
RNA-Binding Proteins: Splicing Factors and Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
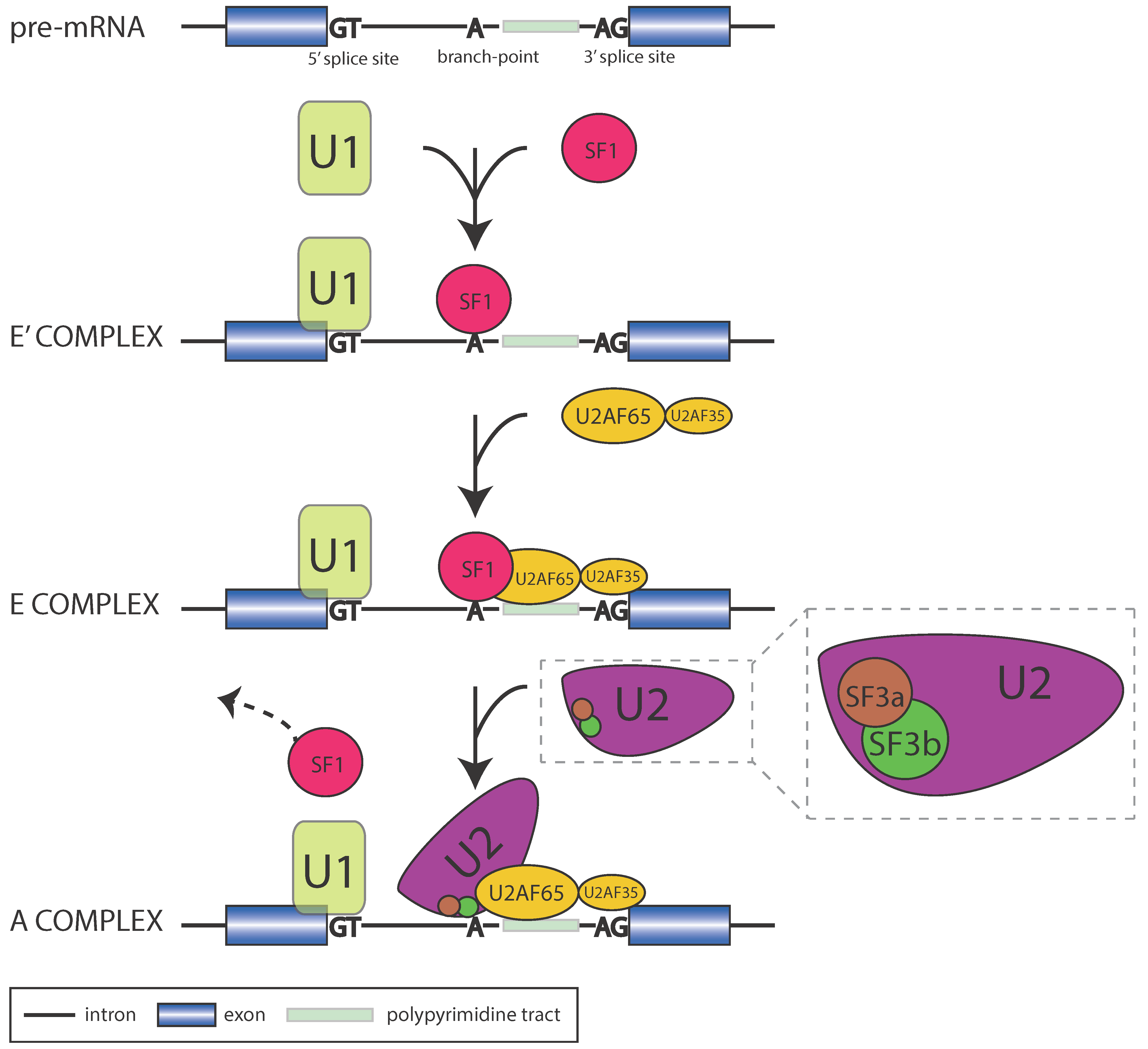
1.1. The Core Spliceosome

1.2. hnRNPs, SR Proteins, and Other Splicing Factors
2. Three Mechanisms of RBP-Related Splicing Dysregulation
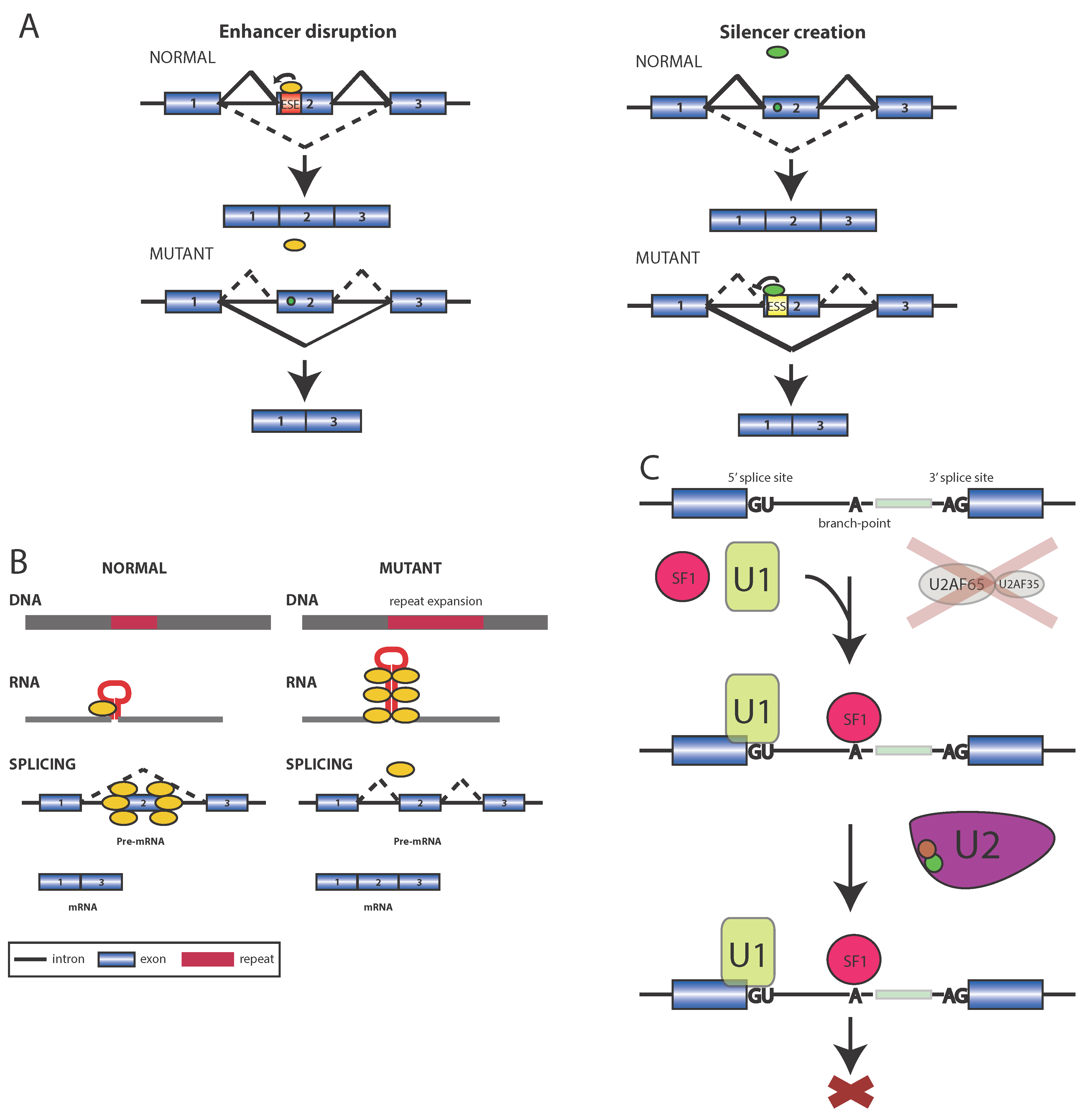
2.1. Mechanism I: Disruption of a Splicing Element

2.2. Mechanisms II: Toxic RNA
2.3. Mechanism III: Mutations that Affect Splicing Factors
3. Developing Tools Predicting Causal SNPs
4. Functionally Validating Individual Variants
5. Conclusions and Future Directions in Therapeutic Interventions for Splicing Disorders
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Merkin, J.J.; Chen, P.; Alexis, M.S.; Hautaniemi, S.K.; Burge, C.B. Origins and impacts of new mammalian exons. Cell Rep. 2015, 10, 1992–2005. [Google Scholar] [CrossRef] [PubMed]
- Osborne, R.J.; Thornton, C.A. RNA-dominant diseases. Hum. Mol. Genet. 2006, 15, R162–R169. [Google Scholar] [CrossRef] [PubMed]
- O'Rourke, J.R.; Swanson, M.S. Mechanisms of RNA-mediated disease. J. Biol. Chem. 2009, 284, 7419–7423. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.; Cooper, T.A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 2006, 29, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Ferraris, L.; Filloux, M.E.; Raphael, B.J.; Fairbrother, W.G. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl. Acad. Sci. USA 2011, 108, 11093–11098. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14. [Google Scholar] [CrossRef]
- Roca, X.; Akerman, M.; Gaus, H.; Berdeja, A.; Bennett, C.F.; Krainer, A.R. Widespread recognition of 5' splice sites by noncanonical base-pairing to U1 snRNA involving bulged nucleotides. Genes Dev. 2012, 26, 1098–109. [Google Scholar] [CrossRef] [PubMed]
- Hartmuth, K.; Urlaub, H.; Vornlocher, H.P.; Will, C.L.; Gentzel, M.; Wilm, M.; Luhrmann, R. Protein composition of human prespliceosomes isolated by a tobramycin affinity-selection method. Proc. Natl. Acad. Sci. USA 2002, 99, 16719–16724. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Licklider, L.J.; Gygi, S.P.; Reed, R. Comprehensive proteomic analysis of the human spliceosome. Nature 2002, 419, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Licklider, L.J.; Gygi, S.R.; Grigorieff, N.; Moore, M.J. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 2002, 8, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W. The spliceosome: The most complex macromolecular machine in the cell? Bioessays 2003, 25, 1147–1149. [Google Scholar] [CrossRef] [PubMed]
- Kent, O.A.; Ritchie, D.B.; Macmillan, A.M. Characterization of a U2AF-independent commitment complex (E') in the mammalian spliceosome assembly pathway. Mol. Cell. Biol. 2005, 25, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Madl, T.; Bagdiul, I.; Kern, T.; Kang, H.S.; Zou, P.; Mausbacher, N.; Sieber, S.A.; Kramer, A.; Sattler, M. Structure, phosphorylation and U2AF65 binding of the N-terminal domain of splicing factor 1 during 3'-splice site recognition. Nucleic Acids Res. 2013, 41, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.W.; Cheng, S.C. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev. 2015, 29, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Perriman, R.; Ares, M., Jr. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol. Cell 2010, 38, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Mohlmann, S.; Mathew, R.; Neumann, P.; Schmitt, A.; Luhrmann, R.; Ficner, R. Structural and functional analysis of the human spliceosomal DEAD-box helicase Prp28. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, X.; Hill, R.C.; Qiu, Y.; Zhang, W.; Hansen, K.C.; Zhao, R. Brr2 plays a role in spliceosomal activation in addition to U4/U6 unwinding. Nucleic Acids Res. 2015, 43, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Nancollis, V.; Ruckshanthi, J.P.; Frazer, L.N.; O'Keefe, R.T. The U5 snRNA internal loop 1 is a platform for Brr2, Snu114 and Prp8 protein binding during U5 snRNP assembly. J. Cell. Biochem. 2013, 114, 2770–2784. [Google Scholar] [CrossRef] [PubMed]
- Wlodaver, A.M.; Staley, J.P. The DExD/H-box ATPase Prp2p destabilizes and proofreads the catalytic RNA core of the spliceosome. RNA 2014, 20, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Lardelli, R.M.; Thompson, J.X.; Yates, J.R., 3rd; Stevens, S.W. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA 2010, 16, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Galej, W.P.; Oubridge, C.; Newman, A.J.; Nagai, K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature 2013, 493, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Fica, S.M.; Tuttle, N.; Novak, T.; Li, N.S.; Lu, J.; Koodathingal, P.; Dai, Q.; Staley, J.P.; Piccirilli, J.A. RNA catalyses nuclear pre-mRNA splicing. Nature 2013, 503, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, L.L.; Corona, D.; Onorati, M.C. Emerging roles for hnRNPs in post-transcriptional regulation: What can we learn from flies? Chromosoma 2014, 123, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Matunis, E.L.; Matunis, M.J.; Dreyfuss, G. Characterization of the major hnRNP proteins from Drosophila melanogaster. J. Cell Biol. 1992, 116, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.F.; Revil, T.; Chabot, B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007, 623, 123–147. [Google Scholar] [PubMed]
- Han, S.P.; Tang, Y.H.; Smith, R. Functional diversity of the hnRNPs: Past, present and perspectives. Biochem. J. 2010, 430, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Li, W.; Zhang, M. The function of the RNA-binding protein hnRNP in cancer metastasis. J. Cancer Res. Ther. 2013, 9, S129–S134. [Google Scholar] [PubMed]
- Rollins, C.; Levengood, J.D.; Rife, B.D.; Salemi, M.; Tolbert, B.S. Thermodynamic and phylogenetic insights into hnRNP A1 recognition of the HIV-1 exon splicing silencer 3 element. Biochemistry 2014, 53, 2172–2184. [Google Scholar] [CrossRef] [PubMed]
- Rooke, N.; Markovtsov, V.; Cagavi, E.; Black, D.L. Roles for SR proteins and hnRNP A1 in the regulation of c-src exon N1. Mol. Cell. Biol. 2003, 23, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Zahler, A.M.; Lane, W.S.; Stolk, J.A.; Roth, M.B. SR proteins: A conserved family of pre-mRNA splicing factors. Genes Dev. 1992, 6, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.B.; Murphy, C.; Gall, J.G. A monoclonal antibody that recognizes a phosphorylated epitope stains lampbrush chromosome loops and small granules in the amphibian germinal vesicle. J. Cell Biol. 1990, 111, 2217–2223. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Zuo, P.; Manley, J.L. Primary structure of the human splicing factor ASF reveals similarities with Drosophila regulators. Cell 1991, 66, 373–382. [Google Scholar] [CrossRef]
- Krainer, A.R.; Mayeda, A.; Kozak, D.; Binns, G. Functional expression of cloned human splicing factor SF2: Homology to RNA-binding proteins, U1 70K, and Drosophila splicing regulators. Cell 1991, 66, 383–394. [Google Scholar] [CrossRef]
- Ge, H.; Manley, J.L. A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell 1990, 62, 25–34. [Google Scholar] [CrossRef]
- Chou, T.B.; Zachar, Z.; Bingham, P.M. Developmental expression of a regulatory gene is programmed at the level of splicing. EMBO J. 1987, 6, 4095–4104. [Google Scholar] [PubMed]
- Boggs, R.T.; Gregor, P.; Idriss, S.; Belote, J.M.; McKeown, M. Regulation of sexual differentiation in D. melanogaster via alternative splicing of RNA from the transformer gene. Cell 1987, 50, 739–747. [Google Scholar]
- Amrein, H.; Gorman, M.; Nothiger, R. The sex-determining gene tra-2 of Drosophila encodes a putative RNA binding protein. Cell 1988, 55, 1025–1035. [Google Scholar] [CrossRef]
- Shepard, P.J.; Hertel, K.J. The SR protein family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Hertel, K.J. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip. Rev. RNA 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Hoang, A.; Sinha, R.; Zhong, X.Y.; Fu, X.D.; Krainer, A.R.; Ghosh, G. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc. Natl. Acad. Sci. USA 2011, 108, 8233–8238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Will, C.L.; Anokhina, M.; Tazi, J.; Urlaub, H.; Luhrmann, R. Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol. Cell 2010, 38, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, S.; Bruzik, J.P. Multiple roles for SR proteins in trans splicing. Mol. Cell. Biol. 2002, 22, 5337–5346. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Girard, C.; Will, C.L.; Peng, J.; Makarov, E.M.; Kastner, B.; Lemm, I.; Urlaub, H.; Hartmuth, K.; Luhrmann, R. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat. Commun. 2012, 3, 994. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.; Coutinho, G.; Nahas, S.; Yeo, G.; Tanouye, R.; Babaei, M.; Dork, T.; Burge, C.; Gatti, R.A. Nonclassical splicing mutations in the coding and noncoding regions of the ATM Gene: Maximum entropy estimates of splice junction strengths. Hum. Mutat. 2004, 23, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Zheng, D.; Pan, Q.; Liu, Z.; Xi, X.; Hu, Z.; Deng, H.; Liu, X.; Jiang, D.; Deng, H.; et al. A novel PRPF31 splice-site mutation in a Chinese family with autosomal dominant retinitis pigmentosa. Mol. Vis. 2004, 10, 361–365. [Google Scholar] [PubMed]
- Hartikainen, J.M.; Pirskanen, M.M.; Arffman, A.H.; Ristonmaa, U.K.; Mannermaa, A.J. A Finnish BRCA1 exon 12 4216-2nt A to G splice acceptor site mutation causes aberrant splicing and frameshift, leading to protein truncation. Hum. Mutat. 2000, 15, 120. [Google Scholar] [CrossRef]
- Sun, H.; Chasin, L.A. Multiple splicing defects in an intronic false exon. Mol. Cell. Biol. 2000, 20, 6414–6425. [Google Scholar] [CrossRef] [PubMed]
- Burrows, N.P.; Nicholls, A.C.; Richards, A.J.; Luccarini, C.; Harrison, J.B.; Yates, J.R.; Pope, F.M. A point mutation in an intronic branch site results in aberrant splicing of COL5A1 and in Ehlers-Danlos syndrome type II in two British families. Am. J. Hum. Genet. 1998, 63, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.B.; Horowitz, D.S. Exon sequences at the splice junctions affect splicing fidelity and alternative splicing. Proc. Natl. Acad. Sci. USA 2009, 106, 18954–18959. [Google Scholar] [CrossRef] [PubMed]
- Maslen, C.; Babcock, D.; Raghunath, M.; Steinmann, B. A rare branch-point mutation is associated with missplicing of fibrillin-2 in a large family with congenital contractural arachnodactyly. Am. J. Hum. Genet. 1997, 60, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Taggart, A.J.; DeSimone, A.M.; Shih, J.S.; Filloux, M.E.; Fairbrother, W.G. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat. Struct. Mol. Biol. 2012, 19, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Chasin, L.A. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004, 18, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Kazan, H.; Cook, K.B.; Weirauch, M.T.; Najafabadi, H.S.; Li, X.; Gueroussov, S.; Albu, M.; Zheng, H.; Yang, A.; et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 2013, 499, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Contreras, R.; Fisette, J.F.; Nasim, F.U.; Madden, R.; Cordeau, M.; Chabot, B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006, 4, e21. [Google Scholar] [CrossRef] [PubMed]
- Kanopka, A.; Muhlemann, O.; Akusjarvi, G. Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature 1996, 381, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Faa, V.; Coiana, A.; Incani, F.; Costantino, L.; Cao, A.; Rosatelli, M.C. A synonymous mutation in the CFTR gene causes aberrant splicing in an italian patient affected by a mild form of cystic fibrosis. J. Mol. Diagn. 2010, 12, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Sterne-Weiler, T.; Howard, J.; Mort, M.; Cooper, D.N.; Sanford, J.R. Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 2011, 21, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Dredge, B.K.; Polydorides, A.D.; Darnell, R.B. The splice of life: Alternative splicing and neurological disease. Nat. Rev. Neurosci. 2001, 2, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Arnold, E.S.; Ling, S.C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Wexler, E.; Wahnich, A.; Friedrich, T.; Vijayendran, C.; Gao, F.; Parikshak, N.; Konopka, G.; Geschwind, D.H. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum. Mol. Genet. 2012, 21, 4171–4186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Manley, J.L. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013, 3, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Brunak, S.; Engelbrecht, J.; Knudsen, S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 1991, 220, 49–65. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeo, G.W.; Yeh, R.; Goldstein, P.; Mawson, M.; Sharp, P.A.; Burge, C.B. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004, 32, W187–W190. [Google Scholar] [CrossRef] [PubMed]
- Goren, A.; Kim, E.; Amit, M.; Vaknin, K.; Kfir, N.; Ram, O.; Ast, G. Overlapping splicing regulatory motifs–combinatorial effects on splicing. Nucleic Acids Res. 2010, 38, 3318–3327. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Fairbrother, W.G. Spliceman—A computational web server that predicts sequence variations in pre-mRNA splicing. Bioinformatics 2012, 28, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [Green Version]
- Cooper, T.A. Use of minigene systems to dissect alternative splicing elements. Methods 2005, 37, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Basler, K.; Siegrist, P.; Hafen, E. The spatial and temporal expression pattern of sevenless is exclusively controlled by gene-internal elements. EMBO J. 1989, 8, 2381–2386. [Google Scholar] [PubMed]
- Soemedi, R.; Vega, H.; Belmont, J.M.; Ramachandran, S.; Fairbrother, W.G. Genetic variation and RNA binding proteins: Tools and techniques to detect functional polymorphisms. Adv. Exp. Med. Biol. 2014, 825, 227–266. [Google Scholar] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Svasti, S.; Suwanmanee, T.; Fucharoen, S.; Moulton, H.M.; Nelson, M.H.; Maeda, N.; Smithies, O.; Kole, R. RNA repair restores hemoglobin expression in IVS2-654 thalassemic mice. Proc. Natl. Acad. Sci. USA 2009, 106, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Hojland, T.; Hansen, B.R.; Persson, R.; Bramsen, J.B.; Kjems, J.; Koch, T.; Wengel, J.; Smith, C.I. Biological activity and biotechnological aspects of locked nucleic acids. Adv. Genet. 2013, 82, 47–107. [Google Scholar] [PubMed]
- Owen, N.; Zhou, H.; Malygin, A.A.; Sangha, J.; Smith, L.D.; Muntoni, F.; Eperon, I.C. Design principles for bifunctional targeted oligonucleotide enhancers of splicing. Nucleic Acids Res. 2011, 39, 7194–7208. [Google Scholar] [CrossRef] [PubMed]
- Disterer, P.; Kryczka, A.; Liu, Y.; Badi, Y.E.; Wong, J.J.; Owen, J.S.; Khoo, B. Development of therapeutic splice-switching oligonucleotides. Hum. Gene Ther. 2014, 25, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.L.; Berniac, J.; Liu, Y.H.; Abato, P.; Jodelka, F.M.; Barthel, L.; Kumar, S.; Dudley, C.; Nelson, M.; Larson, K.; et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci. Transl. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.M.; Sobczak, K.; Lueck, J.D.; Osborne, R.J.; Lin, X.; Dirksen, R.T.; Thornton, C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009, 325, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Mulders, S.A.; van den Broek, W.J.; Wheeler, T.M.; Croes, H.J.; van Kuik-Romeijn, P.; de Kimpe, S.J.; Furling, D.; Platenburg, G.J.; Gourdon, G.; Thornton, C.A.; et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 13915–13920. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Bennett, C.F.; Cooper, T.A. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2012, 109, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Mosquea, L.M.; Clayton, N.P.; Wu, I.H.; Weeden, T.; Nelson, C.A.; Phillips, L.; Roberts, E.; Piepenhagen, P.A.; Cheng, S.H.; et al. Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013, 23, 109–117. [Google Scholar] [PubMed]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Francois, V.; Klein, A.F.; Beley, C.; Jollet, A.; Lemercier, C.; Garcia, L.; Furling, D. Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat. Struct. Mol. Biol. 2011, 18, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, K.; Wheeler, T.M.; Wang, W.; Thornton, C.A. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Ther. 2013, 21, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Warf, M.B.; Nakamori, M.; Matthys, C.M.; Thornton, C.A.; Berglund, J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 18551–18556. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lopez, A.; Llamusi, B.; Orzaez, M.; Perez-Paya, E.; Artero, R.D. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc. Natl. Acad. Sci. USA 2011, 108, 11866–11871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs-Disney, J.L.; Parkesh, R.; Nakamori, M.; Thornton, C.A.; Disney, M.D. Rational design of bioactive, modularly assembled aminoglycosides targeting the RNA that causes myotonic dystrophy type 1. ACS Chem. Biol. 2012, 7, 1984–1993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, Y.; Dong, S.; Choudhury, R.; Jin, Y.; Wang, Z. Treatment of type 1 myotonic dystrophy by engineering site-specific RNA endonucleases that target (CUG)(n) repeats. Mol. Ther. 2014, 22, 312–320. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fredericks, A.M.; Cygan, K.J.; Brown, B.A.; Fairbrother, W.G. RNA-Binding Proteins: Splicing Factors and Disease. Biomolecules 2015, 5, 893-909. https://doi.org/10.3390/biom5020893
Fredericks AM, Cygan KJ, Brown BA, Fairbrother WG. RNA-Binding Proteins: Splicing Factors and Disease. Biomolecules. 2015; 5(2):893-909. https://doi.org/10.3390/biom5020893
Chicago/Turabian StyleFredericks, Alger M., Kamil J. Cygan, Brian A. Brown, and William G. Fairbrother. 2015. "RNA-Binding Proteins: Splicing Factors and Disease" Biomolecules 5, no. 2: 893-909. https://doi.org/10.3390/biom5020893
APA StyleFredericks, A. M., Cygan, K. J., Brown, B. A., & Fairbrother, W. G. (2015). RNA-Binding Proteins: Splicing Factors and Disease. Biomolecules, 5(2), 893-909. https://doi.org/10.3390/biom5020893