Pathways for Genome Integrity in G2 Phase of the Cell Cycle

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Regulation of Cell Cycle Progression through G2 Phase
2.1. Regulation of the Cell Cycle via Cyclin-Dependent Kinases
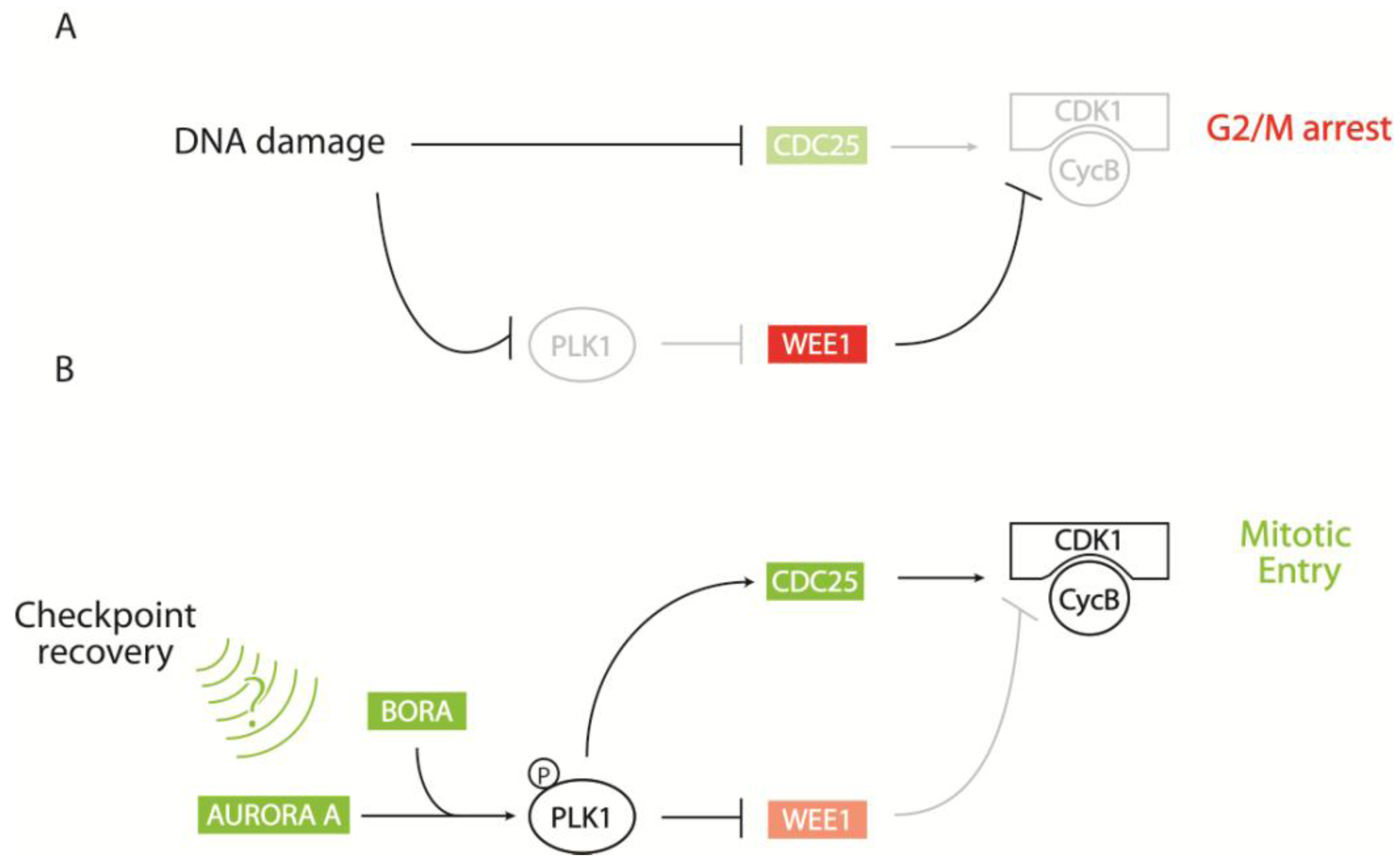
2.2. Regulation of CDK1 Activity at the G2/M Transition
2.2.1. Control of CDK1-Cyclin B via Key Phosphorylation Sites
2.2.2. CDK1-Cyclin B Regulation by the AURORA A/BORA/PLK1 Pathway
3. Cellular Responses to DNA DSBs
3.1. Repair of DNA DSBs
3.1.1. Homologous Recombination
3.1.2. Non-Homologous End-Joining
3.1.3. Alternative Non‐homologous End-Joining
3.1.4. Regulation of DSB Repair
3.2. Detection and Integration of the DNA DSB Signal
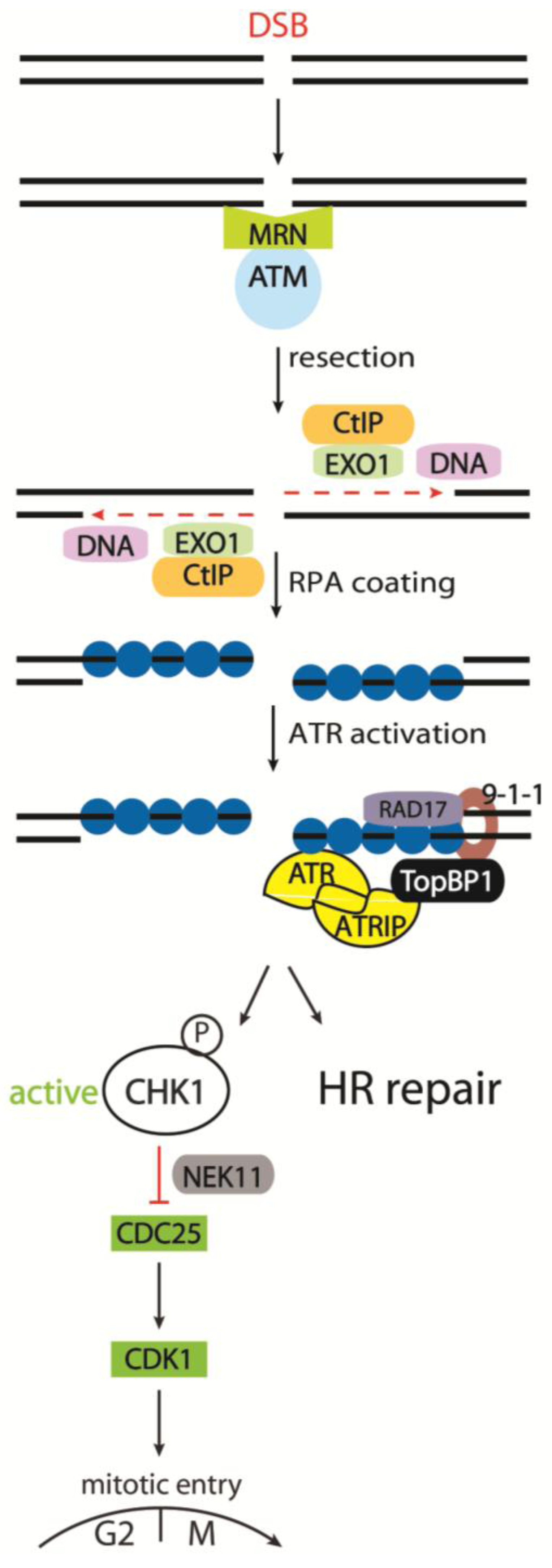
3.2.1. Detecting the DSB - the MRN Complex
3.2.2. Activation of ATM and Its Downstream Targets
3.2.3. The Role of Phosphorylated Histone H2AX
3.2.4. The Molecular Pathways Controlled by ATR and DNA-PKcs
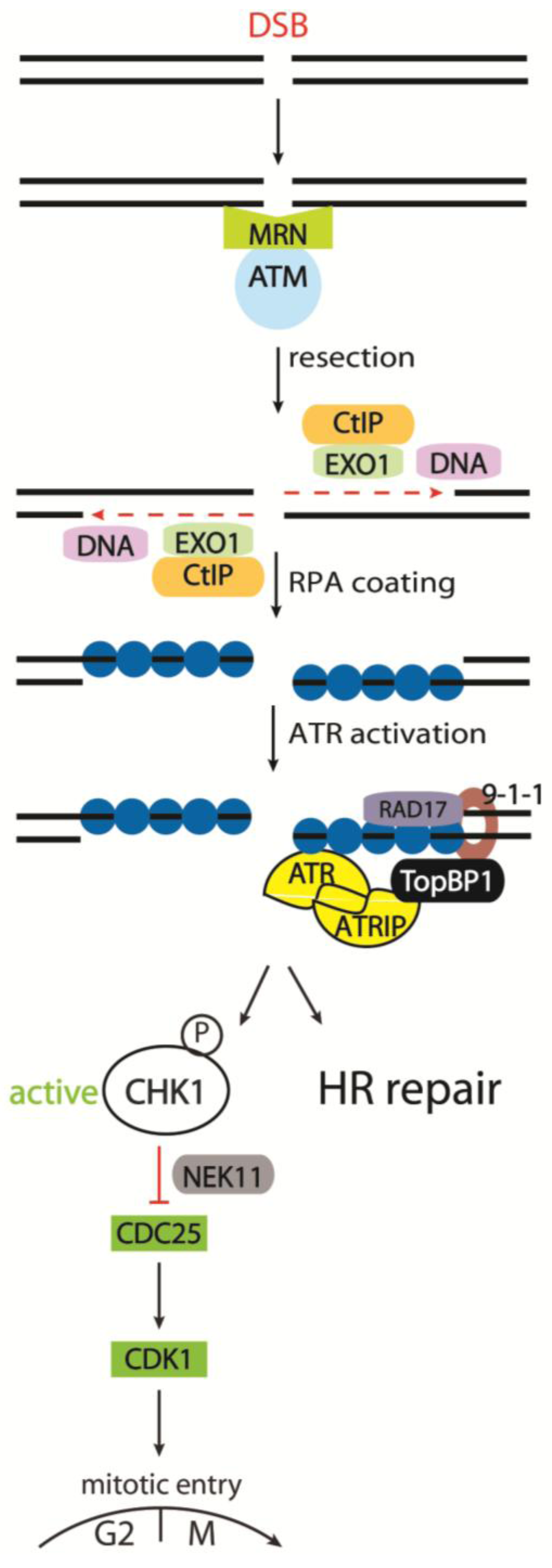
3.2.5. Cell Cycle Kinases CHK1 and CHK2 in Response to IR
3.2.6. The CDC25 Family as CHK1 and CHK2 Targets
4. The Regulation of the IR-Induced G2/M Checkpoint
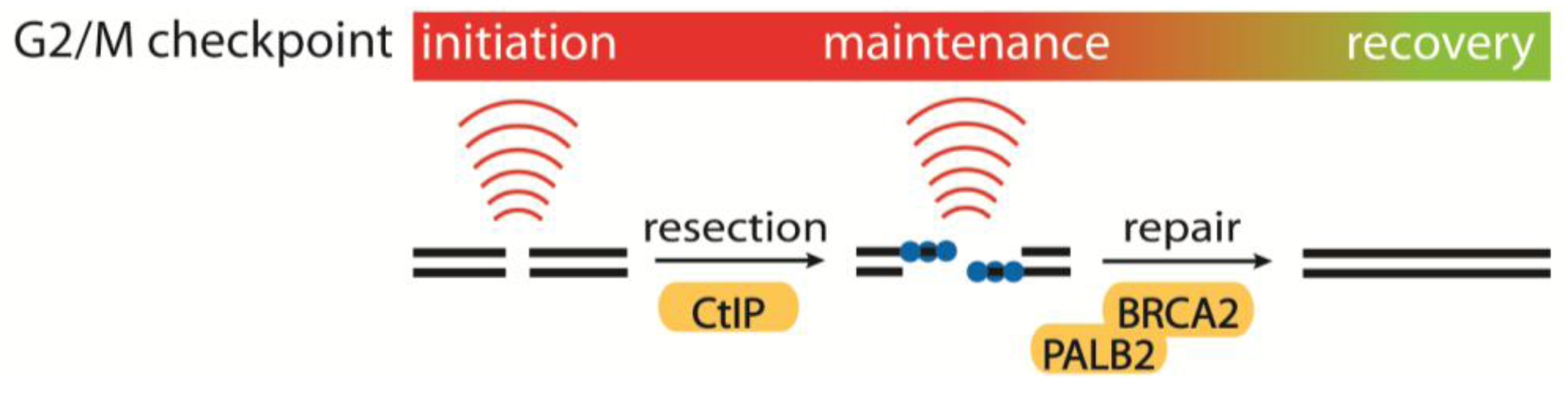
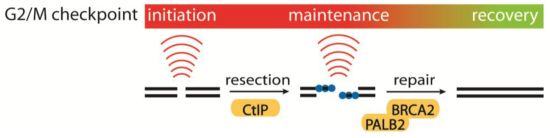
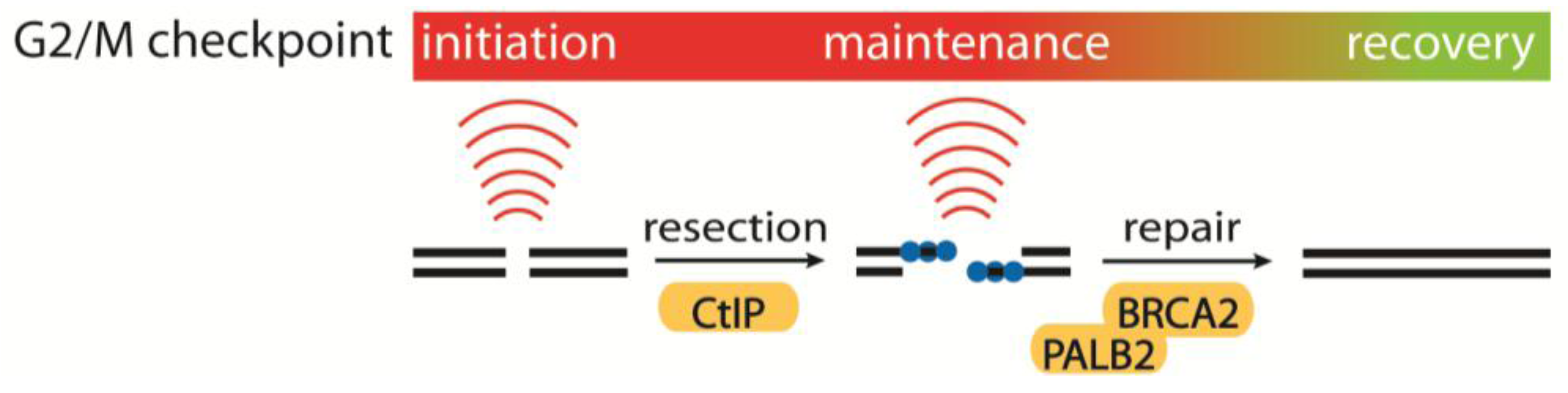
4.1. Initiation, Maintenance and Recovery of the G2/M Checkpoint

5. Perspectives
Supplementary Materials
Supplementary File 1Acknowledgements
References and Notes
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Medema, R.H.; Macůrek, L. Checkpoint control and cancer. Oncogene 2011, 2601–2613. [Google Scholar]
- Cotta-Ramusino, C.; McDonald, E.R.; Hurov, K.; Sowa, M.E.; Harper, J.W.; Elledge, S.J. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 2011, 332, 1313–1317. [Google Scholar] [CrossRef]
- Kousholt, A.N.; Fugger, K.; Hoffmann, S.; Larsen, B.D.; Menzel, T.; Sartori, A.A.; Sørensen, C.S. CtIP-dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. J. Cell Biol. 2012, 197, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Menzel, T.; Nähse-Kumpf, V.; Kousholt, A.N.; Klein, D.K.; Lund-Andersen, C.; Lees, M.; Johansen, J.V.; Syljuåsen, R.G.; Sørensen, C.S. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep. 2011, 12, 705–712. [Google Scholar] [CrossRef]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef]
- Suryadinata, R.; Sadowski, M.; Sarcevic, B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci. Rep. 2010, 30, 243–255. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Laoukili, J.; Alvarez, M.; Meijer, L.A.T.; Stahl, M.; Mohammed, S.; Kleij, L.; Heck, A.J.R.; Medema, R.H. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol. Cell Biol. 2008, 28, 3076–3087. [Google Scholar] [CrossRef]
- Yun, J.; Chae, H.-D.; Choi, T.-S.; Kim, E.-H.; Bang, Y.-J.; Chung, J.; Choi, K.-S.; Mantovani, R.; Shin, D.Y. Cdk2-dependent phosphorylation of the NF-Y transcription factor and its involvement in the p53-p21 signaling pathway. J. Biol. Chem. 2003, 278, 36966–36972. [Google Scholar]
- Saville, M.K.; Watson, R.J. The cell-cycle regulated transcription factor B-Myb is phosphorylated by cyclin A/Cdk2 at sites that enhance its transactivation properties. Oncogene 1998, 17, 2679–2689. [Google Scholar]
- Van Leuken, R.; Clijsters, L.; Wolthuis, R. To cell cycle, swing the APC/C. Biochim. Biophys. Acta 2008, 1786, 49–59. [Google Scholar]
- Lukas, C.; Sørensen, C.S.; Kramer, E.; Santoni-Rugiu, E.; Lindeneg, C.; Peters, J.M.; Bartek, J.; Lukas, J. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature 1999, 401, 815–818. [Google Scholar]
- Hsu, J.Y.; Reimann, J.D.R.; Sørensen, C.S.; Lukas, J.; Jackson, P.K. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat. Cell Biol. 2002, 4, 358–366. [Google Scholar] [CrossRef]
- Stark, G.R.; Taylor, W.R. Control of the G2/M transition. Mol. Biotechnol. 2006, 32, 227–248. [Google Scholar] [CrossRef]
- Lindqvist, A.; Rodríguez-Bravo, V.; Medema, R.H. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J. Cell Biol. 2009, 185, 193–202. [Google Scholar] [CrossRef]
- Hagting, A.; Karlsson, C.; Clute, P.; Jackman, M.; Pines, J. MPF localization is controlled by nuclear export. EMBO J. 1998, 17, 4127–4138. [Google Scholar] [CrossRef]
- Toyoshima, F.; Moriguchi, T.; Wada, A.; Fukuda, M.; Nishida, E. Nuclear export of cyclin B1 and its possible role in the DNA damage-induced G2 checkpoint. EMBO J. 1998, 17, 2728–2735. [Google Scholar] [CrossRef]
- Yang, J.; Song, H.; Walsh, S.; Bardes, E.S.; Kornbluth, S. Combinatorial control of cyclin B1 nuclear trafficking through phosphorylation at multiple sites. J. Biol. Chem. 2001, 276, 3604–3609. [Google Scholar]
- Gavet, O.; Pines, J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J. Cell Biol. 2010, 189, 247–259. [Google Scholar] [CrossRef]
- Jackman, M.; Lindon, C.; Nigg, E.A.; Pines, J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 2003, 5, 143–148. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef]
- Kutay, U.; Hetzer, M.W. Reorganization of the nuclear envelope during open mitosis. Curr. Opin. Cell Biol. 2008, 20, 669–677. [Google Scholar] [CrossRef]
- O’Farrell, P.H. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 2001, 11, 512–519. [Google Scholar] [CrossRef]
- Beck, H.; Nähse, V.; Larsen, M.S.Y.; Groth, P.; Clancy, T.; Lees, M.; Jørgensen, M.; Helleday, T.; Syljuåsen, R.G.; Sørensen, C.S. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J. Cell Biol. 2010, 188, 629–638. [Google Scholar] [CrossRef]
- Booher, R.N.; Holman, P.S.; Fattaey, A. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J. Biol. Chem. 1997, 272, 22300–22306. [Google Scholar] [CrossRef]
- Nakajima, H.; Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem. 2003, 278, 25277–25280. [Google Scholar]
- Lindqvist, A.; Van Zon, W.; Karlsson Rosenthal, C.; Wolthuis, R.M.F. Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS Biol. 2007, 5, 1127–1137. [Google Scholar]
- Deibler, R.W.; Kirschner, M.W. Quantitative reconstitution of mitotic CDK1 activation in somatic cell extracts. Mol. Cell 2010, 37, 753–767. [Google Scholar] [CrossRef]
- Boutros, R.; Dozier, C.; Ducommun, B. The when and wheres of CDC25 phosphatases. Curr. Opin. Cell Biol. 2006, 18, 185–191. [Google Scholar] [CrossRef]
- Mailand, N.; Podtelejnikov, A.V; Groth, A.; Mann, M.; Bartek, J.; Lukas, J. Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002, 21, 5911–5920. [Google Scholar] [CrossRef]
- Baldin, V.; Pelpel, K.; Cazales, M.; Cans, C.; Ducommun, B. Nuclear localization of CDC25B1 and serine 146 integrity are required for induction of mitosis. J. Biol. Chem. 2002, 277, 35176–35182. [Google Scholar]
- Hoffmann, I.; Clarke, P.R.; Marcote, M.J.; Karsenti, E.; Draetta, G. Phosphorylation and activation of human cdc25-C by cdc2--cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J. 1993, 12, 53–63. [Google Scholar]
- Lindqvist, A.; Källström, H.; Lundgren, A.; Barsoum, E.; Rosenthal, C.K. Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J. Cell Biol. 2005, 171, 35–45. [Google Scholar] [CrossRef]
- Barr, A.R.; Gergely, F. Aurora-A: the maker and breaker of spindle poles. J. Cell Sci. 2007, 120, 2987–2996. [Google Scholar] [CrossRef]
- Macůrek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar]
- Seki, A.; Coppinger, J.A.; Jang, C.-Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef]
- Walter, A.O.; Seghezzi, W.; Korver, W.; Sheung, J.; Lees, E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene 2000, 19, 4906–4916. [Google Scholar] [CrossRef]
- Littlepage, L.E.; Wu, H.; Andresson, T.; Deanehan, J.K.; Amundadottir, L.T.; Ruderman, J. V Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc. Natl. Acad. Sci. USA 2002, 99, 15440–15445. [Google Scholar]
- Hutterer, A.; Berdnik, D.; Wirtz-Peitz, F.; Zigman, M.; Schleiffer, A.; Knoblich, J.A. Mitotic activation of the kinase Aurora-A requires its binding partner Bora. Dev. Cell 2006, 11, 147–157. [Google Scholar] [CrossRef]
- Eyers, P.A.; Erikson, E.; Chen, L.G.; Maller, J.L. A novel mechanism for activation of the protein kinase Aurora A. Curr. Biol. 2003, 13, 691–697. [Google Scholar] [CrossRef]
- Hirota, T.; Kunitoku, N.; Sasayama, T.; Marumoto, T.; Zhang, D.; Nitta, M.; Hatakeyama, K.; Saya, H. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 2003, 114, 585–598. [Google Scholar] [CrossRef]
- Chan, E.H.Y.; Santamaria, A.; Silljé, H.H.W.; Nigg, E.A. Plk1 regulates mitotic Aurora A function through βTrCP-dependent degradation of hBora. Chromosoma 2008, 117, 457–469. [Google Scholar] [CrossRef]
- De Luca, M.; Lavia, P.; Guarguaglini, G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle 2006, 5, 296–303. [Google Scholar] [CrossRef]
- Dutertre, S.; Cazales, M.; Quaranta, M.; Froment, C.; Trabut, V.; Dozier, C.; Mirey, G.; Bouché, J.-P.; Theis-Febvre, N.; Schmitt, E.; Monsarrat, B.; Prigent, C.; Ducommun, B. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J. Cell Sci. 2004, 117, 2523–2531. [Google Scholar]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; Van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef]
- Laoukili, J.; Stahl, M.; Medema, R.H. FoxM1: at the crossroads of ageing and cancer. Biochim. Biophys. Acta 2007, 1775, 92–102. [Google Scholar]
- Van Vugt, M.A.T.M.; Brás, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair (Amst) 2004, 3, 817–826. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Löbrich, M. Contribution of DNA repair and cell cycle checkpoint arrest to the maintenance of genomic stability. DNA Repair (Amst) 2006, 5, 1192–1198. [Google Scholar] [CrossRef]
- Krempler, A.; Deckbar, D.; Jeggo, P.A.; Löbrich, M. An imperfect G2M checkpoint contributes to chromosome instability following irradiation of S and G2 phase cells. Cell Cycle 2007, 6, 1682–1686. [Google Scholar] [CrossRef]
- Deckbar, D.; Jeggo, P.A.; Löbrich, M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 271–283. [Google Scholar] [CrossRef]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Geuting, V.; Löbrich, M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother. Oncol. 2011, 101, 7–12. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Symington, L.S. DNA end resection: many nucleases make light work. DNA Repair (Amst) 2009, 8, 983–995. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Hartlerode, A.J.; Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009, 423, 157–168. [Google Scholar] [CrossRef]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef]
- Riballo, E.; Kühne, M.; Rief, N.; Doherty, A.; Smith, G.C.M.; Recio, M.-J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; Parker, A.R.; Jackson, S.P.; Gennery, A.; Jeggo, P.A.; Löbrich, M. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Löbrich, M.; Jeggo, P.A. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Kinoshita, E.; Van der Linden, E.; Sanchez, H.; Wyman, C. RAD50, an SMC family member with multiple roles in DNA break repair: how does ATP affect function? Chromosome Res. 2009, 17, 277–288. [Google Scholar] [CrossRef]
- Feeney, K.M.; Wasson, C.W.; Parish, J.L. Cohesin: a regulator of genome integrity and gene expression. Biochem J. 2010, 428, 147–161. [Google Scholar] [CrossRef]
- Williams, R.S.; Williams, J.S.; Tainer, J.A. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin templat. Biochem. Cell Biol. 2007, 85, 509–520. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar]
- Mimitou, E.P.; Symington, L.S. Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 2009, 34, 264–272. [Google Scholar] [CrossRef]
- Gravel, S.; Chapman, J.R.; Magill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef]
- Nimonkar, A.V; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef]
- Paull, T.T.; Gellert, M. The 3’ to 5' exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Garcia, V.; Phelps, S.E.L.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar]
- Stracker, T.H.; Petrini, J.H. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Wold, M.S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Ishibashi, T.; Uchiyama, Y.; Iwabata, K. The multi-replication protein A (RPA) system--a new perspective. FEBS J. 2009, 276, 943–963. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar]
- Davies, O.R.; Pellegrini, L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat. Struct. Mol. Biol. 2007, 14, 475–483. [Google Scholar]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar]
- McIlwraith, M.J.; Mcllwraith, M.J.; Vaisman, A.; Liu, Y.; Fanning, E.; Woodgate, R.; West, S.C. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol. Cell 2005, 20, 783–792. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.C.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006, 5, 1042–1048. [Google Scholar] [CrossRef]
- Roberts, S.A.; Strande, N.; Burkhalter, M.D.; Strom, C.; Havener, J.M.; Hasty, P.; Ramsden, D.A. Ku is a 5’-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 2010, 464, 1214–1217. [Google Scholar]
- Kim, J.-S.; Krasieva, T.B.; Kurumizaka, H.; Chen, D.J.; Taylor, A.M.R.; Yokomori, K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J. Cell Biol. 2005, 170, 341–347. [Google Scholar] [CrossRef]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The life and death of DNA-PK. Oncogene 2005, 24, 949–961. [Google Scholar] [CrossRef]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: the means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, W.; Ding, Q.; Ye, R.; Chen, D.; Merkle, D.; Schriemer, D.; Meek, K.; Lees-Miller, S.P. DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair (Amst) 2003, 2, 1239–1252. [Google Scholar] [CrossRef]
- Yu, Y.; Mahaney, B.L.; Yano, K.-I.; Ye, R.; Fang, S.; Douglas, P.; Chen, D.J.; Lees-Miller, S.P. DNA-PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair (Amst) 2008, 7, 1680–1692. [Google Scholar] [CrossRef]
- Douglas, P.; Gupta, S.; Morrice, N.; Meek, K.; Lees-Miller, S.P. DNA-PK-dependent phosphorylation of Ku70/80 is not required for non-homologous end joining. DNA Repair (Amst) 2005, 4, 1006–1018. [Google Scholar] [CrossRef]
- Meek, K.; Gupta, S.; Ramsden, D.A.; Lees-Miller, S.P. The DNA-dependent protein kinase: the director at the end. Immunol. Rev. 2004, 200, 132–141. [Google Scholar] [CrossRef]
- Ding, Q.; Reddy, Y.V.R.; Wang, W.; Woods, T.; Douglas, P.; Ramsden, D.A.; Lees-Miller, S.P.; Meek, K. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell Biol. 2003, 23, 5836–5848. [Google Scholar] [CrossRef]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Chen, L.; Trujillo, K.; Sung, P.; Tomkinson, A.E. Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J. Biol. Chem. 2000, 275, 26196–26205. [Google Scholar]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst) 2006, 5, 1021–1029. [Google Scholar] [CrossRef]
- Yan, C.T.; Boboila, C.; Souza, E.K.; Franco, S.; Hickernell, T.R.; Murphy, M.; Gumaste, S.; Geyer, M.; Zarrin, A.A.; Manis, J.P.; Rajewsky, K.; Alt, F.W. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 2007, 449, 478–482. [Google Scholar]
- Soulas-Sprauel, P.; Le Guyader, G.; Rivera-Munoz, P.; Abramowski, V.; Olivier-Martin, C.; Goujet-Zalc, C.; Charneau, P.; De Villartay, J.-P. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. J. Exp. Med. 2007, 204, 1717–1727. [Google Scholar] [CrossRef]
- Perrault, R.; Wang, H.; Wang, M.; Rosidi, B.; Iliakis, G. Backup pathways of NHEJ are suppressed by DNA-PK. J. Cell. Biochem. 2004, 92, 781–794. [Google Scholar] [CrossRef]
- Lee, K.; Lee, S.E. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics 2007, 176, 2003–2014. [Google Scholar] [CrossRef]
- Ma, J.; Kim, E.M.; Haber, J.E.; Lee, S.E. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell. Biol. 2003, 23, 8820–8828. [Google Scholar] [CrossRef]
- Aylon, Y.; Liefshitz, B.; Kupiec, M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004, 23, 4868–4875. [Google Scholar] [CrossRef]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; Haber, J.E.; Foiani, M. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar]
- Ahmad, A.; Robinson, A.R.; Duensing, A.; Van Drunen, E.; Beverloo, H.B.; Weisberg, D.B.; Hasty, P.; Hoeijmakers, J.H.J.; Niedernhofer, L.J. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol. 2008, 28, 5082–5092. [Google Scholar] [CrossRef]
- Daley, J.M.; Wilson, T.E. Rejoining of DNA double-strand breaks as a function of overhang length. Mol. Cell. Biol. 2005, 25, 896–906. [Google Scholar] [CrossRef]
- Liang, L.; Deng, L.; Nguyen, S.C.; Zhao, X.; Maulion, C.D.; Shao, C.; Tischfield, J.A. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008, 36, 3297–3310. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef]
- Wu, D.; Topper, L.M.; Wilson, T.E. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics 2008, 178, 1237–1249. [Google Scholar] [CrossRef]
- Shim, E.Y.; Chung, W.-H.; Nicolette, M.L.; Zhang, Y.; Davis, M.; Zhu, Z.; Paull, T.T.; Ira, G.; Lee, S.E. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010, 29, 3370–3380. [Google Scholar] [CrossRef]
- Foster, S.S.; Balestrini, A.; Petrini, J.H.J. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol. Cell. Biol. 2011, 31, 4379–4389. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Symington, L.S. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010, 29, 3358–3369. [Google Scholar] [CrossRef]
- Delacôte, F.; Han, M.; Stamato, T.D.; Jasin, M.; Lopez, B.S. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res. 2002, 30, 3454–3463. [Google Scholar] [CrossRef]
- Yu, X.; Baer, R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J. Biol. Chem. 2000, 275, 18541–18549. [Google Scholar] [CrossRef]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef]
- Shakya, R.; Reid, L.J.; Reczek, C.R.; Cole, F.; Egli, D.; Lin, C.-S.; DeRooij, D.G.; Hirsch, S.; Ravi, K.; Hicks, J.B.; Szabolcs, M.; Jasin, M.; Baer, R.; Ludwig, T. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 2011, 334, 525–528. [Google Scholar]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; Haffty, B.G.; Tommiska, J.; Blomqvist, C.; Drapkin, R.; Adams, D.J.; Nevanlinna, H.; Bartek, J.; Tarsounas, M.; Ganesan, S.; Jonkers, J. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; Xu, X.; Deng, C.-X.; Finkel, T.; Nussenzweig, M.; Stark, J.M.; Nussenzweig, A. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–54. [Google Scholar] [CrossRef]
- De Jager, M.; Van Noort, J.; Van Gent, D.C.; Dekker, C.; Kanaar, R.; Wyman, C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell 2001, 8, 1129–1135. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Craig, L.; Moncalian, G.; Zinkel, R.A.; Usui, T.; Owen, B.A.L.; Karcher, A.; Henderson, B.; Bodmer, J.-L.; McMurray, C.T.; Carney, J.P.; Petrini, J.H.J.; Tainer, J.A. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 2002, 418, 562–566. [Google Scholar]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; Moiani, D.; Carney, J.P.; Russell, P.; Tainer, J.A. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef]
- Lukas, C.; Melander, F.; Stucki, M.; Falck, J.; Bekker-Jensen, S.; Goldberg, M.; Lerenthal, Y.; Jackson, S.P.; Bartek, J.; Lukas, J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004, 23, 2674–2683. [Google Scholar] [CrossRef]
- Celeste, A.; Fernandez-Capetillo, O.; Kruhlak, M.J.; Pilch, D.R.; Staudt, D.W.; Lee, A.; Bonner, R.F.; Bonner, W.M.; Nussenzweig, A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 2003, 5, 675–679. [Google Scholar] [CrossRef]
- Perry, J.; Kleckner, N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell 2003, 112, 151–155. [Google Scholar] [CrossRef]
- Shiloh, Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu. Rev. Genet. 1997, 31, 635–662. [Google Scholar] [CrossRef]
- Xu, B.; Kim, S.-T.; Lim, D.-S.; Kastan, M.B. Two Molecularly Distinct G2/M Checkpoints Are Induced by Ionizing Irradiation. Mol. Cell. Biol. 2002, 22, 1049–1059. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Kozlov, S.V.; Graham, M.E.; Peng, C.; Chen, P.; Robinson, P.J.; Lavin, M.F. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006, 25, 3504–3514. [Google Scholar] [CrossRef]
- Cerosaletti, K.; Wright, J.; Concannon, P. Active role for nibrin in the kinetics of atm activation. Mol. Cell. Biol 2006, 26, 1691–1699. [Google Scholar] [CrossRef]
- Iijima, K.; Ohara, M.; Seki, R.; Tauchi, H. Dancing on damaged chromatin: functions of ATM and the RAD50/MRE11/NBS1 complex in cellular responses to DNA damage. J. Radiat. Res. 2008, 49, 451–464. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef]
- Lavin, M.F. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 2007, 26, 7749–7758. [Google Scholar] [CrossRef]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; Shiloh, Y.; Gygi, S.P.; Elledge, S.J. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J. Biol. Chem. 2010, 285, 1097–1104. [Google Scholar] [CrossRef]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; Redon, C.; Pilch, D.R.; Olaru, A.; Eckhaus, M.; Camerini-Otero, R.D.; Tessarollo, L.; Livak, F.; Manova, K.; Bonner, W.M.; Nussenzweig, M.C.; Nussenzweig, A. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar]
- Bassing, C.H.; Chua, K.F.; Sekiguchi, J.; Suh, H.; Whitlow, S.R.; Fleming, J.C.; Monroe, B.C.; Ciccone, D.N.; Yan, C.; Vlasakova, K.; Livingston, D.M.; Ferguson, D.O.; Scully, R.; Alt, F.W. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA 2002, 99, 8173–8178. [Google Scholar]
- Xie, A.; Puget, N.; Shim, I.; Odate, S.; Jarzyna, I.; Bassing, C.H.; Alt, F.W.; Scully, R. Control of sister chromatid recombination by histone H2AX. Mol. Cell 2004, 16, 1017–1025. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Sakaguchi, K.; Ullrich, S.J.; Appella, E.; Anderson, C.W. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol. Cell. Biol. 1992, 12, 5041–5049. [Google Scholar]
- Callén, E.; Jankovic, M.; Wong, N.; Zha, S.; Chen, H.-T.; Difilippantonio, S.; Di Virgilio, M.; Heidkamp, G.; Alt, F.W.; Nussenzweig, A.; Nussenzweig, M. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol. Cell 2009, 34, 285–297. [Google Scholar] [CrossRef]
- Li, J.; Stern, D.F. Regulation of CHK2 by DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 12041–12050. [Google Scholar] [CrossRef]
- Paulsen, R.D.; Cimprich, K.A. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007, 6, 953–66. [Google Scholar] [CrossRef]
- O’Neill, T.; Dwyer, A.J.; Ziv, Y.; Chan, D.W.; Lees-Miller, S.P.; Abraham, R.H.; Lai, J.H.; Hill, D.; Shiloh, Y.; Cantley, L.C.; Rathbun, G.A. Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J. Biol. Chem. 2000, 275, 22719–22727. [Google Scholar]
- Kim, S.T.; Lim, D.S.; Canman, C.E.; Kastan, M.B. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 1999, 274, 37538–37543. [Google Scholar] [CrossRef]
- Bennetzen, M.V; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteomics 2010, 9, 1314–1323. [Google Scholar] [CrossRef]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.M.; Iwasaki, H.; Russell, P.; Tainer, J.A. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.J.; Lee, A.Y.-L.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Burrows, A.E.; Elledge, S.J. How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes Dev. 2008, 22, 1416–1421. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Cuadrado, M.; Martinez-Pastor, B.; Murga, M.; Toledo, L.I.; Gutierrez-Martinez, P.; Lopez, E.; Fernandez-Capetillo, O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med. 2006, 203, 297–303. [Google Scholar] [CrossRef]
- Myers, J.S.; Cortez, D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar] [CrossRef]
- Gatei, M.; Sloper, K.; Sorensen, C.; Syljuäsen, R.; Falck, J.; Hobson, K.; Savage, K.; Lukas, J.; Zhou, B.-B.; Bartek, J.; Khanna, K.K. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J. Biol. Chem. 2003, 278, 14806–14811. [Google Scholar]
- Ahn, J.; Urist, M.; Prives, C. The Chk2 protein kinase. DNA Repair (Amst) 2004, 3, 1039–1047. [Google Scholar] [CrossRef]
- Shibata, A.; Barton, O.; Noon, A.T.; Dahm, K.; Deckbar, D.; Goodarzi, A.A.; Löbrich, M.; Jeggo, P.A. Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest. Mol. Cell. Biol. 2010, 30, 3371–3383. [Google Scholar] [CrossRef]
- Hirao, A.; Kong, Y.Y.; Matsuoka, S.; Wakeham, A.; Ruland, J.; Yoshida, H.; Liu, D.; Elledge, S.J.; Mak, T.W. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000, 287, 1824–1827. [Google Scholar] [CrossRef]
- Takai, H.; Naka, K.; Okada, Y.; Watanabe, M.; Harada, N.; Saito, S.; Anderson, C.W.; Appella, E.; Nakanishi, M.; Suzuki, H.; Nagashima, K.; Sawa, H.; Ikeda, K.; Motoyama, N. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 2002, 21, 5195–5205. [Google Scholar]
- Lossaint, G.; Besnard, E.; Fisher, D.; Piette, J.; Dulić, V. Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATM/p53/p21 pathway is functional. Oncogene 2011, 30, 4261–4274. [Google Scholar] [CrossRef]
- Jin, J.; Ang, X.L.; Ye, X.; Livingstone, M.; Harper, J.W. Differential roles for checkpoint kinases in DNA damage-dependent degradation of the Cdc25A protein phosphatase. J. Biol. Chem. 2008, 283, 19322–19328. [Google Scholar]
- Bartek, J.; Lukas, J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 2002, 99, 14795–14800. [Google Scholar]
- Sørensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.-B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef]
- Busino, L.; Donzelli, M.; Chiesa, M.; Guardavaccaro, D.; Ganoth, D.; Dorrello, N.V; Hershko, A.; Pagano, M.; Draetta, G.F. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature 2003, 426, 87–91. [Google Scholar]
- Jin, J.; Shirogane, T.; Xu, L.; Nalepa, G.; Qin, J.; Elledge, S.J.; Harper, J.W. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev 2003, 17, 3062–3074. [Google Scholar] [CrossRef]
- Chen, M.S.; Hurov, J.; White, L.S.; Woodford-Thomas, T.; Piwnica-Worms, H. Absence of apparent phenotype in mice lacking Cdc25C protein phosphatase. Mol. Cell. Biol. 2001, 21, 3853–3861. [Google Scholar] [CrossRef]
- Lincoln, A.J.; Wickramasinghe, D.; Stein, P.; Schultz, R.M.; Palko, M.E.; De Miguel, M.P.; Tessarollo, L.; Donovan, P.J. Cdc25b phosphatase is required for resumption of meiosis during oocyte maturation. Nat. Genet. 2002, 30, 446–449. [Google Scholar]
- Melixetian, M.; Klein, D.K.; Sørensen, C.S.; Helin, K. NEK11 regulates CDC25A degradation and the IR-induced G2/M checkpoint. Nat. Cell Biol. 2009, 11, 1247–1253. [Google Scholar] [CrossRef]
- MacDougall, C.A.; Byun, T.S.; Van, C.; Yee, M.; Cimprich, K.A. The structural determinants of checkpoint activation. Genes Dev. 2007, 21, 898–903. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef]
- Theunissen, J.-W.F.; Kaplan, M.I.; Hunt, P.A.; Williams, B.R.; Ferguson, D.O.; Alt, F.W.; Petrini, J.H.J. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol. Cell 2003, 12, 1511–1523. [Google Scholar] [CrossRef]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef]
- Huertas, P.; Jackson, S.P.S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef]
- Li, S.; Ting, N.S.; Zheng, L.; Chen, P.L.; Ziv, Y.; Shiloh, Y.; Lee, E.Y.; Lee, W.H. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 2000, 406, 210–215. [Google Scholar]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar]
- Lee, J.; Kim, J.A.; Barbier, V.; Fotedar, A.; Fotedar, R. DNA damage triggers p21WAF1-dependent Emi1 down-regulation that maintains G2 arrest. Mol. Biol. Cell 2009, 20, 1891–1902. [Google Scholar] [CrossRef]
- Tategu, M.; Nakagawa, H.; Sasaki, K.; Yamauchi, R.; Sekimachi, S.; Suita, Y.; Watanabe, N.; Yoshid, K. Transcriptional regulation of human polo-like kinases and early mitotic inhibitor. J. Genet. Genomics 2008, 35, 215–224. [Google Scholar] [CrossRef]
- Van Vugt, M.A.; Medema, R.H. Getting in and out of mitosis with Polo-like kinase-1. Oncogene 2005, 24, 2844–2859. [Google Scholar] [CrossRef]
- Watanabe, N.; Arai, H.; Iwasaki, J.-I.; Shiina, M.; Ogata, K.; Hunter, T.; Osada, H. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 11663–11668. [Google Scholar]
- Watanabe, N.; Arai, H.; Nishihara, Y.; Taniguchi, M.; Watanabe, N.; Hunter, T.; Osada, H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA 2004, 101, 4419–4424. [Google Scholar]
- Elia, A.E.H.; Cantley, L.C.; Yaffe, M.B. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 2002, 3, 341–348. [Google Scholar] [CrossRef]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004, 5, 739–751. [Google Scholar] [CrossRef]
- Smits, V.A.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2000, 2, 672–676. [Google Scholar] [CrossRef]
- Tsvetkov, L.; Stern, D.F. Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage. Cell Cycle 2005, 4, 166–171. [Google Scholar] [CrossRef]
- Jang, Y.-J.; Ji, J.-H.; Choi, Y.-C.; Ryu, C.J.; Ko, S.-Y. Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J. Biol. Chem. 2007, 282, 2473–2482. [Google Scholar]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; Orntoft, T.; Lukas, J.; Bartek, J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar]
- Kuntz, K.; O’Connell, M.J. The G(2) DNA damage checkpoint: could this ancient regulator be the Achilles heel of cancer? Cancer Biol. Ther. 2009, 8, 1433–1439. [Google Scholar]
- Abbott, D.W.; Freeman, M.L.; Holt, J.T. Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J. Natl. Cancer Inst. 1998, 90, 978–985. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar]
- Russell, K.J.; Wiens, L.W.; Demers, G.W.; Galloway, D.A.; Plon, S.E.; Groudine, M. Abrogation of the G2 checkpoint results in differential radiosensitization of G1 checkpoint-deficient and G1 checkpoint-competent cells. Cancer Res. 1995, 55, 1639–1642. [Google Scholar]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kousholt, A.N.; Menzel, T.; Sørensen, C.S. Pathways for Genome Integrity in G2 Phase of the Cell Cycle. Biomolecules 2012, 2, 579-607. https://doi.org/10.3390/biom2040579
Kousholt AN, Menzel T, Sørensen CS. Pathways for Genome Integrity in G2 Phase of the Cell Cycle. Biomolecules. 2012; 2(4):579-607. https://doi.org/10.3390/biom2040579
Chicago/Turabian StyleKousholt, Arne Nedergaard, Tobias Menzel, and Claus Storgaard Sørensen. 2012. "Pathways for Genome Integrity in G2 Phase of the Cell Cycle" Biomolecules 2, no. 4: 579-607. https://doi.org/10.3390/biom2040579
APA StyleKousholt, A. N., Menzel, T., & Sørensen, C. S. (2012). Pathways for Genome Integrity in G2 Phase of the Cell Cycle. Biomolecules, 2(4), 579-607. https://doi.org/10.3390/biom2040579