
Structural and Biochemical Investigation of Bacteriophage N4-Encoded RNA Polymerases

Abstract
:1. Introduction
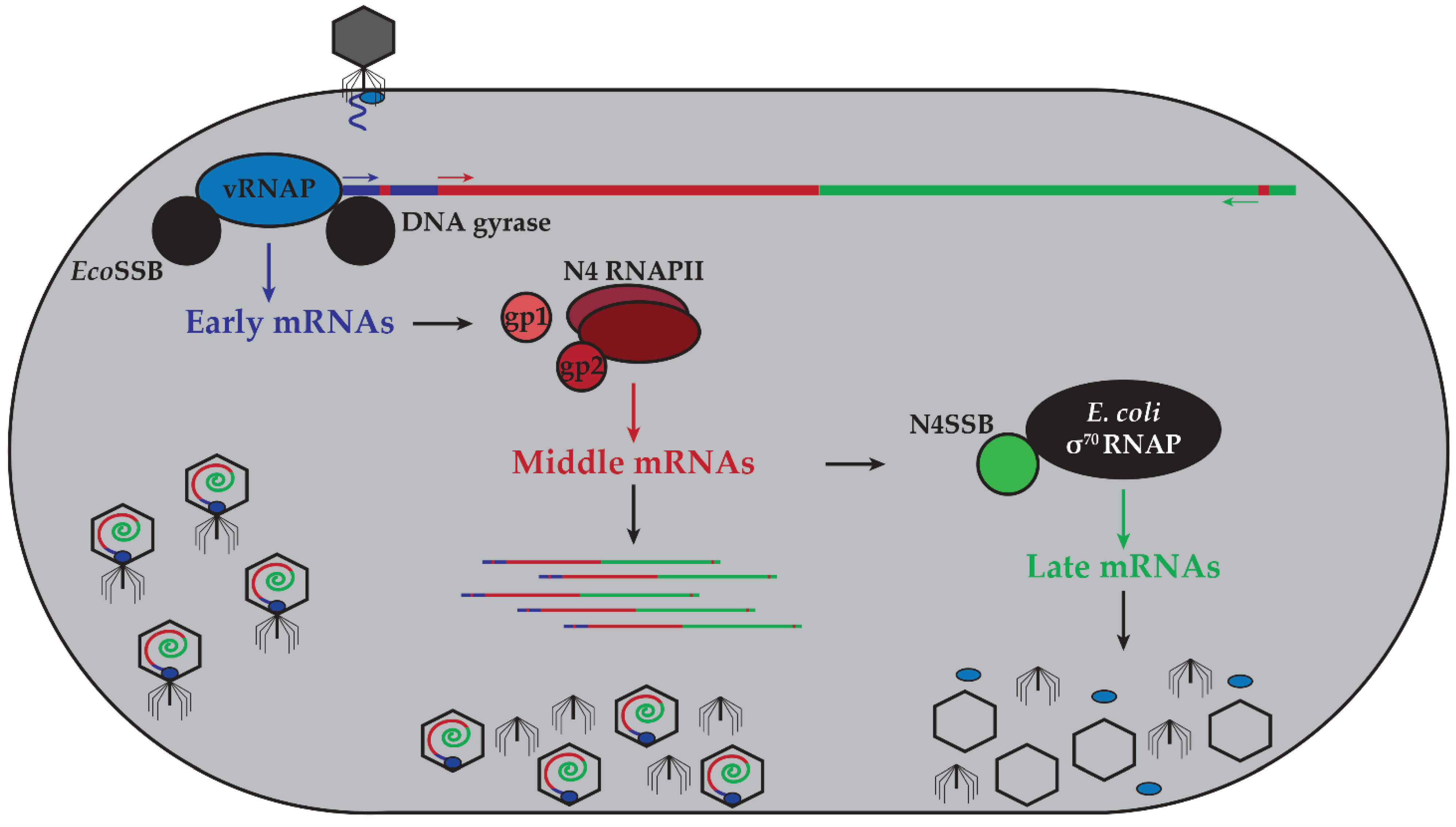
2. N4 Transcriptional Architecture
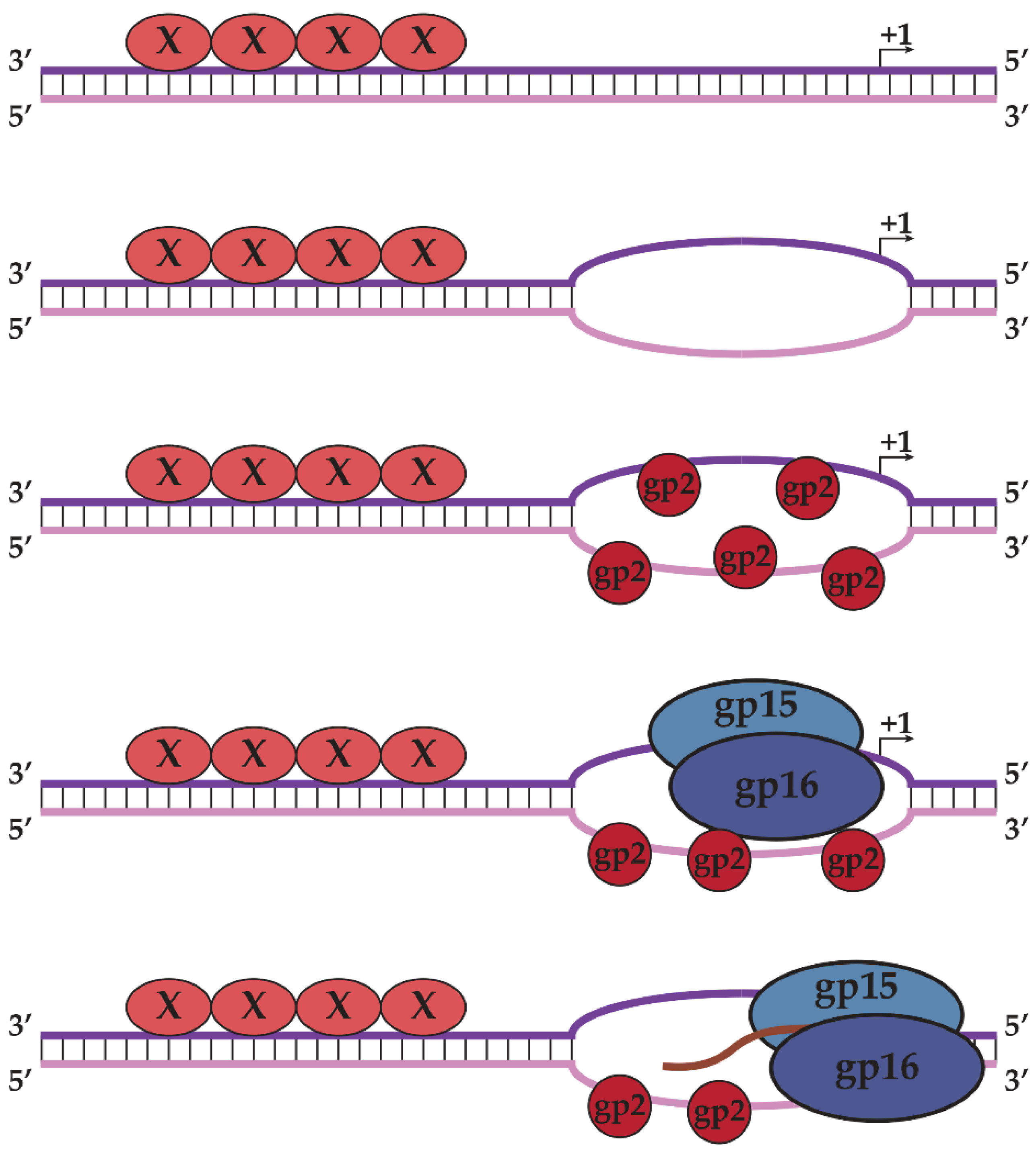
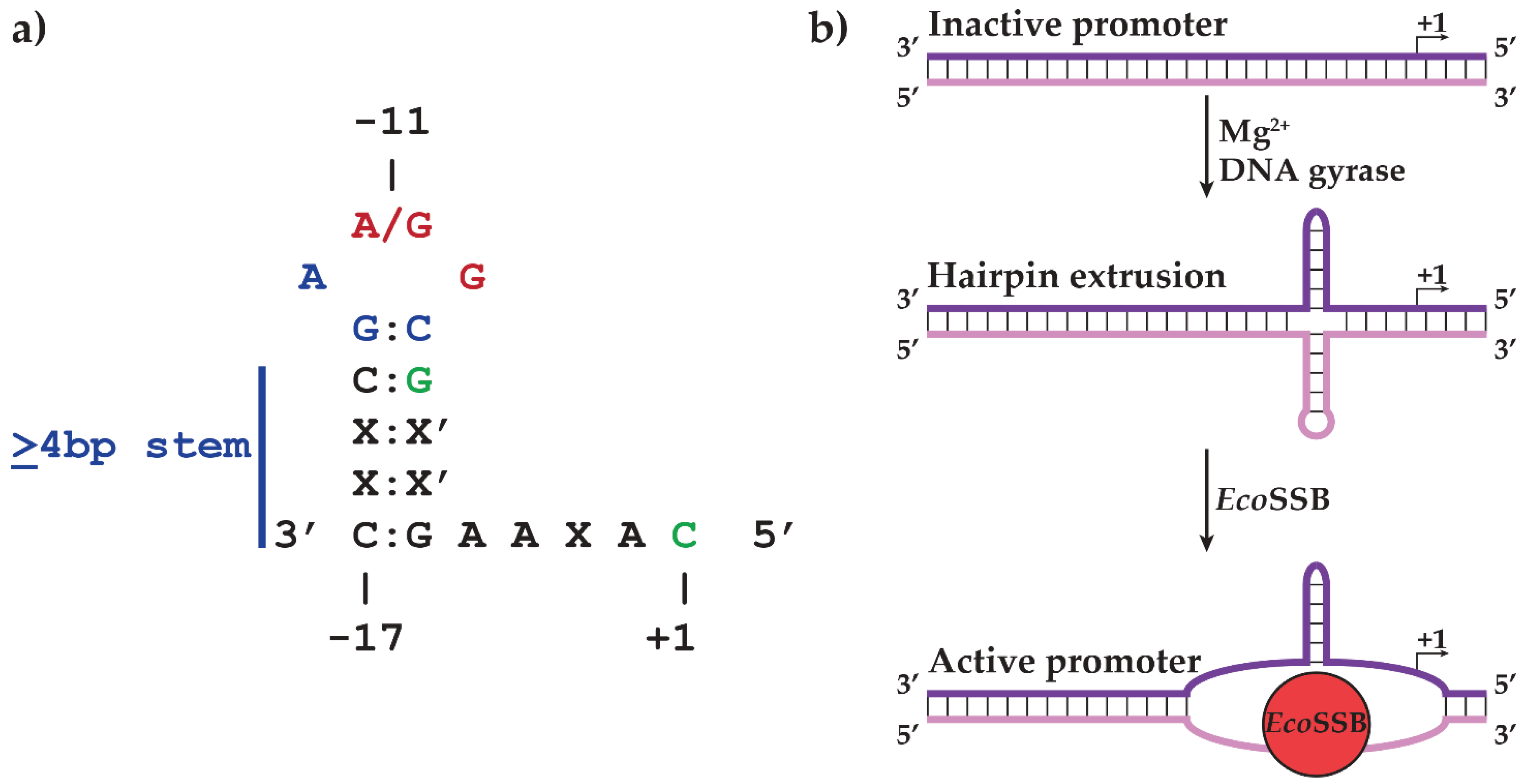
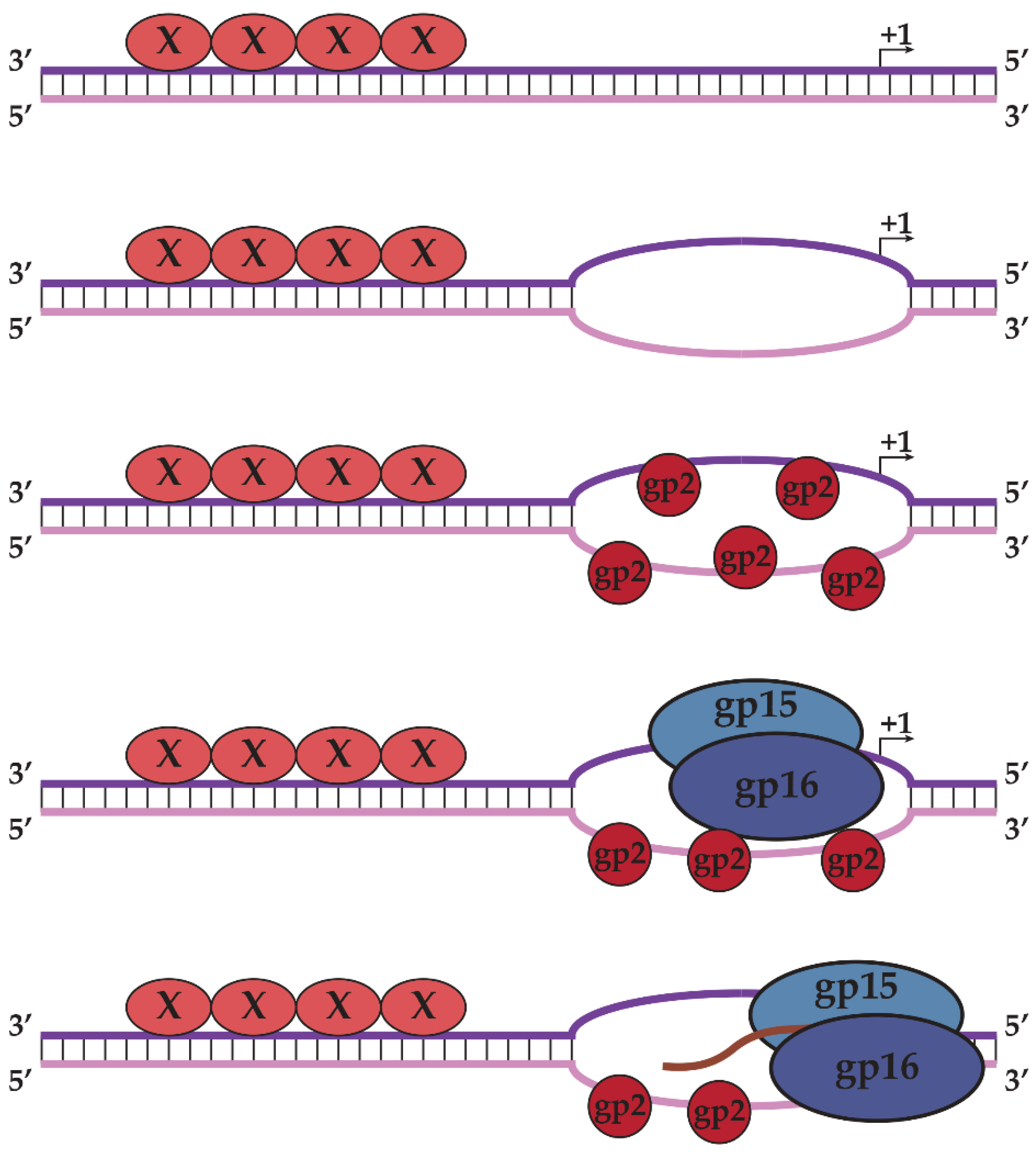
2.1. N4 vRNAP Synthesizes N4 Early RNAs






2.2. N4 RNAPII Synthesizes N4 Middle RNAs
2.3. N4SSB Directs E. coli σ70-RNAP to Synthesize N4 Late RNAs
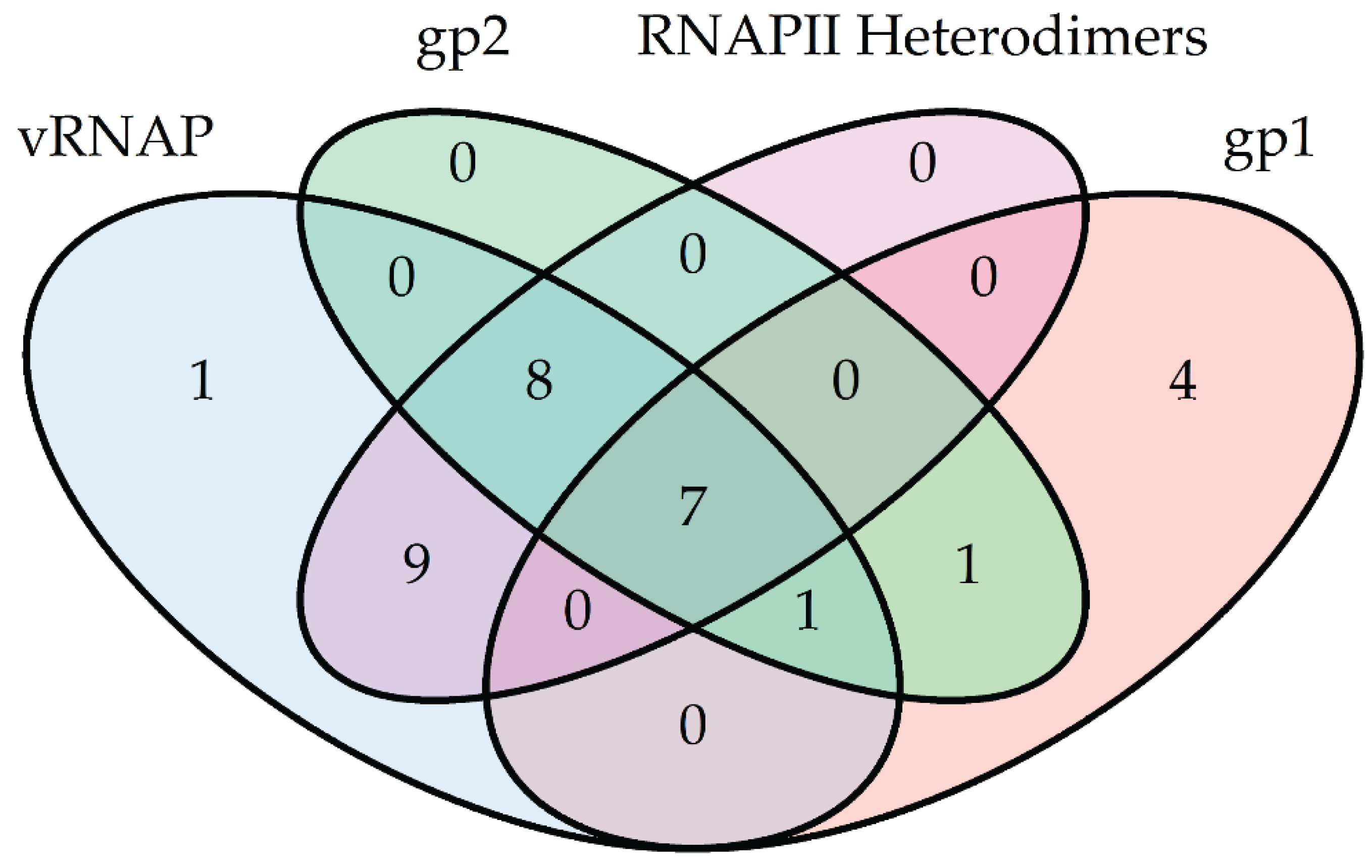
3. N4 RNAPs Have Multiple Roles in Phage Development
4. Phylogenetic Analysis of N4-Like Phage Proteins Involved in Transcription

5. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | RNAP | Cofactor(s) |
|---|---|---|
| T7 | T7 RNAP | None |
| Saccharomyces cerevisiae mitochondria | Rpo41 | Mtf1p |
| Homo sapiens mitochondria | POLRMT | TFAM and TFB2M |
| N4 | N4 vRNAP | EcoSSB |
| N4 | N4 RNAPII | gp1 and gp2 |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Santangelo, T.J.; Artsimovitch, I. Termination and antitermination: RNA polymerase runs a stop sign. Nat. Rev. Microbiol. 2011, 9, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.M. Transcriptional control in the prereplicative phase of T4 development. Virol. J. 2010. [Google Scholar] [CrossRef]
- Losick, R.; Pero, J. Cascade of Sigma Factors. Cell 1981, 25, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, M.; Calles, B.; Mencía, M.; Salas, M.; Rojo, F. Transcription activation or repression by phage phi 29 protein p4 depends on the strength of the RNA polymerase-promoter interactions. Mol. Cell 1997, 1, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Bacteriophage T7. Science 1972, 176, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.; Davis, E.; Brown, D.; Campbell, E.A.; Wigneshweraraj, S.; Darst, S.A. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ70 domain 1.1. Proc. Natl. Acad. Sci. USA 2013, 110, 19772–19777. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shadrin, A.; Sheppard, C.; Mekler, V.; Xu, Y.; Severinov, K.; Matthews, S.; Wigneshweraraj, S. A bacteriophage transcription regulator inhibits bacterial transcription initiation by σ-factor displacement. Nucleic Acids Res. 2014, 42, 4294–4305. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Djordjevic, M.; Shralman, B.; Severinov, K. The tale of two RNA polymerases: Transcription profiling and gene expression strategy of bacteriophage Xp10. Mol. Microbiol. 2005, 55, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Minakhin, L.; van den Bossche, A.; Yakunina, M.; Klimuk, E.; Blasdel, B.; de Smet, J.; Noben, J.-P.; Bläsi, U.; Severinov, K.; Lavigne, R. Development of giant bacteriophage ϕKZ is independent of the host transcription apparatus. J. Virol. 2014, 88, 10501–10510. [Google Scholar] [CrossRef] [PubMed]
- Rothman-Denes, L.B.; Schito, G.C. Novel transcribing activities in N4-infected Escherichia coli. Virology 1974, 60, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Vander Laan, K.; Falco, S.C.; Rothman-Denes, L.B. The program of RNA synthesis in N4-infected Escherichia coli. Virology 1977, 76, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Falco, S.C.; Vander Laan, K.; Rothman-Denes, L.B. Virion-associated RNA polymerase required for bacteriophage N4 development. Proc. Natl. Acad. Sci. USA 1977, 74, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Falco, S.C.; Zehring, W.; Rothman-Denes, L.B. DNA-dependent RNA polymerase from bacteriophage N4 virions. Purification and characterization. J. Biol. Chem. 1980, 255, 4339–4347. [Google Scholar] [PubMed]
- Falco, S.C.; Zivin, R.; Rothman-Denes, L.B. Novel template requirements of N4 virion RNA polymerase. Proc. Natl. Acad. Sci. USA 1978, 75, 3220–3224. [Google Scholar] [CrossRef] [PubMed]
- Brody, E.N.; Geiduschek, E.P. Transcription of the bacteriophage T4 template. Detailed comparison of in vitro and in vivo transcripts. Biochemistry 1970, 9, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Chamberlin, M.; Ring, J. Characterization of T7-specific ribonucleic acid polymerase. J. Biol. Chem. 1973, 248, 2235–2244. [Google Scholar] [PubMed]
- Gellert, M.; Mizuuchi, K.; O’Dea, M.H.; Nash, H.A. DNA gyrase: An enzyme that introduces superhelical turns into DNA. Proc. Natl. Acad. Sci. USA 1976, 73, 3872–3876. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; O’Dea, M. H.; Itoh, T.; Tomizawa, J. Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc. Natl. Acad. Sci. USA 1976, 73, 4474–4478. [Google Scholar] [CrossRef] [PubMed]
- Zivin, R.; Zehring, W.; Rothman-Denes, L.B. Transcriptional map of bacteriophage N4. Location and polarity of N4 RNAs. J. Mol. Biol. 1981, 152, 335–356. [Google Scholar] [CrossRef] [PubMed]
- Zivin, R.; Malone, C.; Rothman-Denes, L.B. Physical map of coliphage N4 DNA. Virology 1980, 104, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.L.; Rothman-Denes, L.B. N4 virion RNA polymerase sites of transcription initiation. Cell 1985, 41, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Glucksmann, M.A.; Markiewicz, P.; Malone, C.; Rothman-Denes, L.B. Specific sequences and a hairpin structure in the template strand are required for N4 virion RNA polymerase promoter recognition. Cell 1992, 70, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Sinden, R.R.; Carlson, J.O.; Pettijohn, D.E. Torsional tension in the DNA double helix measured with trimethylpsoralen in living E. coli cells: Analogous measurements in insect and human cells. Cell 1980, 21, 773–783. [Google Scholar] [CrossRef] [PubMed]
- McClellan, J.A.; Boublíková, P.; Palecek, E.; Lilley, D.M. Superhelical torsion in cellular DNA responds directly to environmental and genetic factors. Proc. Natl. Acad. Sci. USA 1990, 87, 8373–8377. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Greizerstein, M.B.; Nadas-Chinni, K.; Rothman-Denes, L.B. Supercoil-induced extrusion of a regulatory DNA hairpin. Proc. Natl. Acad. Sci. USA 1997, 94, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Kloster, M.; Rothman-Denes, L.B. Sequence-dependent extrusion of a small DNA hairpin at the N4 virion RNA polymerase promoters. J. Mol. Biol. 1998, 283, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Rothman-Denes, L.B. Sequence and DNA structural determinants of N4 virion RNA polymerase-promoter recognition. Genes Dev. 1998, 12, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Davydova, E.K.; Santangelo, T.J.; Rothman-Denes, L.B. Bacteriophage N4 virion RNA polymerase interaction with its promoter DNA hairpin. Proc. Natl. Acad. Sci. USA 2007, 104, 7033–7038. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, P.; Malone, C.; Chase, J.W.; Rothman-Denes, L.B. Escherichia coli single-stranded DNA-binding protein is a supercoiled template-dependent transcriptional activator of N4 virion RNA polymerase. Genes Dev. 1992, 6, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Glucksmann-Kuis, M.A.; Dai, X.; Markiewicz, P.; Rothman-Denes, L.B. E. coli SSB activates N4 virion RNA polymerase promoters by stabilizing a DNA hairpin required for promoter recognition. Cell 1996, 84, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Chase, J. Single-stranded DNA binding proteins required for DNA replication. Annu. Rev. Biochem. 1986, 55, 103–136. [Google Scholar] [CrossRef] [PubMed]
- Davydova, E.K.; Rothman-Denes, L.B. Escherichia coli single-stranded DNA-binding protein mediates template recycling during transcription by bacteriophage N4 virion RNA polymerase. Proc. Natl. Acad. Sci. USA 2003, 100, 9250–9255. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A. The structural basis of the transition from initiation to elongation phases of transcription, as well as translocation and strand separation, by T7 RNA polymerase. Curr. Opin. Struct. Biol. 2004, 14, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak, K.M.; Davydova, E.K.; Mustaev, A.A.; Rothman-Denes, L.B. The phage N4 virion RNA polymerase catalytic domain is related to single-subunit RNA polymerases. EMBO J. 2002, 21, 5815–5823. [Google Scholar] [CrossRef] [PubMed]
- Cermakian, N.; Ikeda, T.M.; Miramontes, P.; Lang, B.F.; Gray, M.W.; Cedergren, R. On the evolution of the single-subunit RNA polymerases. J. Mol. Evol. 1997, 45, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.S.; Davydova, E.K.; Rothman-Denes, L.B. X-ray crystal structure of the polymerase domain of the bacteriophage N4 virion RNA polymerase. Proc. Natl. Acad. Sci. USA 2008, 105, 5046–5051. [Google Scholar] [CrossRef] [PubMed]
- Jeruzalmi, D.; Steitz, T.A. Structure of T7 RNA polymerase complexed to the transcriptional inhibitor T7 lysozyme. EMBO J. 1998, 17, 4101–4113. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, G.M.; Steitz, T.A. Structure of a transcribing T7 RNA polymerase initiation complex. Science 1999, 286, 2305–2309. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, G.M.; Jeruzalmi, D.; Steitz, T.A. Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature 1999, 399, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Gleghorn, M.L.; Davydova, E.K.; Rothman-Denes, L.B.; Murakami, K.S. Structural basis for DNA-hairpin promoter recognition by the bacteriophage N4 virion RNA polymerase. Mol. Cell 2008, 32, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Imburgio, D.; Rong, M.; Ma, K.; McAllister, W.T. Studies of promoter recognition and start site selection by T7 RNA polymerase using a comprehensive collection of promoter variants. Biochemistry 2000, 39, 10419–10430. [Google Scholar] [CrossRef] [PubMed]
- Rong, M.; He, B.; McAllister, W.T.; Durbin, R.K. Promoter specificity determinants of T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 1998, 95, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.A.; Burgess, R.R. Construction of bacteriphage T7 late promoters with point mutations and characterization by in vitro transcription properties. Nucleic Acids Res. 1987, 15, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ho, H.H.; Maslak, M.; Schick, C.; Martin, C.T. Major groove recognition elements in the middle of the T7 RNA polymerase promoter. Biochemistry 1996, 35, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, W.P.; Momand, J.R.; Yin, Y.W. Mechanism for de novo RNA Synthesis and initiating nucleotide specificity by T7 RNA polymerase. J. Mol. Biol. 2007, 370, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Gleghorn, M.L.; Davydova, E.K.; Basu, R.; Rothman-Denes, L.B.; Murakami, K.S. X-ray crystal structures elucidate the nucleotidyl transfer reaction of transcript initiation using two nucleotides. Proc. Natl. Acad. Sci. USA 2011, 108, 3566–3571. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.S.; Murakami, K.S. Watching the bacteriophage N4 RNA polymerase transcription by time-dependent soak-trigger-freeze X-ray crystallography. J. Biol. Chem. 2013, 288, 3305–3311. [Google Scholar] [CrossRef] [PubMed]
- Falco, S.C.; Rothman-Denes, L.B. Bacteriophage N4-induced transcribing activities in Escherichia coli I. Detection and characterization in cell extracts. Virology 1979, 95, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.H.; Kazmierczak, K.M.; Carter, R.H.; Rothman-Denes, L.B. N4 RNA polymerase II, a heterodimeric RNA polymerase with homology to the single-subunit family of RNA polymerases. J. Bacteriol. 2002, 184, 4952–4961. [Google Scholar] [CrossRef] [PubMed]
- Falco, S.C.; Rothman-Denes, L.B. Bacteriophage transcribing activities in Escherichia coli II. Association of the N4 transcriptional apparatus with the cytoplasmic membrane. Virology 1979, 95, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Zehring, W.A.; Falco, S.C.; Malone, C.; Rothman-Denes, L.B. Bacteriophage N4-induced transcribing activities in E. coli. III. A third cistron required for N4 RNA polymerase II activity. Virology 1983, 126, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Zehring, W.A.; Rothman-Denes, L.B. Purification and characterization of coliphage N4 RNA polymerase II activity from infected cell extracts. J. Biol. Chem. 1983, 258, 8074–8080. [Google Scholar] [PubMed]
- Abravaya, K.; Rothman-Denes, L.B. In vitro requirements for N4 RNA polymerase II-specific Initiation. J. Biol. Chem. 1989, 264, 12695–12699. [Google Scholar] [PubMed]
- Abravaya, K.; Rothman-Denes, L.B. N4 RNA polymerase II sites of transcription initiation. J. Mol. Biol. 1990, 211, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.; Lenneman, B.R.; Rothman-Denes, L.B. Determinants of bacteriophage N4 RNAPII promoter specificity. Unpublished data. 2015. [Google Scholar]
- Sousa, R. Structural and mechanistic relationships between nucleic acid polymerases. Trends Biochem. Sci. 1996, 21, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.H.; Demidenko, A.A.; Hattingh-Willis, S.; Rothman-Denes, L.B. Phage N4 RNA polymerase II recruitment to DNA by a single-stranded DNA-binding protein. Genes Dev. 2003, 17, 2334–2345. [Google Scholar] [CrossRef] [PubMed]
- Guinta, D.; Stambouly, J.; Falco, S.C.; Rist, J.K.; Rothman-Denes, L.B. Host and phage-coded functions required for coliphage N4 DNA replication. Virology 1986, 150, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, G.K.; Rist, J.K.; Kunkel, T.A.; Sugino, A.; Rothman-Denes, L.B. Purification and characterization of bacteriophage N4-induced DNA polymerase. J. Biol. Chem. 1988, 263, 11319–11326. [Google Scholar] [PubMed]
- Lindberg, G.; Kowalczykowski, S.C.; Rist, J.K.; Sugino, A.; Rothman-Denes, L.B. Purification and characterization of the coliphage N4-coded single-stranded DNA binding protein. J. Biol. Chem. 1989, 264, 12700–12708. [Google Scholar] [PubMed]
- Choi, M.; Miller, A.; Cho, N.Y.; Rothman-Denes, L.B. Identification, cloning, and characterization of the bacteriophage N4 gene encoding the single-stranded DNA-binding protein: A protein required for phage replication, recombination, and late transcription. J. Biol. Chem. 1995, 270, 22541–22547. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.Y.; Choi, M.; Rothman-Denes, L.B. The bacteriophage N4-coded single-stranded DNA-binding protein (N4SSB) is the transcriptional activator of Escherichia coli RNA polymerase at N4 late promoters. J. Mol. Biol. 1995, 246, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Dai, X.; Choi, M.; Glucksmann-Kuis, M.A.; Rothman-Denes, L.B. Single-stranded DNA-binding proteins as transcriptional activators. Methods Enzymol. 1996, 274, 9–20. [Google Scholar] [PubMed]
- Choi, K.H.; Mcpartland, J.; Kaganman, I.; Bowman, V.D.; Rothman-Denes, L.B.; Rossmann, M.G. Insight into DNA and protein transport in double-stranded DNA viruses: The structure of bacteriophage N4. J. Mol. Biol. 2009, 378, 726–736. [Google Scholar] [CrossRef]
- Kiino, D.R.; Rothman-Denes, L.B. Genetic analysis of bacteriophage N4 adsorption. J. Bacteriol. 1989, 171, 4595–4602. [Google Scholar] [PubMed]
- Kiino, D.R.; Singer, M.S.; Rothman-Denes, L.B. Two overlapping genes encoding membrane proteins required for bacteriophage N4 adsorption. J. Bacteriol. 1993, 175, 7081–7085. [Google Scholar] [PubMed]
- Kiino, D.R.; Licudine, R.; Wilt, K.; Yang, D.H.C.; Rothman-Denes, L.B. A cytoplasmic protein, NfrC, is required for bacteriophage N4 adsorption. J. Bacteriol. 1993, 175, 7074–7080. [Google Scholar] [PubMed]
- McPartland, J.; Rothman-Denes, L.B. The tail sheath of bacteriophage N4 interacts with the Escherichia coli receptor. J. Bacteriol. 2009, 191, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, A.A.; Rothman-Denes, L.B. Bacteriophage N4 genome injection is driven by the phage transcription systems. Unpublished data. 2015. [Google Scholar]
- Hsieh, L.S.; Rouviere-Yaniv, J.; Drlica, K. Bacterial DNA supercoiling and [ATP]/[ADP] ratio: Changes associated with salt shock. J. Bacteriol. 1991, 173, 3914–3917. [Google Scholar] [PubMed]
- Hsieh, L.S.; Burger, R.M.; Drlica, K. Bacterial DNA supercoiling and [ATP]/[ADP]. Changes associated with a transition to anaerobic growth. J. Mol. Biol. 1991, 219, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.T.; Rothman-Denes, L.B. A phage-encoded inhibitor of Escherichia coli DNA replication targets the DNA polymerase clamp loader. Mol. Microbiol. 2011, 79, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Stojković, E.A.; Rothman-Denes, L.B. Coliphage N4 N-acetylmuramidase defines a new family of murein hydrolases. J. Mol. Biol. 2007, 366, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Khuong, N.; Loose, M.; Yano, S.; Rothman-Denes, L.B. Coliphage N4 inhibits cell division by targeting FtsZ and FtsA. Proc. Natl. Acad. Sci. USA 2015. submitted for publication. [Google Scholar]
- Schito, G.C. Development of coliphage N4: Ultrastructural studies. J. Virol. 1974, 13, 186–196. [Google Scholar] [PubMed]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Kaganman, I.; Davydova, E.K.; Kazmierczak, K.M.; Rothman-Denes, L.B. The N-terminal and C-terminal domains of bacteriophage N4 vRNAP are required for genome injection and encapsidation respectively. Unpublished data. 2015. [Google Scholar]
- Morozov, Y.I.; Agaronyan, K.; Cheung, A.C.M.; Anikin, M.; Cramer, P.; Temiakov, D. A novel intermediate in transcription initiation by human mitochondrial RNA polymerase. Nucleic Acids Res. 2014, 42, 3884–3893. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.S.; Stohl, L.L.; Clayton, D.A. Yeast mitochondrial RNA polymerase is homologous to those encoded by bacteriophages T3 and T7. Cell 1987, 51, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.P.; Patel, S.S. Mechanism of transcription initiation by the yeast mitochondrial RNA polymerase. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 930–938. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenneman, B.R.; Rothman-Denes, L.B. Structural and Biochemical Investigation of Bacteriophage N4-Encoded RNA Polymerases. Biomolecules 2015, 5, 647-667. https://doi.org/10.3390/biom5020647
Lenneman BR, Rothman-Denes LB. Structural and Biochemical Investigation of Bacteriophage N4-Encoded RNA Polymerases. Biomolecules. 2015; 5(2):647-667. https://doi.org/10.3390/biom5020647
Chicago/Turabian StyleLenneman, Bryan R., and Lucia B. Rothman-Denes. 2015. "Structural and Biochemical Investigation of Bacteriophage N4-Encoded RNA Polymerases" Biomolecules 5, no. 2: 647-667. https://doi.org/10.3390/biom5020647
APA StyleLenneman, B. R., & Rothman-Denes, L. B. (2015). Structural and Biochemical Investigation of Bacteriophage N4-Encoded RNA Polymerases. Biomolecules, 5(2), 647-667. https://doi.org/10.3390/biom5020647