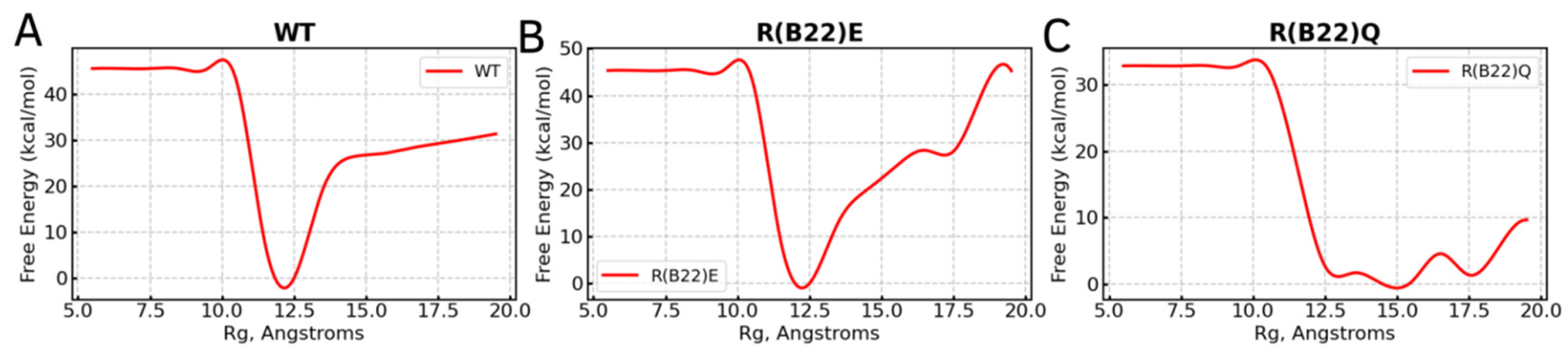
3.1. WT Proinsulin Shows a Stable Minimum Around Compact Conformations
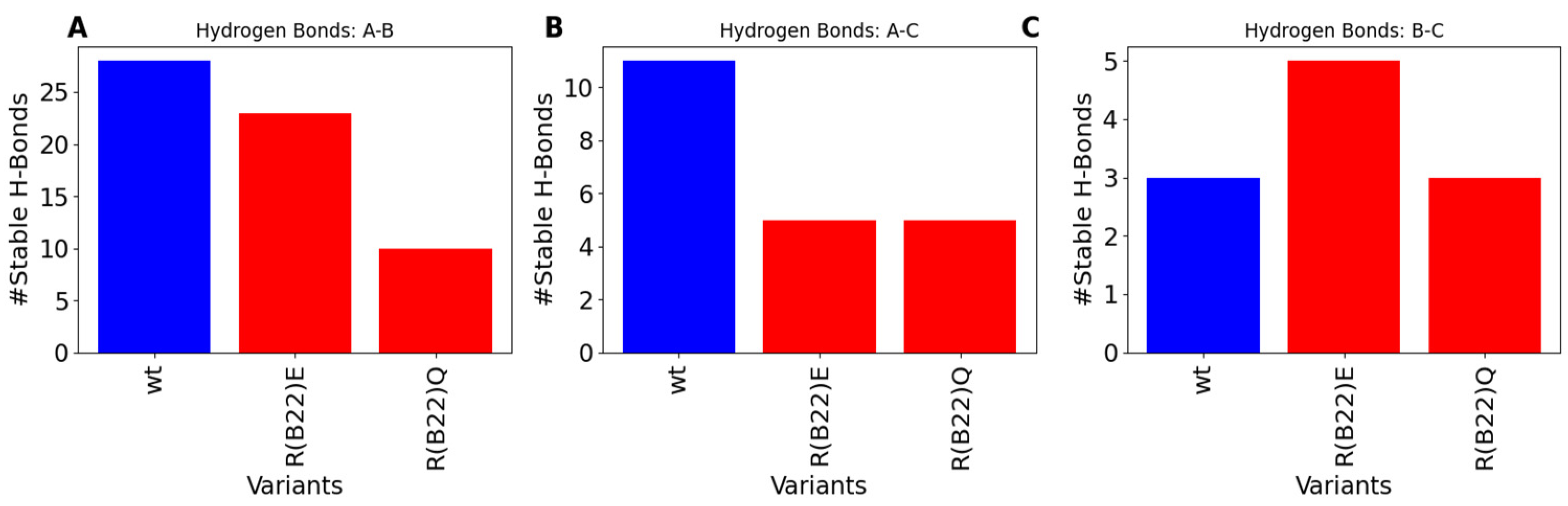
3.2. Disruption of Key A-Chain–B-Chain Interaction Leads to a Disruption of C-Peptide-Mediated Interactions
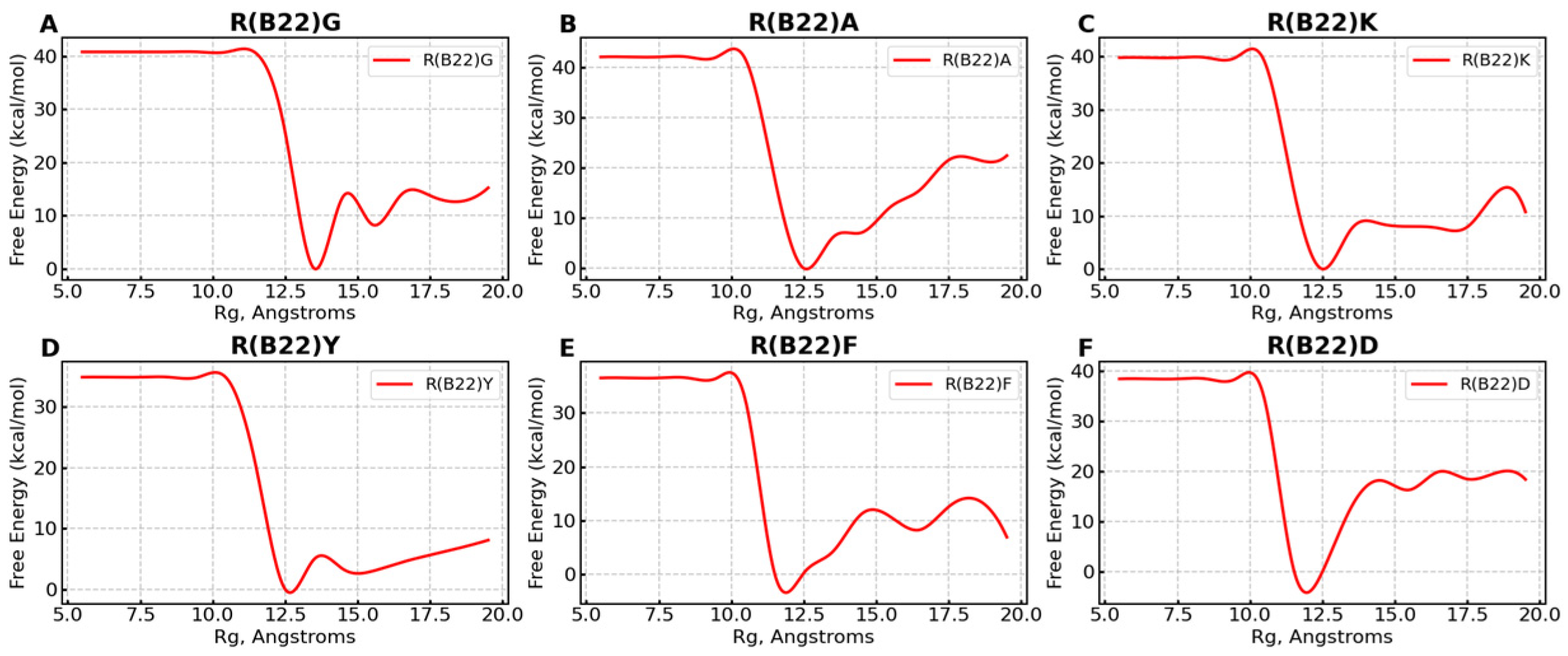
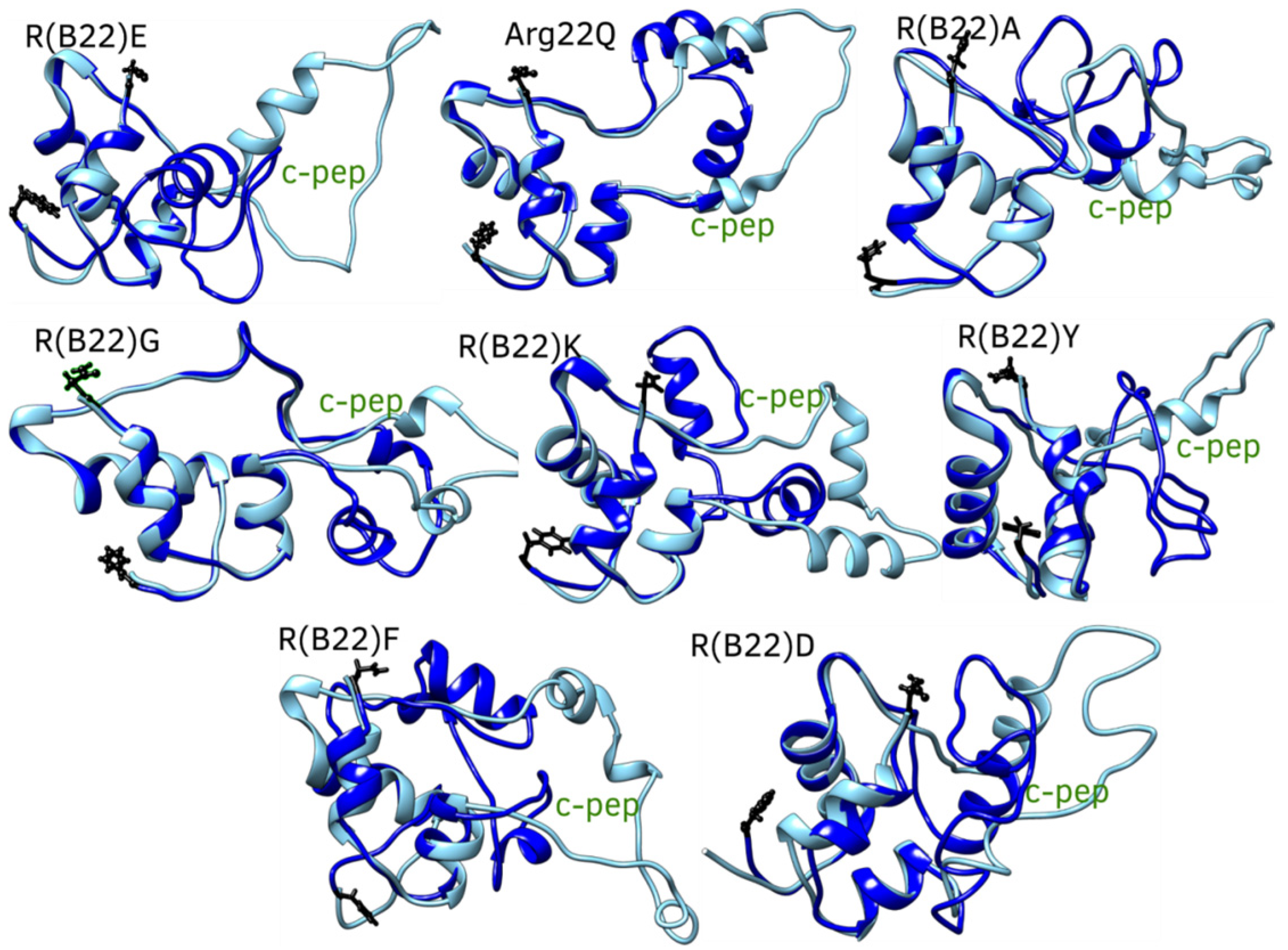
3.3. Non Q/E, R(B22)-Variants Display Increased Conformational Flexibility
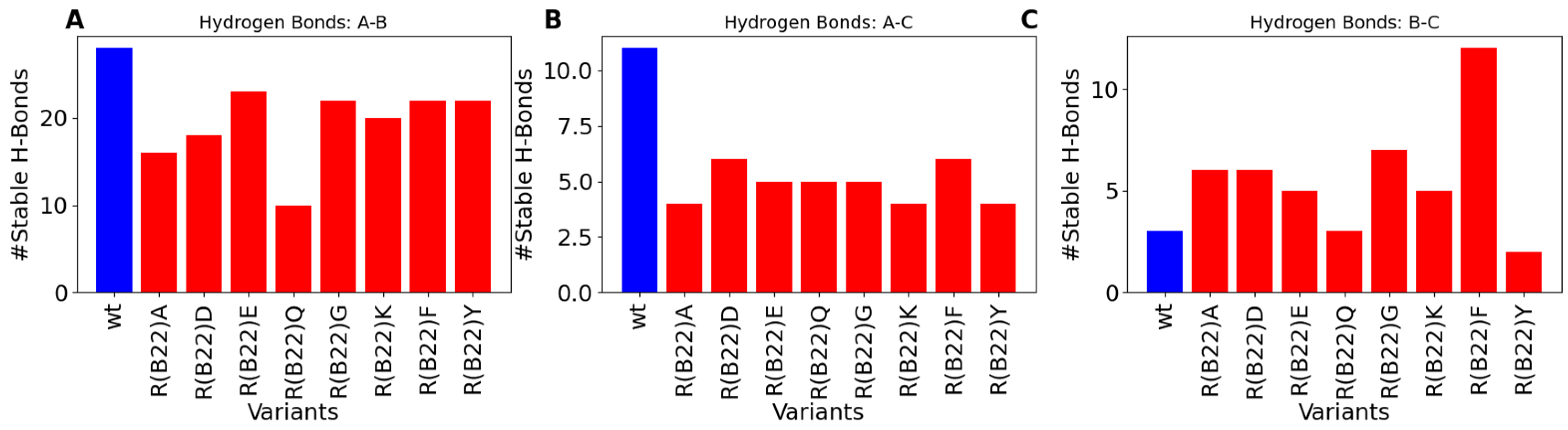
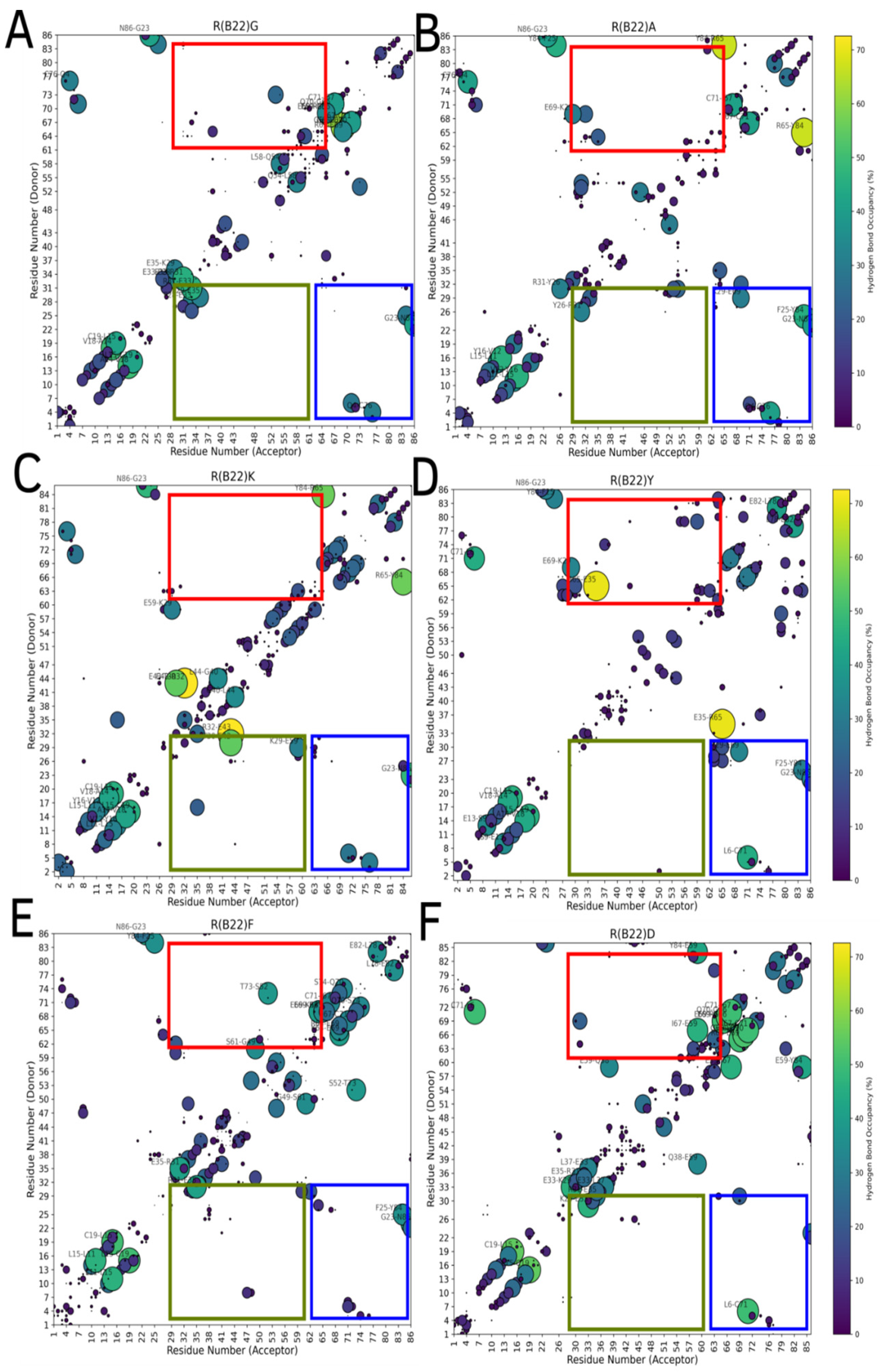
3.4. Non-Q/E, R(B22)-Variants Display Consistent Disruption in A-Chain–B-Chain Interactions Compared to WT
4. Discussion
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Chiang, J.L.; Maahs, D.M.; Garvey, K.C.; Hood, K.K.; Laffel, L.M.; Weinzimer, S.A.; Wolfsdorf, J.I.; Schatz, D. Type 1 Diabetes in Children and Adolescents: A Position Statement by the American Diabetes Association. Diabetes Care 2018, 41, 2026–2044. [Google Scholar] [CrossRef] [PubMed]
- Ardisson Korat, A.V.; Willett, W.C.; Hu, F.B. Diet, Lifestyle, and Genetic Risk Factors for Type 2 Diabetes: A Review from the Nurses’ Health Study, Nurses’ Health Study 2, and Health Professionals’ Follow-Up Study. Curr. Nutr. Rep. 2014, 3, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Antal, Z. Maturity-Onset Diabetes of the Young (MODY): Genetic Causes, Clinical Characteristics, Considerations for Testing, and Treatment Options. Endocrines 2021, 2, 485–501. [Google Scholar] [CrossRef]
- Molven, A.; Ringdal, M.; Nordbø, A.M.; Ræder, H.; Støy, J.; Lipkind, G.M.; Steiner, D.F.; Philipson, L.H.; Bergmann, I.; Aarskog, D.; et al. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes 2008, 57, 1131–1135. [Google Scholar] [CrossRef]
- Støy, J.; Olsen, J.; Park, S.-Y.; Gregersen, S.; Hjørringgaard, C.U.; Bell, G.I. In vivo measurement and biological characterisation of the diabetes-associated mutant insulin p.R46Q (GlnB22-insulin). Diabetologia 2017, 60, 1423–1431. [Google Scholar] [CrossRef]
- Wang, J.; Takeuchi, T.; Tanaka, S.; Kubo, S.-K.; Kayo, T.; Lu, D.; Takata, K.; Koizumi, A.; Izumi, T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the Mody mouse. J. Clin. Investig. 1999, 103, 27–37. [Google Scholar] [CrossRef]
- Alam, M.; Arunagiri, A.; Haataja, L.; Torres, M.; Larkin, D.; Kappler, J.; Jin, N.; Arvan, P. Predisposition to Proinsulin Misfolding as a Genetic Risk to Diet-Induced Diabetes. Diabetes 2021, 70, 2580–2594. [Google Scholar] [CrossRef]
- Haataja, L.; Manickam, N.; Soliman, A.; Tsai, B.; Liu, M.; Arvan, P. Disulfide Mispairing During Proinsulin Folding in the Endoplasmic Reticulum. Diabetes 2016, 65, 1050–1060. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Progress. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Li, D.; Liu, M.S.; Ji, B. Mapping the Dynamics Landscape of Conformational Transitions in Enzyme: The Adenylate Kinase Case. Biophys. J. 2015, 109, 647–660. [Google Scholar] [CrossRef]
- Anoop, A.; Ranganathan, S.; Dhaked, B.D.; Jha, N.N.; Pratihar, S.; Ghosh, S.; Sahay, S.; Kumar, S.; Das, S.; Kombrabail, M.; et al. Elucidating the role of disulfide bond on amyloid formation and fibril reversibility of somatostatin-14: Relevance to its storage and secretion. J. Biol. Chem. 2014, 289, 16884–16903. [Google Scholar] [CrossRef]
- Lukauskis, D.; Samways, M.L.; Aureli, S.; Cossins, B.P.; Taylor, R.D.; Gervasio, F.L. Open Binding Pose Metadynamics: An Effective Approach for the Ranking of Protein-Ligand Binding Poses. J. Chem. Inf. Model. 2022, 62, 6209–6216. [Google Scholar] [CrossRef]
- Brown, A.M.; Bevan, D.R. Molecular Dynamics Simulations of Amyloid beta-Peptide (1–42): Tetramer Formation and Membrane Interactions. Biophys. J. 2016, 111, 937–949. [Google Scholar] [CrossRef]
- Klimov, D.K.; Thirumalai, D. Dissecting the assembly of Abeta16-22 amyloid peptides into antiparallel beta sheets. Structure 2003, 11, 295–307. [Google Scholar] [CrossRef]
- Snell, C.R.; Smyth, D.G. Proinsulin: A proposed three-dimensional structure. J. Biol. Chem. 1975, 250, 6291–6295. [Google Scholar] [CrossRef] [PubMed]
- Arquilla, E.R.; Bromer, W.W.; Mercola, D. Immunology conformation and biological activity of insulin. Diabetes 1969, 18, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Landreh, M.; Johansson, J.; Wahren, J.; Jornvall, H. The structure, molecular interactions and bioactivities of proinsulin C-peptide correlate with a tripartite molecule. Biomol. Concepts 2014, 5, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Arunagiri, A.; Hassan, A.; Regan, K.; Tsai, B.; Dhayalan, B.; Weiss, M.A.; Liu, M.; Arvan, P. Distinct states of proinsulin misfolding in MIDY. Cell Mol. Life Sci. 2021, 78, 6017–6031. [Google Scholar] [CrossRef]
- Liu, M.; Haataja, L.; Wright, J.; Wickramasinghe, N.P.; Hua, Q.-X.; Phillips, N.F.; Barbetti, F.; Weiss, M.A.; Arvan, P. Mutant INS-gene induced diabetes of youth: Proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS ONE 2010, 5, e13333. [Google Scholar] [CrossRef]
- Arunagiri, A.; Alam, M.; Haataja, L.; Draz, H.; Alasad, B.; Samy, P.; Sadique, N.; Tong, Y.; Cai, Y.; Shakeri, H.; et al. Proinsulin folding and trafficking defects trigger a common pathological disturbance of endoplasmic reticulum homeostasis. Protein Sci. 2024, 33, e4949. [Google Scholar] [CrossRef]
- Izumi, T.; Yokota-Hashimoto, H.; Zhao, S.; Wang, J.; Halban, P.A.; Takeuchi, T. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes 2003, 52, 409–416. [Google Scholar] [CrossRef]
- Wang, H.; Saint-Martin, C.; Xu, J.; Ding, L.; Wang, R.; Feng, W.; Liu, M.; Shu, H.; Fan, Z.; Haataja, L.; et al. Biological behaviors of mutant proinsulin contribute to the phenotypic spectrum of diabetes associated with insulin gene mutations. Mol. Cell Endocrinol. 2020, 518, 111025. [Google Scholar] [CrossRef]
- Arunagiri, A.; Haataja, L.; Pottekat, A.; Pamenan, F.; Kim, S.; Zeltser, L.M.; Paton, A.W.; Paton, J.C.; Tsai, B.; Itkin-Ansari, P.; et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife 2019, 8, e44532. [Google Scholar] [CrossRef]
- Rege, N.K.; Liu, M.; Yang, Y.; Dhayalan, B.; Wickramasinghe, N.P.; Chen, Y.-S.; Rahimi, L.; Guo, H.; Haataja, L.; Sun, J.; et al. Evolution of insulin at the edge of foldability and its medical implications. Proc. Natl. Acad. Sci. USA 2020, 117, 29618–29628. [Google Scholar] [CrossRef]
- Dhayalan, B.; Chatterjee, D.; Chen, Y.S.; Weiss, M.A. Structural Lessons From the Mutant Proinsulin Syndrome. Front. Endocrinol. 2021, 12, 754693. [Google Scholar] [CrossRef] [PubMed]
- Gorr, S.U.; Huang, X.F.; Cowley, D.J.; Kuliawat, R.; Arvan, P. Disruption of disulfide bonds exhibits differential effects on trafficking of regulated secretory proteins. Am. J. Physiol. 1999, 277, C121–C131. [Google Scholar] [CrossRef] [PubMed]
- Huston, A.L.; Haeggstrom, J.Z.; Feller, G. Cold adaptation of enzymes: Structural, kinetic and microcalorimetric characterizations of an aminopeptidase from the Arctic psychrophile Colwellia psychrerythraea and of human leukotriene A(4) hydrolase. Biochim. Biophys. Acta 2008, 1784, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Ban, X.; Lahiri, P.; Dhoble, A.S.; Li, D.; Gu, Z.; Li, C.; Cheng, L.; Hong, Y.; Li, Z.; Kaustubh, B. Evolutionary Stability of Salt Bridges Hints Its Contribution to Stability of Proteins. Comput. Struct. Biotechnol. J. 2019, 17, 895–903. [Google Scholar] [CrossRef]
- Panja, A.S.; Maiti, S.; Bandyopadhyay, B. Protein stability governed by its structural plasticity is inferred by physicochemical factors and salt bridges. Sci. Rep. 2020, 10, 1822. [Google Scholar] [CrossRef]
- Balboa, D.; Saarimäki-Vire, J.; Borshagovski, D.; Survila, M.; Lindholm, P.; Galli, E.; Eurola, S.; Ustinov, J.; Grym, H.; Huopio, H.; et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. eLife 2018, 7, e38519. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranganathan, S.; Arunagiri, A. The B22 Dilemma: Structural Basis for Conformational Differences in Proinsulin B-Chain Arg22 Mutants. Biomolecules 2025, 15, 577. https://doi.org/10.3390/biom15040577
Ranganathan S, Arunagiri A. The B22 Dilemma: Structural Basis for Conformational Differences in Proinsulin B-Chain Arg22 Mutants. Biomolecules. 2025; 15(4):577. https://doi.org/10.3390/biom15040577
Chicago/Turabian StyleRanganathan, Srivastav, and Anoop Arunagiri. 2025. "The B22 Dilemma: Structural Basis for Conformational Differences in Proinsulin B-Chain Arg22 Mutants" Biomolecules 15, no. 4: 577. https://doi.org/10.3390/biom15040577
APA StyleRanganathan, S., & Arunagiri, A. (2025). The B22 Dilemma: Structural Basis for Conformational Differences in Proinsulin B-Chain Arg22 Mutants. Biomolecules, 15(4), 577. https://doi.org/10.3390/biom15040577