
Activation of Proinflammatory Responses in Cells of the Airway Mucosa by Particulate Matter: Oxidant- and Non-Oxidant-Mediated Triggering Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Some General Principles of Particle Toxicology
3. Molecular Initiating Events: Triggering Mechanisms for Particle-Induced Activation of Proinflammatory Genes
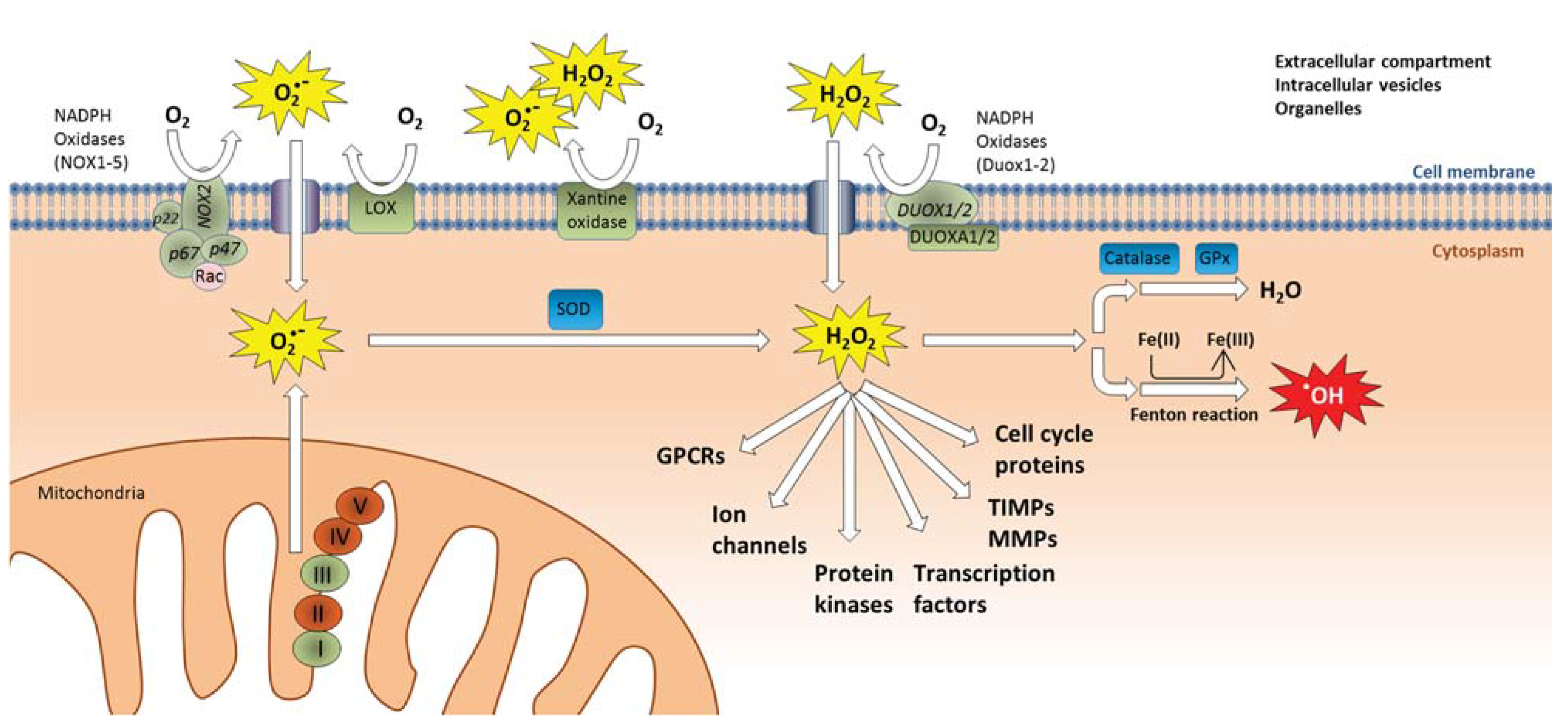
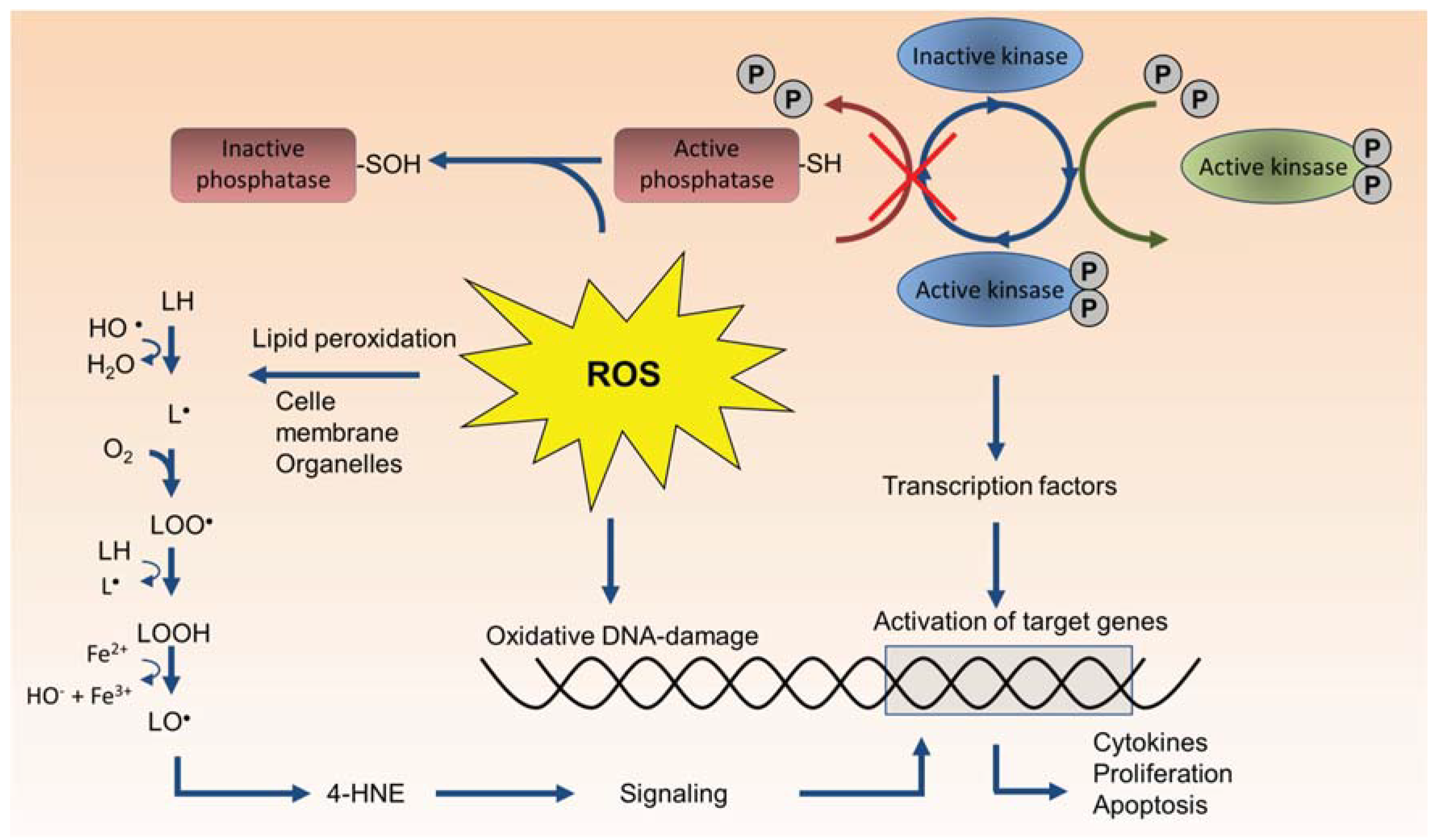
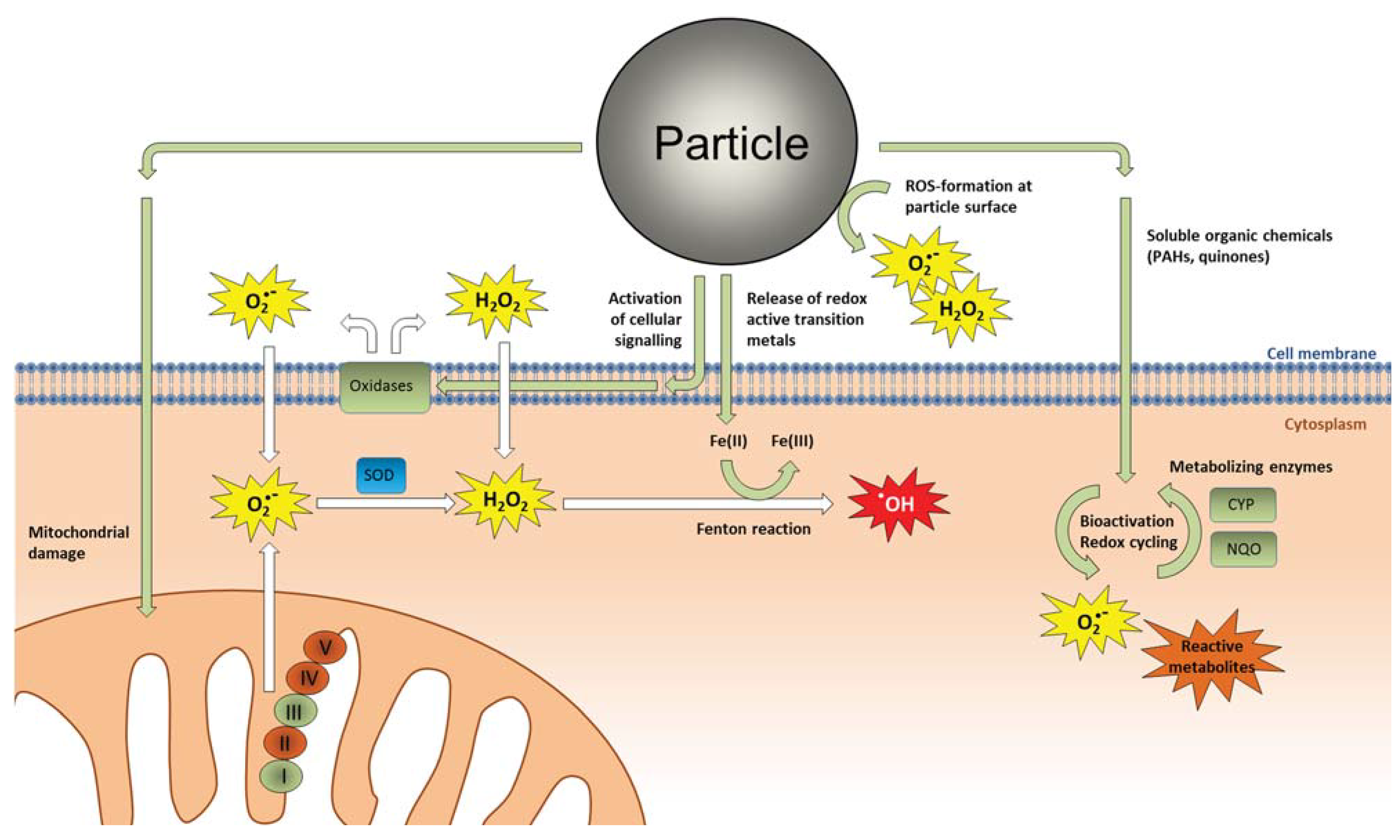
3.1. Oxidative Stress and Direct ROS Formation by Particles
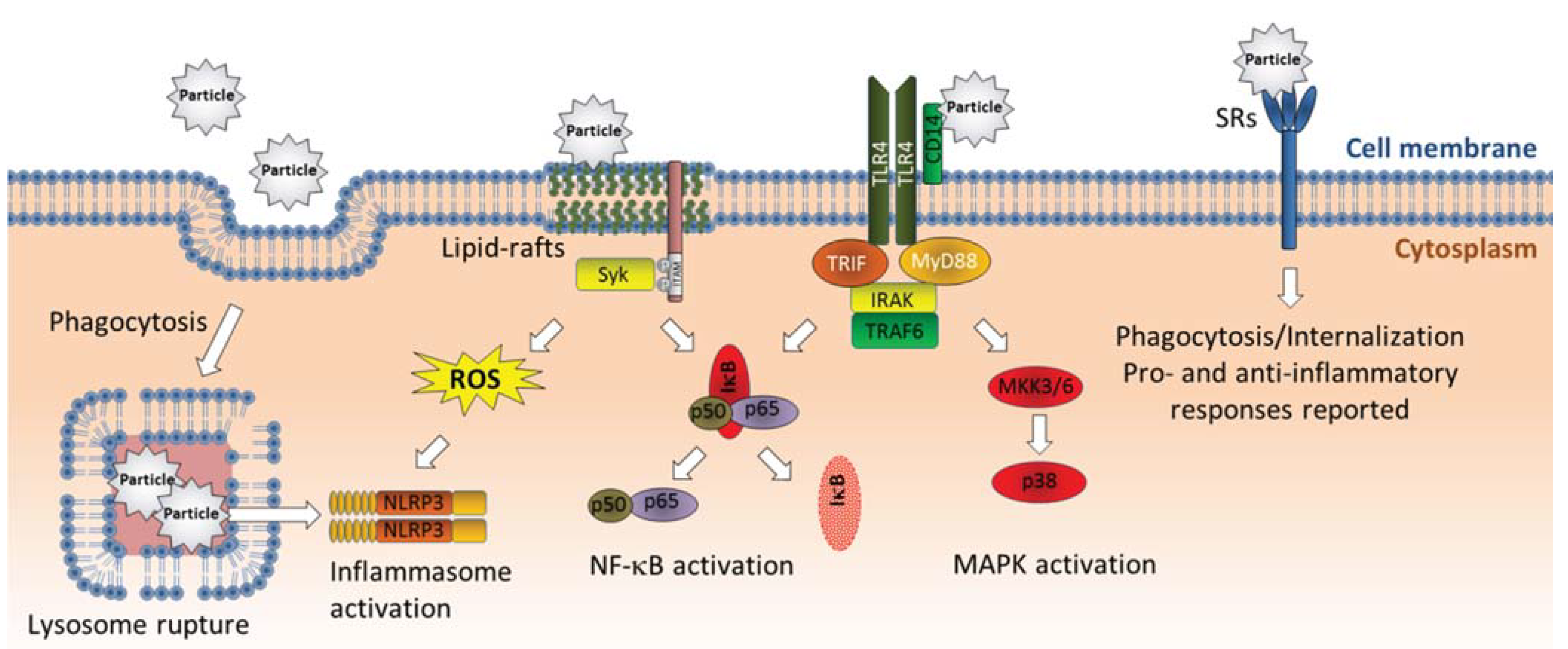
3.2. Interactions with Cellular Membranes

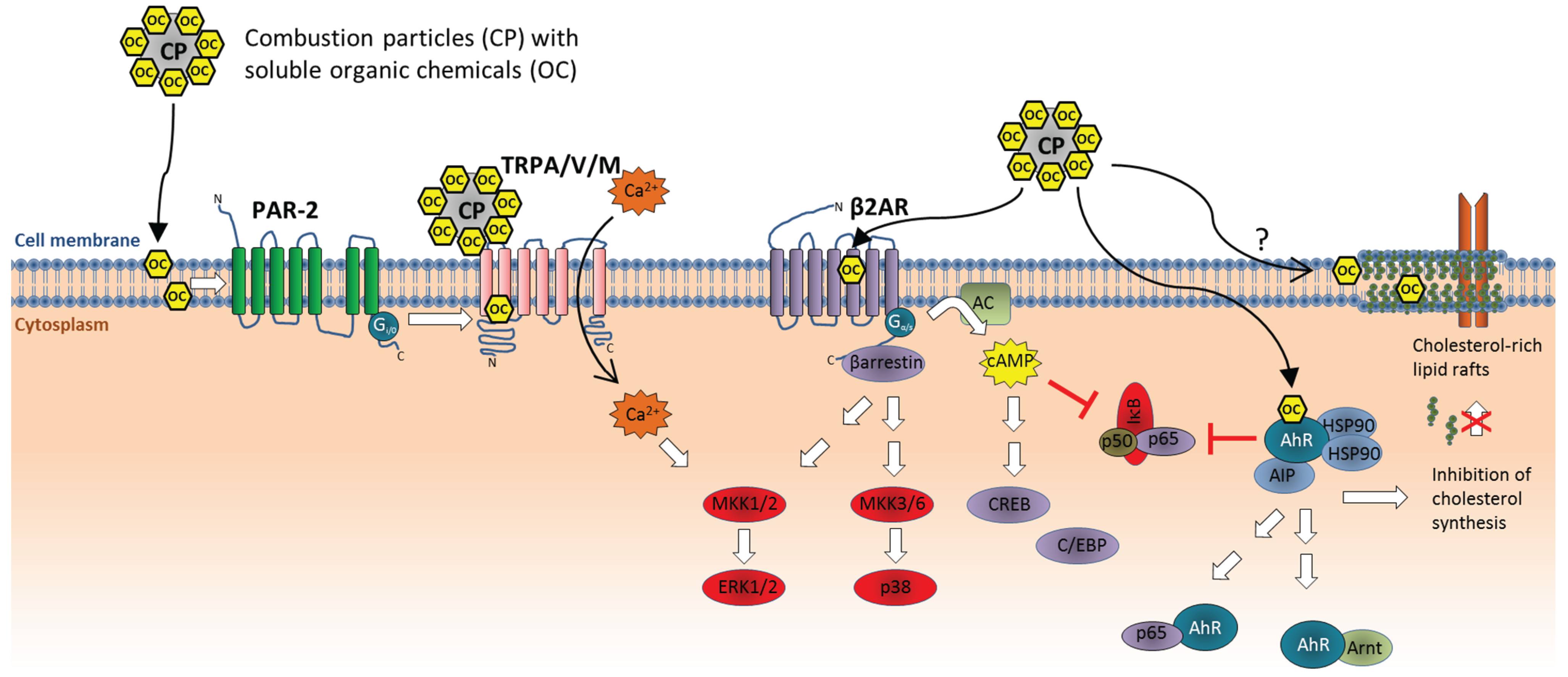
3.3. Activation of Cell Surface Receptors

3.4. Intracellular Molecular Targets
4. Central Signaling Pathways and Processes Involved in Particle-Induced Activation of Proinflammatory Genes
4.1. Role of Endogenous ROS Formation in Cellular Signaling and Inflammation


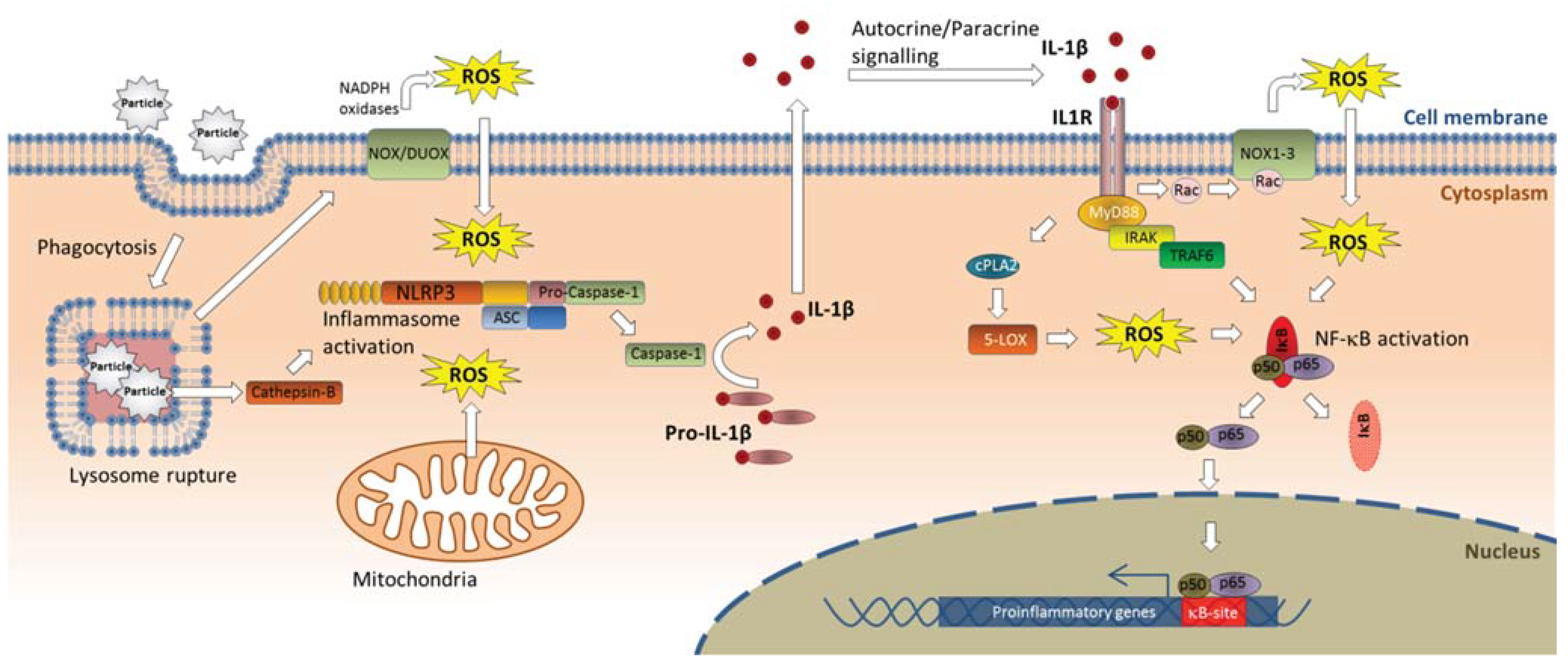
4.2. Inflammasome Activation—Linking Lysosomal Rupture to Proinflammatory Responses

4.3. The EGF Receptor—A Regulator of the Magnitude of Particle-Induced Inflammation?
4.4. Proinflammatory Transcription Factors Activated by Particle Exposure
4.5. Cytotoxicity as Triggering Mechanism for Proinflammatory Responses
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Borm, P.J. Particle toxicology: From coal mining to nanotechnology. Inhal. Toxicol. 2002, 14, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, K.; Seaton, A. A short history of the toxicology of inhaled particles. Part. Fibre Toxicol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Manning, C.B.; Vallyathan, V.; Mossman, B.T. Diseases caused by asbestos: Mechanisms of injury and disease development. Int. Immunopharmacol. 2002, 2, 191–200. [Google Scholar] [CrossRef]
- Kelly, F.J.; Fussell, J.C. Air pollution and airway disease. Clin. Exp. Allergy 2011, 41, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Grahame, T.J.; Klemm, R.; Schlesinger, R.B. Public health and components of particulate matter: The changing assessment of black carbon. J. Air Waste Manage. Assoc. 2014, 64, 620–660. [Google Scholar] [CrossRef]
- Kim, K.H.; Kabir, E.; Kabir, S. A review on the human health impact of airborne particulate matter. Environ. Int. 2015, 74, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, P.E.; Øvrevik, J.; Låg, M.; Refsnes, M.; Nafstad, P.; Hetland, R.B.; Dybing, E. Particulate matter properties and health effects: Consistency of epidemiological and toxicological studies. Hum. Exp. Toxicol. 2006, 25, 559–579. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Franklin, B.; Cascio, W.; Hong, Y.; Howard, G.; Lipsett, M.; Luepker, R.; Mittleman, M.; Samet, J.M.; Smith, S.C., Jr.; Tager, I. Air pollution and cardiovascular disease: A statement for healthcare professionals from the expert panel on population and prevention science of the American Heart Association. Circulation 2004, 109, 2655–2671. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Yue, P.; Deiuliis, J.A.; Lumeng, C.N.; Kampfrath, T.; Mikolaj, M.B.; Cai, Y.; Ostrowski, M.C.; Lu, B.; Parthasarathy, S.; et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 2009, 119, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yavar, Z.; Verdin, M.; Ying, Z.; Mihai, G.; Kampfrath, T.; Wang, A.; Zhong, M.; Lippmann, M.; Chen, L.C.; et al. Effect of early particulate air pollution exposure on obesity in mice: Role of p47phox. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garciduenas, L.; Solt, A.C.; Henriquez-Roldan, C.; Torres-Jardon, R.; Nuse, B.; Herritt, L.; Villarreal-Calderon, R.; Osnaya, N.; Stone, I.; Garcia, R.; et al. Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol. Pathol. 2008, 36, 289–310. [Google Scholar] [PubMed]
- Donaldson, K.; Borm, P.J.; Castranova, V.; Gulumian, M. The limits of testing particle-mediated oxidative stress in vitro in predicting diverse pathologies; relevance for testing of nanoparticles. Part. Fibre Toxicol. 2009, 6, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Holgate, S.T. Mechanisms of particulate matter toxicity. Clin. Exp. Allergy 1999, 29, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, K.; Stone, V.; Seaton, A.; MacNee, W. Ambient particle inhalation and the cardiovascular system: Potential mechanisms. Environ. Health Perspect. 2001, 109, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Diaz-Sanchez, D.; Li, N. The role of particulate pollutants in pulmonary inflammation and asthma: Evidence for the involvement of organic chemicals and oxidative stress. Curr. Opin. Pulm. Med. 2001, 7, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, K.; Tran, C.L. Inflammation caused by particles and fibers. Inhal. Toxicol. 2002, 14, 5–27. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xia, T.; Nel, A.E. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic. Biol. Med. 2008, 44, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Schins, R.P. Mechanisms of genotoxicity of particles and fibers. Inhal. Toxicol. 2002, 14, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.; Harder, V.; Ibald-Mulli, A.; Khandoga, A.; Koenig, W.; Krombach, F.; Radykewicz, R.; Stampfl, A.; Thorand, B.; Peters, A. Cardiovascular effects of fine and ultrafine particles. J. Aerosol. Med. 2005, 18, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Terzano, C.; di Stefano, F.; Conti, V.; Graziani, E.; Petroianni, A. Air pollution ultrafine particles: Toxicity beyond the lung. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 809–821. [Google Scholar] [PubMed]
- Grunig, G.; Marsh, L.M.; Esmaeil, N.; Jackson, K.; Gordon, T.; Reibman, J.; Kwapiszewska, G.; Park, S.H. Perspective: Ambient air pollution: Inflammatory response and effects on the lung’s vasculature. Pulm. Circ. 2014, 4, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Dittrich-Breiholz, O.; Holtmann, H.; Kracht, M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002, 72, 847–855. [Google Scholar] [PubMed]
- Kato, A.; Schleimer, R.P. Beyond inflammation: Airway epithelial cells are at the interface of innate and adaptive immunity. Curr. Opin. Immunol. 2007, 19, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Perkins, T.N.; Shukla, A.; Peeters, P.M.; Steinbacher, J.L.; Landry, C.C.; Lathrop, S.A.; Steele, C.; Reynaert, N.L.; Wouters, E.F.; Mossman, B.T. Differences in gene expression and cytokine production by crystalline vs. amorphous silica in human lung epithelial cells. Part. Fibre Toxicol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Merget, R.; Bauer, T.; Kupper, H.U.; Philippou, S.; Bauer, H.D.; Breitstadt, R.; Bruening, T. Health hazards due to the inhalation of amorphous silica. Arch. Toxicol. 2002, 75, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Maricq, M.M. Chemical characterization of particulate emissions from diesel engines: A review. Aerosol. Science 2007, 38, 1079–1118. [Google Scholar] [CrossRef]
- Totlandsdal, A.I.; Øvrevik, J.; Cochran, R.E.; Herseth, J.I.; Bølling, A.K.; Låg, M.; Schwarze, P.; Lilleaas, E.; Holme, J.A.; Kubatova, A. The occurrence of polycyclic aromatic hydrocarbons and their derivatives and the proinflammatory potential of fractionated extracts of diesel exhaust and wood smoke particles. J. Environ. Sci. Health. A Tox. Hazard. Subst. Environ. Eng. 2014, 49, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Bonvallot, V.; BaezaSquiban, A.; Baulig, A.; Brulant, S.; Boland, S.; Muzeau, F.; Barouki, R.; Marano, F. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am. J. Respir. Cell. Mol. Biol. 2001, 25, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, R.; Takano, H.; Inoue, K.I.; Ichinose, T.; Sadakane, K.; Yoshino, S.; Yamaki, K.; Yoshikawa, T.; Hayakawa, K. Components of diesel exhaust particles differentially affect Th1/Th2 response in a murine model of allergic airway inflammation. Clin. Exp. Allergy 2006, 36, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Yanagisawa, R.; Inoue, K. Components of diesel exhaust particles diversely enhance a variety of respiratory diseases related to infection or allergy: Extracted organic chemicals and the residual particles after extraction differently affect respiratory diseases. J. Clin. Biochem. Nutr. 2007, 40, 101–107. [Google Scholar] [CrossRef] [PubMed]
- De Marini, D.M.; Brooks, L.R.; Warren, S.H.; Kobayashi, T.; Gilmour, M.I.; Singh, P. Bioassay-directed fractionation and salmonella mutagenicity of automobile and forklift diesel exhaust particles. Environ. Health Perspect. 2004, 112, 814–819. [Google Scholar] [CrossRef]
- Devouassoux, G.; Saxon, A.; Metcalfe, D.D.; Prussin, C.; Colomb, M.G.; Brambilla, C.; Diaz-Sanchez, D. Chemical constituents of diesel exhaust particles induce IL-4 production and histamine release by human basophils. J. Allergy Clin. Immunol. 2002, 109, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Totlandsdal, A.I.; Herseth, J.I.; Bølling, A.K.; Kubatova, A.; Braun, A.; Cochran, R.E.; Refsnes, M.; Øvrevik, J.; Låg, M. Differential effects of the particle core and organic extract of diesel exhaust particles. Toxicol. Lett. 2012, 208, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Totlandsdal, A.I.; Låg, M.; Lilleaas, E.; Cassee, F.; Schwarze, P. Differential proinflammatory responses induced by diesel exhaust particles with contrasting PAH and metal content. Environ. Toxicol. 2013, 30, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hao, M.; Phalen, R.F.; Hinds, W.C.; Nel, A.E. Particulate air pollutants and asthma: A paradigm for the role of oxidative stress in PM-induced adverse health effects. Clin. Immunol. 2003, 109, 250–265. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Ding, M.; Chen, F.; Wang, L.; Rojanasakul, Y.; Vallyathan, V.; Castranova, V. Reactive oxygen species and molecular mechanism of silica-induced lung injury. J. Environ. Pathol. Toxicol. Oncol. 2001, 20, 85–93. [Google Scholar] [PubMed]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Rad. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef]
- Shukla, A.; Gulumian, M.; Hei, T.K.; Kamp, D.; Rahman, Q.; Mossman, B.T. Multiple roles of oxidants in the pathogenesis of asbestos-induced diseases. Free Rad. Biol. Med. 2003, 34, 1117–1129. [Google Scholar] [CrossRef]
- Donaldson, K.; Stone, V.; Borm, P.J.A.; Jimenez, L.A.; Gilmour, P.S.; Schins, R.P.F.; Knaapen, A.M.; Rahman, I.; Faux, S.P.; Brown, D.M.; et al. Oxidative stress and calcium signaling in the adverse effects of environmental particles (PM10). Free Radical Biol. Med. 2003, 34, 1369–1382. [Google Scholar] [CrossRef]
- Gonzalez-Flecha, B. Oxidant mechanisms in response to ambient air particles. Mol. Aspects. Med. 2004, 25, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Jacobsen, N.R.; Folkmann, J.K.; Danielsen, P.H.; Mikkelsen, L.; Hemmingsen, J.G.; Vesterdal, L.K.; Forchhammer, L.; Wallin, H.; Loft, S. Role of oxidative damage in toxicity of particulates. Free Radic. Res. 2010, 44, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Mazzoli-Rocha, F.; Fernandes, S.; Einicker-Lamas, M.; Zin, W.A. Roles of oxidative stress in signaling and inflammation induced by particulate matter. Cell. Biol. Toxicol. 2010, 26, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Somanathan, R. Nanoparticles: Toxicity, radicals, electron transfer, and antioxidants. Methods Mol. Biol. 2013, 1028, 15–35. [Google Scholar] [PubMed]
- Nel, A.; Xia, T.; Madler, L.; Li, N. Toxic potential of materials at the nanolevel. Science 2006, 311, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E. Implementation of alternative test strategies for the safety assessment of engineered nanomaterials. J. Intern. Med. 2013, 274, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Danielsen, P.H.; Karottki, D.G.; Jantzen, K.; Roursgaard, M.; Klingberg, H.; Jensen, D.M.; Christophersen, D.V.; Hemmingsen, J.G.; Cao, Y. Oxidative stress and inflammation generated DNA damage by exposure to air pollution particles. Mutat. Res. Rev. Mutat. Res. 2014, 762, 133–166. [Google Scholar] [CrossRef] [PubMed]
- Borm, P.J.; Kelly, F.; Kunzli, N.; Schins, R.P.; Donaldson, K. Oxidant generation by particulate matter: From biologically effective dose to a promising, novel metric. Occup. Environ. Med. 2007, 64, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Ayres, J.G.; Borm, P.; Cassee, F.R.; Castranova, V.; Donaldson, K.; Ghio, A.; Harrison, R.M.; Hider, R.; Kelly, F.; Kooter, I.M.; et al. Evaluating the toxicity of airborne particulate matter and nanoparticles by measuring oxidative stress potential—A workshop report and consensus statement. Inhal. Toxicol. 2008, 20, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.F., Jr.; Thakur, S.A.; Holian, A. Silica binding and toxicity in alveolar macrophages. Free Radic. Biol. Med. 2008, 44, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Shvedova, A.A.; Pietroiusti, A.; Fadeel, B.; Kagan, V.E. Mechanisms of carbon nanotube-induced toxicity: Focus on oxidative stress. Toxicol. Appl. Pharmacol. 2012, 261, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Venkatesan, M.I.; Miguel, A.; Kaplan, R.; Gujuluva, C.; Alam, J.; Nel, A. Induction of heme oxygenase-1 expression in macrophages by diesel exhaust particle chemicals and quinones via the antioxidant-responsive element. J. Immunol. 2000, 165, 3393–3401. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Charrier, J.G.; Kodani, S.D.; Vogel, C.F.; Kado, S.Y.; Anderson, D.S.; Anastasio, C.; Van Winkle, L.S. Combustion-derived flame generated ultrafine soot generates reactive oxygen species and activates Nrf2 antioxidants differently in neonatal and adult rat lungs. Part. Fibre Toxicol. 2013, 10, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Ohtoshi, T.; Kawasaki, S.; Kohyama, T.; Desaki, M.; Kasama, T.; Kobayashi, K.; Nakahara, K.; Yamamoto, K.; Matsushima, K.; et al. Diesel exhaust particles induce NF-kappa B activation in human bronchial epithelial cells in vitro: Importance in cytokine transcription. J. Immunol. 1999, 162, 4705–4711. [Google Scholar] [PubMed]
- Desaki, M.; Takizawa, H.; Kasama, T.; Kobayashi, K.; Morita, Y.; Yamamoto, K. Nucklear factor kappa b activation in silica-induced interleukin-8 production by human bronchial epithelial cells. Cytokine 2000, 12, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Shi, X.L.; Lu, Y.J.; Huang, C.S.; Leonard, S.; Roberts, J.; Antonini, J.; Castranova, V.; Vallyathan, V. Induction of activator protein-1 through reactive oxygen species by crystalline silica in JB6 cells. J. Biol. Chem. 2001, 276, 9108–9114. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Timblin, C.R.; Hubbard, A.K.; Bravman, J.; Mossman, B.T. Silica-induced activation of c-Jun-NH2-terminal amino kinases, protracted expression of the activator protein-1 proto-oncogene, fra-1, and S-phase alterations are mediated via oxidative stress. Cancer Res. 2001, 61, 1791–1795. [Google Scholar] [PubMed]
- Albrecht, C.; Borm, P.J.A.; Adolf, B.; Timblin, C.R.; Mossman, B.T. In vitro and in vivo activation of extracellular signal-regulated kinases by coal dusts and quartz silica. Toxicol. Appl. Pharmacol. 2002, 184, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Boland, S.; Baeza-Squiban, A.; Hamel, R.; Thomassen, L.C.; Martens, J.A.; Billon-Galland, M.A.; Fleury-Feith, J.; Moisan, F.; Pairon, J.C.; et al. Oxidative stress and proinflammatory effects of carbon black and titanium dioxide nanoparticles: role of particle surface area and internalized amount. Toxicology 2009, 260, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Weissenberg, A.; Sydlik, U.; Peuschel, H.; Schroeder, P.; Schneider, M.; Schins, R.P.; Abel, J.; Unfried, K. Reactive oxygen species as mediators of membrane-dependent signaling induced by ultrafine particles. Free Radic. Biol. Med. 2010, 49, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Hetland, R.B.; Myhre, O.; Låg, M.; Hongve, D.; Schwarze, P.E.; Refsnes, M. Importance of soluble metals and reactive oxygen species for cytokine release induced by mineral particles. Toxicology 2001, 165, 133–144. [Google Scholar] [CrossRef]
- Øvrevik, J.; Hetland, R.B.; Schins, R.P.; Myran, T.; Schwarze, P.E. Iron release and ROS generation from mineral particles are not related to cytokine release or apoptosis in exposed A549 cells. Toxicol. Lett. 2006, 165, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Vallyathan, V. Generation of oxygen radicals by minerals and its correlation to cytotoxicity. Environ. Health Perspect. 1994, 102, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Clouter, A.; Brown, D.; Hohr, D.; Borm, P.; Donaldson, K. Inflammatory effects of respirable quartz collected in workplaces versus standard DQ12 quartz: Particle surface correlates. Toxicol. Sci. 2001, 63, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, G.D.; Schins, R.P.; Shi, T.; Fenoglio, I.; Fubini, B.; Borm, P.J. In vitro genotoxicity assessment of commercial quartz flours in comparison to standard DQ12 quartz. Int. J. Hyg. Environ. Health 2004, 207, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, I.; Croce, A.; DiRenzo, F.; Tiozzo, R.; Fubini, B. Pure-silica zeolites (porosils) as model solids for the evaluation of the physicochemical features determining silica toxicity to macrophages. Chem. Res. Toxicol. 2000, 13, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Fubini, B.; Fenoglio, I.; Ceschino, R.; Ghiazza, M.; Martra, G.; Tomatis, M.; Borm, P.; Schins, R.; Bruch, J. Relationship between the state of the surface of four commercial quartz flours and their biological activity in vitro and in vivo. Int. J. Hyg. Environ. Health 2004, 207, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Veranth, J.M.; Kaser, E.G.; Veranth, M.M.; Koch, M.; Yost, G.S. Cytokine responses of human lung cells (BEAS-2B) treated with micron-sized and nanoparticles of metal oxides compared to soil dusts. Part. Fibre Toxicol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Monteiller, C.; Tran, L.; MacNee, W.; Faux, S.P.; Jones, A.D.; Miller, B.G.; Donaldson, K. The pro-inflammatory effects of low solubility low toxicity particles, nanoparticles and fine particles, on epithelial cells in vitro: The role of surface area. Occup. Environ. Med. 2007, 64, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Duffin, R.; Poland, C.; Daly, P.; Murphy, F.; Drost, E.; MacNee, W.; Stone, V.; Donaldson, K. Efficacy of simple short-term in vitro assays for predicting the potential of metal oxide nanoparticles to cause pulmonary inflammation. Environ. Health Perspect. 2009, 117, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Steenhof, M.; Gosens, I.; Strak, M.; Godri, K.J.; Hoek, G.; Cassee, F.R.; Mudway, I.S.; Kelly, F.J.; Harrison, R.M.; Lebret, E. In vitro toxicity of particulate matter (PM) collected at different sites in the Netherlands is associated with PM composition, size fraction and oxidative potential—the RAPTES project. Part. Fibre Toxicol. 2011, 8, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Su, S.; Jin, W.; Wang, B.; Li, N.; Shen, H.; Li, W.; Huang, Y.; Chen, H.; Zhang, Y.; et al. Characteristics and cellular effects of ambient particulate matter from Beijing. Environ. Pollut. 2014, 191, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Gerlofs-Nijland, M.E.; Totlandsdal, A.I.; Tzamkiozis, T.; Leseman, D.L.; Samaras, Z.; Låg, M.; Schwarze, P.; Ntziachristos, L.; Cassee, F.R. Cell toxicity and oxidative potential of engine exhaust particles: Impact of using particulate filter or biodiesel fuel blend. Environ. Sci. Technol. 2013, 47, 5931–5938. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Arlt, V.M.; Oya, E.; Nagy, E.; Mollerup, S.; Phillips, D.H.; Låg, M.; Holme, J.A. Differential effects of nitro-PAHs and amino-PAHs on cytokine and chemokine responses in human bronchial epithelial BEAS-2B cells. Toxicol. Appl. Pharmacol. 2010, 242, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Janssen, N.A.; Yang, A.; Strak, M.; Steenhof, M.; Hellack, B.; Gerlofs-Nijland, M.E.; Kuhlbusch, T.; Kelly, F.; Harrison, R.; Brunekreef, B. Oxidative potential of particulate matter collected at sites with different source characteristics. Sci. Total. Environ. 2014, 472, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Steenhof, M.; Janssen, N.A.; Strak, M.; Hoek, G.; Gosens, I.; Mudway, I.S.; Kelly, F.J.; Harrison, R.M.; Pieters, R.H.; Cassee, F.R.; et al. Air pollution exposure affects circulating white blood cell counts in healthy subjects: The role of particle composition, oxidative potential and gaseous pollutants—the RAPTES project. Inhal. Toxicol. 2014, 26, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Steenhof, M.; Mudway, I.S.; Gosens, I.; Hoek, G.; Godri, K.J.; Kelly, F.J.; Harrison, R.M.; Pieters, R.H.; Cassee, F.R.; Lebret, E.; et al. Acute nasal pro-inflammatory response to air pollution depends on characteristics other than particle mass concentration or oxidative potential: The RAPTES project. Occup. Environ. Med. 2013, 70, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Strak, M.; Hoek, G.; Godri, K.J.; Gosens, I.; Mudway, I.S.; van Oerle, R.; Spronk, H.M.; Cassee, F.R.; Lebret, E.; Kelly, F.J.; et al. Composition of PM affects acute vascular inflammatory and coagulative markers—the RAPTES project. PLoS ONE 2013, 8, e58944. [Google Scholar] [CrossRef] [PubMed]
- Strak, M.; Hoek, G.; Steenhof, M.; Kilinc, E.; Godri, K.J.; Gosens, I.; Mudway, I.S.; van Oerle, R.; Spronk, H.M.; Cassee, F.R.; et al. Components of ambient air pollution affect thrombin generation in healthy humans: The RAPTES project. Occup. Environ. Med. 2013, 70, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Strak, M.; Janssen, N.A.; Godri, K.J.; Gosens, I.; Mudway, I.S.; Cassee, F.R.; Lebret, E.; Kelly, F.J.; Harrison, R.M.; Brunekreef, B.; et al. Respiratory health effects of airborne particulate matter: the role of particle size, composition, and oxidative potential—the RAPTES project. Environ. Health Perspect. 2012, 120, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Janssen, N.A.; Strak, M.; Yang, A.; Hellack, B.; Kelly, F.J.; Kuhlbusch, T.A.; Harrison, R.M.; Brunekreef, B.; Cassee, F.R.; Steenhof, M.; et al. Associations between three specific a-cellular measures of the oxidative potential of particulate matter and markers of acute airway and nasal inflammation in healthy volunteers. Occup. Environ. Med. 2015, 72, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tonne, C.; Yanosky, J.D.; Beevers, S.; Wilkinson, P.; Kelly, F.J. PM mass concentration and PM oxidative potential in relation to carotid intima-media thickness. Epidemiology 2012, 23, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Canova, C.; Minelli, C.; Dunster, C.; Kelly, F.; Shah, P.L.; Caneja, C.; Tumilty, M.K.; Burney, P. PM10 oxidative properties and asthma and COPD. Epidemiology 2014, 25, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.D.; Margolis, J. Haemolytic activity of colloidal silica. Nature 1961, 189, 1010–1011. [Google Scholar] [CrossRef] [PubMed]
- Nash, T.; Allison, A.C.; Harington, J.S. Physico-chemical properties of silica in relation to its toxicity. Nature 1966, 210, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Stalder, K.; Stober, W. Haemolytic activity of suspensions of different silica modifications and inert dusts. Nature 1965, 207, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Macnab, G.; Harington, J.S. Haemolytic activity of asbestos and other mineral dusts. Nature 1967, 214, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Harington, J.S.; Birbeck, M. An examination of the cytotoxic effects of silica on macrophages. J. Exp. Med. 1966, 124, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Harington, J.S.; Allison, A.C. Lysosomal enzymes in relation to the toxicity of silica. Med. Lav. 1965, 56, 471–484. [Google Scholar] [PubMed]
- Sahai, N. Biomembrane phospholipid-oxide surface interactions: Crystal chemical and thermodynamic basis. J. Colloid Interface Sci. 2002, 252, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, G.; Rita, G.A. Molecular basis of gouty inflammation: interaction of monosodium urate crystals with lysosomes and liposomes. Nat. New Biol. 1972, 240, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C. Lysosomes and the toxicity of particulate pollutants. Arch. Intern. Med. 1971, 128, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Warheit, D.B.; Webb, T.R.; Colvin, V.L.; Reed, K.L.; Sayes, C.M. Pulmonary bioassay studies with nanoscale and fine-quartz particles in rats: toxicity is not dependent upon particle size but on surface characteristics. Toxicol. Sci. 2007, 95, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.S.; Duffin, R.; Bradley, M.; Megson, I.L.; MacNee, W.; Lee, J.K.; Jeong, J.; Donaldson, K. Predictive value of in vitro assays depends on the mechanism of toxicity of metal oxide nanoparticles. Part. Fibre Toxicol. 2013, 10, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Pavan, C.; Rabolli, V.; Tomatis, M.; Fubini, B.; Lison, D. Why does the hemolytic activity of silica predict its pro-inflammatory activity? Part. Fibre Toxicol. 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Refsnes, M.; Namork, E.; Becher, R.; Sandnes, D.; Schwarze, P.E.; Låg, M. Mechanisms of silica-induced IL-8 release from A549 cells: initial kinase-activation does not require EGFR activation or particle uptake. Toxicology 2006, 227, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Ng, G.; Sharma, K.; Ward, S.M.; Desrosiers, M.D.; Stephens, L.A.; Schoel, W.M.; Li, T.; Lowell, C.A.; Ling, C.C.; Amrein, M.W.; et al. Receptor-independent, direct membrane binding leads to cell-surface lipid sorting and Syk kinase activation in dendritic cells. Immunity 2008, 29, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, S.; Miyazaki, Y.; Yoshii, C.; Nakaya, M.; Ozaki, N.; Toda, S.; Kuroda, E.; Ishibashi, K.; Yasuda, T.; Natsuaki, Y.; et al. An ITAM-Syk-CARD9 signalling axis triggers contact hypersensitivity by stimulating IL-1 production in dendritic cells. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Flach, T.L.; Ng, G.; Hari, A.; Desrosiers, M.D.; Zhang, P.; Ward, S.M.; Seamone, M.E.; Vilaysane, A.; Mucsi, A.D.; Fong, Y.; et al. Alum interaction with dendritic cell membrane lipids is essential for its adjuvanticity. Nat. Med. 2011, 17, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Peuschel, H.; Sydlik, U.; Grether-Beck, S.; Felsner, I.; Stockmann, D.; Jakob, S.; Kroker, M.; Haendeler, J.; Gotic, M.; Bieschke, C.; et al. Carbon nanoparticles induce ceramide- and lipid raft-dependent signalling in lung epithelial cells: A target for a preventive strategy against environmentally-induced lung inflammation. Part. Fibre Toxicol. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Vallyathan, V.; Shi, X.; Castranova, V. Reactive oxygen species: Their relation to pneumoconiosis and carcinogenesis. Environ. Health Perspect. 1998, 106, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Leroueil, P.R.; Hong, S.; Mecke, A.; Baker, J.R., Jr.; Orr, B.G.; Banaszak Holl, M.M. Nanoparticle interaction with biological membranes: Does nanotechnology present a Janus face? Acc. Chem. Res. 2007, 40, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, M.S.; Samelko, L.; McAllister, K.; Jacobs, J.J.; Hallab, N.J. Increasing both CoCrMo-alloy particle size and surface irregularity induces increased macrophage inflammasome activation in vitro potentially through lysosomal destabilization mechanisms. J. Orthop. Res. 2013, 31, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Vaine, C.A.; Patel, M.K.; Zhu, J.; Lee, E.; Finberg, R.W.; Hayward, R.C.; Kurt-Jones, E.A. Tuning innate immune activation by surface texturing of polymer microparticles: The role of shape in inflammasome activation. J. Immunol. 2013, 190, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Tekpli, X.; Holme, J.A.; Sergent, O.; Lagadic-Gossmann, D. Role for membrane remodeling in cell death: Implication for health and disease. Toxicology 2013, 304, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Tekpli, X.; Holme, J.A.; Sergent, O.; Lagadic-Gossmann, D. Importance of plasma membrane dynamics in chemical-induced carcinogenesis. Recent Pat. Anticancer Drug Discov. 2011, 6, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Liland, N.S.; Simonsen, A.C.; Duelund, L.; Torstensen, B.E.; Berntssen, M.H.; Mouritsen, O.G. Polyaromatic hydrocarbons do not disturb liquid-liquid phase coexistence, but increase the fluidity of model membranes. Chem. Phys. Lipids 2014, 184, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Korchowiec, B.; Corvis, Y.; Viitala, T.; Feidt, C.; Guiavarch, Y.; Corbier, C.; Rogalska, E. Interfacial approach to polyaromatic hydrocarbon toxicity: Phosphoglyceride and cholesterol monolayer response to phenantrene, anthracene, pyrene, chrysene, and benzo[a]pyrene. J. Phys. Chem. B 2008, 112, 13518–13531. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Dolganiuc, A.; Dai, Q.; Pruett, S.B. TLR4, ethanol, and lipid rafts: A new mechanism of ethanol action with implications for other receptor-mediated effects. J. Immunol. 2007, 178, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Resnick, D.; Freedman, N.J.; Xu, S.; Krieger, M. Secreted extracellular domains of macrophage scavenger receptors form elongated trimers which specifically bind crocidolite asbestos. J. Biol. Chem. 1993, 268, 3538–3545. [Google Scholar] [PubMed]
- Kobzik, L. Lung macrophage uptake of unopsonized environmental particulates: Role of scavenger-type receptors. J. Immunol. 1995, 155, 367–376. [Google Scholar] [PubMed]
- Iyer, R.; Hamilton, R.F.; Li, L.; Holian, A. Silica-induced apoptosis mediated via scavenger receptor in human alveolar macrophages. Toxicol. Appl. Pharmacol. 1996, 141, 84–92. [Google Scholar] [CrossRef]
- Chao, S.L.; Hamilton, R.F.; Pau, J.C.; Holian, A. Cell surface regulation of silica-induced apoptosis by the SR-A scavenger receptor in a murine lung macrophage cell line (MH-S). Toxicol. Appl. Pharmacol. 2001, 174, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.M.; Rich, A.; Krieger, M. Polynucleotide binding to macrophage scavenger receptors depends on the formation of base-quartet-stabilized four-stranded helices. J. Biol. Chem. 1993, 268, 3546–3554. [Google Scholar] [PubMed]
- Peiser, L.; Mukhopadhyay, S.; Gordon, S. Scavenger receptors in innate immunity. Curr. Opin. Immunol. 2002, 14, 123–128. [Google Scholar] [CrossRef]
- Biswas, R.; Hamilton, R.F., Jr.; Holian, A. Role of lysosomes in silica-induced inflammasome activation and inflammation in absence of MARCO. J. Immunol. Res. 2014, 2014, e304180. [Google Scholar] [CrossRef] [PubMed]
- Beamer, C.A.; Holian, A. Silica suppresses Toll-like receptor ligand-induced dendritic cell activation. FASEB J. 2008, 22, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Swindle, E.J.; Kushnir-Sukhov, N.M.; Holian, A.; Metcalfe, D.D. Silica-directed mast cell activation is enhanced by scavenger receptors. Am. J. Respir. Cell. Mol. Biol. 2007, 36, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Zanella, C.L.; Timblin, C.R.; Cummins, A.; Jung, M.; Goldberg, J.; Raabe, R.; Tritton, T.R.; Mossman, B.T. Asbestos-induced phosphorylation of epidermal growth factor receptor is linked to c-fos and apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 277, L684–L693. [Google Scholar]
- Zanella, C.L.; Posada, J.; Tritton, T.R.; Mossman, B.T. Asbestos causes stimulation of the extracellular signal-regulated kinase 1 mitogen-activated protein kinase cascade after phosphorylation of the epidermal growth factor receptor. Cancer Res. 1996, 56, 5334–5338. [Google Scholar] [PubMed]
- Wu, W.; Samet, J.M.; Ghio, A.J.; Devlin, R.B. Activation of the EGF receptor signaling pathway in airway epithelial cells exposed to Utah Valley PM. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L483–L489. [Google Scholar] [PubMed]
- Scapoli, L.; Ramos-Nino, M.E.; Martinelli, M.; Mossman, B.T. Src-dependent ERK5 and Src/EGFR-dependent ERK1/2 activation is required for cell proliferation by asbestos. Oncogene 2004, 23, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, S.; Ramgolam, K.; Baulig, A.; Marano, F.; Baeza-Squiban, A. Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2004, 30, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Refsnes, M.; Totlandsdal, A.I.; Holme, J.A.; Schwarze, P.E.; Låg, M. TACE/TGF-alpha/EGFR regulates CXCL8 in bronchial epithelial cells exposed to particulate matter components. Eur. Respir. J. 2011, 38, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Skuland, T.; Øvrevik, J.; Låg, M.; Schwarze, P.; Refsnes, M. Silica nanoparticles induce cytokine responses in lung epithelial cells through activation of a p38/TACE/TGF-alpha/EGFR-pathway and NF-kappaBeta signalling. Toxicol. Appl. Pharmacol. 2014, 279, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Parnia, S.; Hamilton, L.M.; Puddicombe, S.M.; Holgate, S.T.; Frew, A.J.; Davies, D.E. Autocrine ligands of the epithelial growth factor receptor mediate inflammatory responses to diesel exhaust particles. Respir. Res. 2014, 15, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.; Ma, H.; Viriyakosol, S.; Terkeltaub, R.; Liu-Bryan, R. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J. Immunol. 2006, 177, 6370–6378. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, B.; Oortgiesen, M.; Carter, J.D.; Devlin, R.B. Particulate matter initiates inflammatory cytokine release by activation of capsaicin and acid receptors in a human bronchial epithelial cell line. Toxicol. Appl. Pharmacol. 1999, 154, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, B.; Oortgiesen, M.; Roy, J.; Carter, J.D.; Simon, S.A.; Gavett, S.H. Vanilloid (capsaicin) receptors influence inflammatory sensitivity in response to particulate matter. Toxicol. Appl. Pharmacol. 2000, 169, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, B.; Haar, C.D.; Lee, L.; Oortgiesen, M. The surface charge of visible particulate matter predicts biological activation in human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 2002, 178, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kanju, P.; Patterson, M.; Chew, W.L.; Cho, S.H.; Gilmour, I.; Oliver, T.; Yasuda, R.; Ghio, A.; Simon, S.A.; Liedtke, W. TRPV4-Mediated Calcium Influx into Human Bronchial Epithelia upon Exposure to Diesel Exhaust Particles. Environ. Health Perspect. 2011, 119, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Fariss, M.W.; Gilmour, M.I.; Reilly, C.A.; Liedtke, W.; Ghio, A.J. Emerging mechanistic targets in lung injury induced by combustion-generated particles. Toxicol. Sci. 2013, 132, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; le Ferrec, E.; Lagadic-Gossmann, D.; Fardel, O. Aryl hydrocarbon receptor-independent up-regulation of intracellular calcium concentration by environmental polycyclic aromatic hydrocarbons in human endothelial HMEC-1 cells. Environ. Toxicol. 2012, 27, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; Levoin, N.; Paris, H.; N'Diaye, M.; Courtois, A.; Uriac, P.; Lagadic-Gossmann, D.; Fardel, O.; Le, F.E. Induction of intracellular calcium concentration by environmental benzo(a)pyrene involves a beta2-adrenergic receptor/adenylyl cyclase/Epac-1/inositol 1,4,5-trisphosphate pathway in endothelial cells. J. Biol. Chem. 2012, 287, 4041–4052. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; Le, F.E.; Holme, J.A.; Fardel, O.; Lagadic-Gossmann, D.; Øvrevik, J. Calcium signaling and beta2-adrenergic receptors regulate 1-nitropyrene induced CXCL8 responses in BEAS-2B cells. Toxicol. in Vitro 2014, 28, 1153–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhang, Y.; Guo, Y.; Zhang, Y.; Xu, M.; He, B. beta2-Adrenoceptor involved in smoking-induced airway mucus hypersecretion through beta-arrestin-dependent signaling. PLoS ONE 2014, 9, e97788. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Haas, J.; Panettieri, R.A., Jr.; Johnson, M.; Johnston, S.L. Corticosteroids and beta2 agonists differentially regulate rhinovirus-induced interleukin-6 via distinct cis-acting elements. J. Biol. Chem. 2007, 282, 15366–15375. [Google Scholar] [CrossRef] [PubMed]
- Holden, N.; Rider, C.F.; Bell, M.J.; Velayudhan, J.; King, E.M.; Kaur, M.; Salmon, M.; Giembycz, M.A.; Newton, R. Enhancement of inflammatory mediator release by beta(2)-adrenoceptor agonists in airway epithelial cells is reversed by glucocorticoid action. Br. J. Pharmacol. 2010, 160, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Karoly, E.D.; Dailey, L.A.; Schmitt, M.T.; Silbajoris, R.; Graff, D.W.; Devlin, R.B. Comparison of gene expression profiles induced by coarse, fine, and ultrafine particulate matter. J. Toxicol. Environ. Health A 2011, 74, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Monn, C.; Becker, S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10–2.5) in outdoor and indoor air. Toxicol. Appl. Pharmacol. 1999, 155, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Osornio-Vargas, A.R.; Bonner, J.C.; Alfaro-Moreno, E.; Martinez, L.; Garcia-Cuellar, C.; Ponce-de-Leon, R.S.; Miranda, J.; Rosas, I. Proinflammatory and cytotoxic effects of Mexico City air pollution particulate matter in vitro are dependent on particle size and composition. Environ. Health Perspect. 2003, 111, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Schins, R.P.F.; Lightbody, J.H.; Borm, P.J.A.; Shi, T.; Donaldson, K.; Stone, V. Inflammatory effects of coarse and fine particulate matter in relation to chemical and biological constituents. Toxicol. Appl. Pharmacol. 2004, 195, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Dailey, L.; Soukup, J.M.; Silbajoris, R.; Devlin, R.B. TLR-2 is involved in airway epithelial cell response to air pollution particles. Toxicol. Appl. Pharmacol. 2005, 203, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, M.; Øvrevik, J.; Holme, J.A.; Perrone, M.G.; Bolzacchini, E.; Schwarze, P.E.; Camatini, M. Differences in cytotoxicity vs. pro-inflammatory potency of different PM fractions in human epithelial lung cells. Toxicol. In Vitro 2010, 24, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Steerenberg, P.A.; Withagen, C.E.T.; van Dalen, W.J.; Dormans, J.A.M.A.; Cassee, F.R.; Heisterkamp, S.H.; van Loveren, H. Adjuvant activity of ambient particulate matter of different sites, sizes, and seasons in a respiratory allergy mouse model. Toxicol. Appl. Pharmacol. 2004, 200, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Hetland, R.; Cassee, F.; Låg, M.; Refsnes, M.; Dybing, E.; Schwarze, P. Cytokine release from alveolar macrophages exposed to ambient particulate matter: Heterogeneity in relation to size, city and season. Part. Fibre Toxicol. 2005, 2, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.H.; Chhowalla, M.; Iqbal, Z.; Sesti, F. Single-walled carbon nanotubes are a new class of ion channel blockers. J. Biol. Chem. 2003, 278, 50212–50216. [Google Scholar] [CrossRef] [PubMed]
- Sargent, L.M.; Shvedova, A.A.; Hubbs, A.F.; Salisbury, J.L.; Benkovic, S.A.; Kashon, M.L.; Lowry, D.T.; Murray, A.R.; Kisin, E.R.; Friend, S.; et al. Induction of aneuploidy by single-walled carbon nanotubes. Environ. Mol. Mutagen. 2009, 50, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Holt, B.D.; Short, P.A.; Rape, A.D.; Wang, Y.L.; Islam, M.F.; Dahl, K.N. Carbon nanotubes reorganize actin structures in cells and ex vivo. ACS Nano 2010, 4, 4872–4878. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Sioutas, C.; Cho, A.; Schmitz, D.; Misra, C.; Sempf, J.; Wang, M.; Oberley, T.; Froines, J.; Nel, A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 2003, 111, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Totlandsdal, A.I.; Cassee, F.R.; Schwarze, P.; Refsnes, M.; Låg, M. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells. Part. Fibre Toxicol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Andrysik, Z.; Vondracek, J.; Marvanova, S.; Ciganek, M.; Neca, J.; Pencikova, K.; Mahadevan, B.; Topinka, J.; Baird, W.M.; Kozubik, A.; et al. Activation of the aryl hydrocarbon receptor is the major toxic mode of action of an organic extract of a reference urban dust particulate matter mixture: The role of polycyclic aromatic hydrocarbons. Mutat. Res. 2011, 714, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Fardel, O. Cytokines as molecular targets for aryl hydrocarbon receptor ligands: Implications for toxicity and xenobiotic detoxification. Expert Opin. Drug Metab. Toxicol. 2013, 9, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di, M.P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Sciullo, E.; Wong, P.; Kuzmicky, P.; Kado, N.; Matsumura, F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ. Health Perspect. 2005, 113, 1536–1541. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.S.; Vogel, C.F.; Kokosinski, K.; Matsumura, F. Arylhydrocarbon receptor activation in NCI-H441 cells and C57BL/6 mice: Possible mechanisms for lung dysfunction. Am. J. Respir. Cell. Mol. Biol. 2010, 42, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Låg, M.; Lecureur, V.; Gilot, D.; Lagadic-Gossmann, D.; Refsnes, M.; Schwarze, P.; Skuland, T.; Becher, R.; Holme, J. AhR and Arnt differentially regulate NF-kappaB signaling and chemokine responses in human bronchial epithelial cells. Cell. Commun. Signal. 2014, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung, H.H.; Lame, M.W.; Gohil, K.; He, G.; Denison, M.S.; Rutledge, J.C.; Wilson, D.W. Comparative gene responses to collected ambient particles in vitro: Endothelial responses. Physiol. Genomics 2011, 43, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Salcido-Neyoy, M.E.; Sanchez-Perez, Y.; Osornio-Vargas, A.R.; Gonsebatt, M.E.; Melendez-Zajgla, J.; Morales-Barcenas, R.; Petrosyan, P.; Molina-Servin, E.D.; Vega, E.; Manzano-Leon, N.; et al. Induction of c-Jun by air particulate matter (PM10) of Mexico city: Participation of polycyclic aromatic hydrocarbons. Environ. Pollut. 2015, 203, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Rabson, A.B.; Gallo, M.A. Ah receptor and NF-kappaB interactions: Mechanisms and physiological implications. Chem. Biol. Interact. 2002, 141, 97–115. [Google Scholar] [CrossRef]
- N'Diaye, M.; Le, F.E.; Lagadic-Gossmann, D.; Corre, S.; Gilot, D.; Lecureur, V.; Monteiro, P.; Rauch, C.; Galibert, M.D.; Fardel, O. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J. Biol. Chem. 2006, 281, 19906–19915. [Google Scholar] [CrossRef] [PubMed]
- Podechard, N.; Lecureur, V.; Le Ferrec, E.; Guenon, I.; Sparfel, L.; Gilot, D.; Gordon, J.R.; Lagente, V.; Fardel, O. Interleukin-8 induction by the environmental contaminant benzo(a)pyrene is aryl hydrocarbon receptor-dependent and leads to lung inflammation. Toxicol. Lett. 2008, 177, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Matsumura, F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem. Pharmacol. 2009, 77, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Sciullo, E.; Matsumura, F. Involvement of RelB in aryl hydrocarbon receptor-mediated induction of chemokines. Biochem. Biophys. Res. Commun. 2007, 363, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.W.; Duckett, C.S. The aryl hydrocarbon nuclear translocator alters CD30-mediated NF-kappaB-dependent transcription. Science 2009, 323, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Chang, H.; Chang, J.T.; Lin, P. Aryl hydrocarbon receptor in association with RelA modulates IL-6 expression in non-smoking lung cancer. Oncogene 2012, 31, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, T.H.; Maggirwar, S.B.; Baglole, C.J.; Lakatos, H.F.; Gasiewicz, T.A.; Phipps, R.P.; Sime, P.J. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am. J. Pathol. 2007, 170, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Naka, T.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med. 2009, 206, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Beamer, C.A.; Seaver, B.P.; Shepherd, D.M. Aryl hydrocarbon receptor (AhR) regulates silica-induced inflammation but not fibrosis. Toxicol. Sci. 2012, 126, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Dorrie, A.; Schmitz, M.L.; Hoffmann, E.; Resch, K.; Kracht, M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 2004, 279, 55633–55643. [Google Scholar] [PubMed]
- Suh, J.; Jeon, Y.J.; Kim, H.M.; Kang, J.S.; Kaminski, N.E.; Yang, K.H. Aryl hydrocarbon receptor-dependent inhibition of AP-1 activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin in activated B cells. Toxicol. Appl. Pharmacol. 2002, 181, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Li, W.; Lok, P.; Matsumura, F.; Vogel, C.F. AhR deficiency impairs expression of LPS-induced inflammatory genes in mice. Biochem. Biophys. Res. Commun. 2011, 410, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Ple, C.; Fan, Y.; Ait, Y.S.; Vorng, H.; Everaere, L.; Chenivesse, C.; Balsamelli, J.; Azzaoui, I.; de Nadai, P.; Wallaert, B.; et al. Polycyclic aromatic hydrocarbons reciprocally regulate IL-22 and IL-17 cytokines in peripheral blood mononuclear cells from both healthy and asthmatic subjects. PLoS ONE 2015, 10, e0122372. [Google Scholar] [CrossRef] [PubMed]
- Van Zijverden, M.; van der Pijl, A.; Bol, M.; van Pinxteren, F.A.; de Haar, C.; Penninks, A.H.; van Loveren, H.; Pieters, R. Diesel exhaust, carbon black, and silica particles display distinct Th1/Th2 modulating activity. Toxicol. Appl. Pharmacol. 2000, 168, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Fahy, O.; Senechal, S.; Pene, J.; Scherpereel, A.; Lassalle, P.; Tonnel, A.B.; Yssel, H.; Wallaert, B.; Tsicopoulos, A. Diesel exposure favors Th2 cell recruitment by mononuclear cells and alveolar macrophages from allergic patients by differentially regulating macrophage-derived chemokine and IFN-{gamma}-induced protein-10 production. J. Immunol. 2002, 168, 5912–5919. [Google Scholar] [CrossRef] [PubMed]
- Ferecatu, I.; Borot, M.C.; Bossard, C.; Leroux, M.; Boggetto, N.; Marano, F.; Baeza-Squiban, A.; Andreau, K. Polycyclic aromatic hydrocarbon components contribute to the mitochondria-antiapoptotic effect of fine particulate matter on human bronchial epithelial cells via the aryl hydrocarbon receptor. Part. Fibre Toxicol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Milnerowicz, H.; Sciskalska, M.; Dul, M. Pro-inflammatory effects of metals in persons and animals exposed to tobacco smoke. J. Trace Elem. Med. Biol. 2015, 29, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Lippmann, M. Effects of metals within ambient air particulate matter (PM) on human health. Inhal. Toxicol. 2009, 21, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Samet, J.M.; Graves, L.M.; Quay, J.; Dailey, L.A.; Devlin, R.B.; Ghio, A.J.; Wu, W.; Bromberg, P.A.; Reed, W. Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 275, L551–L558. [Google Scholar]
- De Moor, J.M.; Koropatnick, D.J. Metals and cellular signaling in mammalian cells. Cell. Mol. Biol. 2000, 46, 367–381. [Google Scholar]
- Kim, Y.M.; Reed, W.; Wu, W.; Bromberg, P.A.; Graves, L.M.; Samet, J.M. Zn2+-induced IL-8 expression involves AP-1, JNK, and ERK activities in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1028–L1035. [Google Scholar] [CrossRef] [PubMed]
- Ansteinsson, V.; Refsnes, M.; Skomedal, T.; Osnes, J.B.; Schiander, I.; Låg, M. Zinc- and copper-induced interleukin-6 release in primary cell cultures from rat heart. Cardiovasc. Toxicol. 2009, 9, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.S.; Harris, G.K.; Shi, X. Metal-induced oxidative stress and signal transduction. Free Radic. Biol. Med. 2004, 37, 1921–1942. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Singh, S.; Siddiqi, N.J. Biomedical implications of heavy metals induced imbalances in redox systems. Biomed. Res. Int. 2014, 2014, e640754. [Google Scholar] [CrossRef] [PubMed]
- Samet, J.M.; Silbajoris, R.; Wu, W.; Graves, L.M. Tyrosine phosphatases as targets in metal-induced signaling in human airway epithelial cells. Am. J. Respir. Cell. Mol. Biol. 1999, 21, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Fauman, E.B.; Saper, M.A. Structure and function of the protein tyrosine phosphatases. Trends Biochem. Sci. 1996, 21, 413–417. [Google Scholar] [CrossRef]
- Hao, Q.; Maret, W. Aldehydes release zinc from proteins: A pathway from oxidative stress/lipid peroxidation to cellular functions of zinc. FEBS J. 2006, 273, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Protein tyrosine phosphatases as targets of the combined insulinomimetic effects of zinc and oxidants. Biometals 2005, 18, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Tal, T.L.; Graves, L.M.; Silbajoris, R.; Bromberg, P.A.; Wu, W.; Samet, J.M. Inhibition of protein tyrosine phosphatase activity mediates epidermal growth factor receptor signaling in human airway epithelial cells exposed to Zn(2+). Toxicol. Appl. Pharmacol. 2006, 214, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Samet, J.M.; Dewar, B.J.; Wu, W.; Graves, L.M. Mechanisms of Zn2+-induced signal initiation through the epidermal growth factor receptor. Toxicol. Appl. Pharmacol. 2003, 191, 86–93. [Google Scholar] [CrossRef]
- Wu, W.; Bromberg, P.A.; Samet, J.M. Zinc ions as effectors of environmental oxidative lung injury. Free Radic. Biol. Med. 2013, 65, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [PubMed]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Torres, M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am. J. Respir. Crit. Care Med. 2002, 166, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Yang, S.R.; Kode, A.; Rajendrasozhan, S.; Caito, S.; Adenuga, D.; Henry, R.; Edirisinghe, I.; Rahman, I. Redox regulation of lung inflammation: Role of NADPH oxidase and NF-kappaB signalling. Biochem. Soc. Trans. 2007, 35, 1151–1155. [Google Scholar] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Ammendola, R.; Faraonio, R.; Russo, T.; Cimino, F. Redox control of signal transduction, gene expression and cellular senescence. Neurochem. Res. 2004, 29, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Touyz, R.M. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31, S170–S180. [Google Scholar] [CrossRef] [PubMed]
- Petry, A.; Weitnauer, M.; Gorlach, A. Receptor activation of NADPH oxidases. Antioxid. Redox Signal. 2010, 13, 467–487. [Google Scholar] [CrossRef] [PubMed]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Polito, L.; Bolognesi, A. Xanthine oxidoreductase in atherosclerosis pathogenesis: Not only oxidative stress. Atherosclerosis 2014, 237, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Vascular signaling through G protein-coupled receptors: New concepts. Curr. Opin. Nephrol. Hypertens. 2009, 18, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Jyrkkanen, H.K.; Levonen, A.L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 2012, 52, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.L.; Kunimoto, M.; Patel, J.A. Autocrine regulation and experimental modulation of interleukin-6 expression by human pulmonary epithelial cells infected with respiratory syncytial virus. J. Virol. 1998, 72, 2496–2499. [Google Scholar] [PubMed]
- Patel, J.A.; Jiang, Z.; Nakajima, N.; Kunimoto, M. Autocrine regulation of interleukin-8 by interleukin-1alpha in respiratory syncytial virus-infected pulmonary epithelial cells in vitro. Immunology 1998, 95, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.G.; Johnston, C.; Oberdorster, G.; Finkelstein, J.N. Silica-induced chemokine expression in alveolar type II cells is mediated by TNF-alpha-induced oxidant stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, L979–L988. [Google Scholar]
- Vila-del, S.V.; Fresno, M. Involvement of TNF and NF-kappa B in the transcriptional control of cyclooxygenase-2 expression by IFN-gamma in macrophages. J. Immunol. 2005, 174, 2825–2833. [Google Scholar] [CrossRef]
- Totlandsdal, A.I.; Refsnes, M.; Låg, M. Mechanisms involved in ultrafine carbon black-induced release of IL-6 from primary rat epithelial lung cells. Toxicol. in Vitro 2010, 24, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.K.; Ye, R.D.; Christiansen, S.C.; Jagels, M.A.; Bokoch, G.M.; Zuraw, B.L. Role of the Rho GTPase in bradykinin-stimulated nuclear factor-kappaB activation and IL-1beta gene expression in cultured human epithelial cells. J. Immunol. 1998, 160, 3038–3045. [Google Scholar] [PubMed]
- Haddad, J.J.; Saade, N.E.; Safieh-Garabedian, B. Redox regulation of TNF-alpha biosynthesis: augmentation by irreversible inhibition of gamma-glutamylcysteine synthetase and the involvement of an IkappaB-alpha/NF-kappaB-independent pathway in alveolar epithelial cells. Cell. Signal. 2002, 14, 211–218. [Google Scholar] [CrossRef]
- Haddad, J.J.; Land, S.C. Redox signaling-mediated regulation of lipopolysaccharide-induced proinflammatory cytokine biosynthesis in alveolar epithelial cells. Antioxid. Redox Signal. 2002, 4, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Dickerson, R.; Khanna, S.; Collard, E.; Gnyawali, U.; Gordillo, G.M.; Sen, C.K. Particulate beta-glucan induces TNF-alpha production in wound macrophages via a redox-sensitive NF-kappabeta-dependent pathway. Wound Repair Regener. 2011, 19, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Gabelloni, M.L.; Sabbione, F.; Jancic, C.; Fuxman, B.J.; Keitelman, I.; Iula, L.; Oleastro, M.; Geffner, J.R.; Trevani, A.S. NADPH oxidase derived reactive oxygen species are involved in human neutrophil IL-1beta secretion but not in inflammasome activation. Eur. J. Immunol. 2013, 43, 3324–3335. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid beta induces IL-1beta processing via production of ROS: implication in Alzheimer’s disease. Cell. Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Oliver, P.; Lancaster, J.R., Jr.; Schwarzenberger, P.O.; Joshi, M.S.; Cork, J.; Kolls, J.K. Reactive oxygen species mediate tumor necrosis factor alpha-converting, enzyme-dependent ectodomain shedding induced by phorbol myristate acetate. FASEB J. 2001, 15, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Trifilieff, A.; Walker, C.; Keller, T.; Kottirsch, G.; Neumann, U. Pharmacological profile of PKF242-484 and PKF241-466, novel dual inhibitors of TNF-alpha converting enzyme and matrix metalloproteinases, in models of airway inflammation. Br. J. Pharmacol. 2002, 135, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Pietri, M.; Schneider, B.; Mouillet-Richard, S.; Ermonval, M.; Mutel, V.; Launay, J.M.; Kellermann, O. Reactive oxygen species-dependent TNF-alpha converting enzyme activation through stimulation of 5-HT2B and alpha1D autoreceptors in neuronal cells. FASEB J. 2005, 19, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Piette, J.; Schoonbroodt, S.; Greimers, R.; Havard, L.; Merville, M.P.; Bours, V. Reactive oxygen intermediate-dependent NF-kappaB activation by interleukin-1beta requires 5-lipoxygenase or NADPH oxidase activity. Mol. Cell. Biol. 1999, 19, 1950–1960. [Google Scholar] [PubMed]
- Deshpande, S.S.; Angkeow, P.; Huang, J.; Ozaki, M.; Irani, K. Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: Dual regulation by reactive oxygen species. FASEB J. 2000, 14, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Schumacker, P.T.; Arch, R.H. Reactive oxygen species are downstream products of TRAF-mediated signal transduction. J. Biol. Chem. 2001, 276, 42728–42736. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Harraz, M.M.; Zhou, W.; Zhang, L.N.; Ding, W.; Zhang, Y.; Eggleston, T.; Yeaman, C.; Banfi, B.; Engelhardt, J.F. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol. Cell. Biol. 2006, 26, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Spencer, N.Y.; Oakley, F.D.; Buettner, G.R.; Engelhardt, J.F. Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxid. Redox Signal. 2009, 11, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, T.; Nadel, J.A.; Ueki, I.F.; Koff, J.L.; Shao, M.X. Regulation of interleukin-8 via an airway epithelial signaling cascade. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1289–L1296. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.X.; Nadel, J.A. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc. Natl. Acad. Sci. USA 2005, 102, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide: Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [PubMed]
- DeYulia, G.J., Jr.; Carcamo, J.M. EGF receptor-ligand interaction generates extracellular hydrogen peroxide that inhibits EGFR-associated protein tyrosine phosphatases. Biochem. Biophys. Res. Commun. 2005, 334, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, K.E.; Howard, B.W.; Carter, J.M.; Janssen, Y.M.; Mossman, B.T.; Isfort, R.J. Mitochondrial-derived oxidants and quartz activation of chemokine gene expression. Adv. Exp. Med. Biol. 2001, 500, 489–496. [Google Scholar] [PubMed]
- Becher, R.; Bucht, A.; Øvrevik, J.; Hongslo, J.K.; Dahlman, H.J.; Samuelsen, J.T.; Schwarze, P.E. Involvement of NADPH oxidase and iNOS in rodent pulmonary cytokine responses to urban air And mineral particles. Inhal. Toxicol. 2007, 19, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Yano, E.; Evans, P.H. Cellular mechanisms of reactive oxygen metabolite generation from human polymorphonuclear leukocytes induced by crocidolite asbestos. Environ. Res. 1997, 75, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Kim, Y.H.; Seok Seo, M.; Kyu Lee, W.; Won Kim, S.; Kim, H.; Lee, K.H.; Shin, I.C.; Han, J.S.; Joong Kim, H.; et al. Mechanism of silica-induced ROS generation in Rat2 fibroblast cells. Toxicol. Lett. 2002, 135, 185–191. [Google Scholar] [CrossRef]
- Mo, Y.; Wan, R.; Chien, S.; Tollerud, D.J.; Zhang, Q. Activation of endothelial cells after exposure to ambient ultrafine particles: the role of NADPH oxidase. Toxicol. Appl. Pharmacol. 2009, 236, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Kampfrath, T.; Maiseyeu, A.; Ying, Z.; Shah, Z.; Deiuliis, J.A.; Xu, X.; Kherada, N.; Brook, R.D.; Reddy, K.M.; Padture, N.P.; et al. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ. Res. 2011, 108, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Magnani, N.D.; Marchini, T.; Tasat, D.R.; Alvarez, S.; Evelson, P.A. Lung oxidative metabolism after exposure to ambient particles. Biochem. Biophys. Res. Commun. 2011, 412, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Nurkiewicz, T.R.; Porter, D.W.; Hubbs, A.F.; Stone, S.; Moseley, A.M.; Cumpston, J.L.; Goodwill, A.G.; Frisbee, S.J.; Perrotta, P.L.; Brock, R.W.; et al. Pulmonary particulate matter and systemic microvascular dysfunction. Res. Rep. Health Eff Inst. 2011, 164, 3–48. [Google Scholar] [PubMed]
- Culcasi, M.; Benameur, L.; Mercier, A.; Lucchesi, C.; Rahmouni, H.; Asteian, A.; Casano, G.; Botta, A.; Kovacic, H.; Pietri, S. EPR spin trapping evaluation of ROS production in human fibroblasts exposed to cerium oxide nanoparticles: Evidence for NADPH oxidase and mitochondrial stimulation. Chem. Biol. Interact. 2012, 199, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Fazzi, F.; Njah, J.; Di, G.M.; Winnica, D.E.; Go, K.; Sala, E.; St. Croix, C.M.; Watkins, S.C.; Tyurin, V.A.; Phinney, D.G.; et al. TNFR1/phox interaction and TNFR1 mitochondrial translocation Thwart silica-induced pulmonary fibrosis. J. Immunol. 2014, 192, 3837–3846. [Google Scholar] [CrossRef] [PubMed]
- Konczol, M.; Weiss, A.; Stangenberg, E.; Gminski, R.; Garcia-Kaufer, M.; Giere, R.; Merfort, I.; Mersch-Sundermann, V. Cell-cycle changes and oxidative stress response to magnetite in A549 human lung cells. Chem. Res. Toxicol. 2013, 26, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Van Berlo, D.; Hullmann, M.; Wessels, A.; Scherbart, A.M.; Cassee, F.R.; Gerlofs-Nijland, M.E.; Albrecht, C.; Schins, R.P. Investigation of the effects of short-term inhalation of carbon nanoparticles on brains and lungs of c57bl/6j and p47(phox-/-) mice. Neurotoxicology 2014, 43, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V. Orbituary: Jürg Tschopp (1951–2011). Nature 2011. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell. Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Pope, R.M.; Tschopp, J. The role of interleukin-1 and the inflammasome in gout: Implications for therapy. Arthritis Rheum 2007, 56, 3183–3188. [Google Scholar] [CrossRef] [PubMed]
- So, A.; De, S.T.; Revaz, S.; Tschopp, J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 2007, 9, eR28. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Cassel, S.L.; Eisenbarth, S.C.; Iyer, S.S.; Sadler, J.J.; Colegio, O.R.; Tephly, L.A.; Carter, A.B.; Rothman, P.B.; Flavell, R.A.; Sutterwala, F.S. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9035–9040. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O'Connor, C.; et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE 2013, 8, e62143. [Google Scholar] [CrossRef] [PubMed]
- Ghaemi-Oskouie, F.; Shi, Y. The role of uric acid as an endogenous danger signal in immunity and inflammation. Curr. Rheumatol. Rep. 2011, 13, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Lunov, O.; Syrovets, T.; Loos, C.; Nienhaus, G.U.; Mailander, V.; Landfester, K.; Rouis, M.; Simmet, T. Amino-functionalized polystyrene nanoparticles activate the NLRP3 inflammasome in human macrophages. ACS Nano 2011, 5, 9648–9657. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Coste, A.; Olagnier, D.; Authier, H.; Lefevre, L.; Dardenne, C.; Bernad, J.; Beraud, M.; Flahaut, E.; Pipy, B. Double-walled carbon nanotubes trigger IL-1beta release in human monocytes through Nlrp3 inflammasome activation. Nanomedicine 2012, 8, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E.; Meindl, C.; Roblegg, E.; Ebner, B.; Absenger, M.; Pieber, T.R. Action of polystyrene nanoparticles of different sizes on lysosomal function and integrity. Part. Fibre Toxicol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Tahara, Y.; Nakamura, M.; Yang, M.; Zhang, M.; Iijima, S.; Yudasaka, M. Lysosomal membrane destabilization induced by high accumulation of single-walled carbon nanohorns in murine macrophage RAW 264.7. Biomaterials 2012, 33, 2762–2769. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Kim, S.; Kim, J.S.; Choi, I.H. Inflammasome formation and IL-1beta release by human blood monocytes in response to silver nanoparticles. Biomaterials 2012, 33, 6858–6867. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, W.J.; Låg, M.; Holme, J.A.; Friede, B.; Gualtieri, M.; Kruszewski, M.; Schwarze, P.E.; Skuland, T.; Refsnes, M. Comparison of non-crystalline silica nanoparticles in IL-1beta release from macrophages. Part. Fibre Toxicol. 2012. [Google Scholar] [CrossRef]
- Peeters, P.M.; Perkins, T.N.; Wouters, E.F.; Mossman, B.T.; Reynaert, N.L. Silica induces NLRP3 inflammasome activation in human lung epithelial cells. Part. Fibre Toxicol. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Sangtian, S.; Anderson, S.M.; Snyder, R.J.; Marshburn, J.D.; Rice, A.B.; Bonner, J.C.; Garantziotis, S. Inflammasome activation in airway epithelial cells after multi-walled carbon nanotube exposure mediates a profibrotic response in lung fibroblasts. Part. Fibre Toxicol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, I.; Lillehoj, E.P.; Lu, W.; Singh, I.S.; Isohama, Y.; Miyata, T.; Kim, K.C. Neutrophil elastase induces IL-8 gene transcription and protein release through p38/NF-κB activation via EGFR transactivation in a lung epithelial cell line. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L407–L416. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Holme, J.A.; Låg, M.; Schwarze, P.E.; Refsnes, M. Differential chemokine induction by 1-nitropyrene and 1-aminopyrene in bronchial epithelial cells: Importance of the TACE/TGF-alpha/EGFR-pathway. Environ. Toxicol. Pharmacol. 2013, 35, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.X.; Nakanaga, T.; Nadel, J.A. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L420–L427. [Google Scholar] [CrossRef] [PubMed]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Nadel, J.A. Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur. Respir. J. 2008, 32, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.R.; Xia, X.Z.; Chen, R.C. Expressions of tumor necrosis factor-converting enzyme and ErbB3 in rats with chronic obstructive pulmonary disease. Chin. Med. J. (Engl.) 2007, 120, 1505–1510. [Google Scholar] [PubMed]
- Castranova, V. Signaling pathways controlling the production of inflammatory mediators in response to crystalline silica exposure: Role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 2004, 37, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.W.; Macara, I.; Mossman, B.T. Cooperativity between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-kappa B—Requirement of Ras/mitogen-activated protein kinases in the activation of NF-kappa B by oxidants. Am. J. Respir. Cell. Mol. Biol. 1999, 20, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Tal, T.L.; Simmons, S.O.; Silbajoris, R.; Dailey, L.; Cho, S.H.; Ramabhadran, R.; Linak, W.; Reed, W.; Bromberg, P.A.; Samet, J.M. Differential transcriptional regulation of IL-8 expression by human airway epithelial cells exposed to diesel exhaust particles. Toxicol. Appl. Pharmacol. 2010, 243, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Samet, J.M.; Silbajoris, R.; Huang, T.; Jaspers, I. Transcription factor activation following exposure of an intact lung preparation to metallic particulate matter. Environ. Health Perspect. 2002, 110, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Cho, M.; Choi, G.; Na, H.; Chung, Y. Dynamic control of Th2 cell responses by STAT3 during allergic lung inflammation in mice. Int. Immunopharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Tal, T.L.; Graves, L.M.; Gilmour, I.; Linak, W.; Reed, W.; Bromberg, P.A.; Samet, J.M. Diesel exhaust particulate-induced activation of Stat3 requires activities of EGFR and Src in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L422–L429. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Abe, S.; Okazaki, H.; Kohyama, T.; Sugawara, I.; Saito, Y.; Ohtoshi, T.; Kawasaki, S.; Desaki, M.; Nakahara, K.; et al. Diesel exhaust particles upregulate eotaxin gene expression in human bronchial epithelial cells via nuclear factor-kappa B-dependent pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L1055–L1062. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Cosio, M.G.; Hoidal, J.R. Cigarette smoke-induced Egr-1 upregulates proinflammatory cytokines in pulmonary epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2006, 35, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Kasteler, S.D.; Cosio, M.G.; Sturrock, A.; Huecksteadt, T.; Hoidal, J.R. RAGE: Developmental expression and positive feedback regulation by Egr-1 during cigarette smoke exposure in pulmonary epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1094–L1101. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Wasley, K.M.; Allison, C.H. Diesel particulate matter induces receptor for advanced glycation end-products (RAGE) expression in pulmonary epithelial cells, and RAGE signaling influences NF-kappaB-mediated inflammation. Environ. Health Perspect. 2011, 119, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Wan, R.; Feng, L.; Chien, S.; Tollerud, D.J.; Zhang, Q. Combination effects of cigarette smoke extract and ambient ultrafine particles on endothelial cells. Toxicol. In Vitro 2012, 26, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Gasse, P.; Riteau, N.; Charron, S.; Girre, S.; Fick, L.; Petrilli, V.; Tschopp, J.; Lagente, V.; Quesniaux, V.F.; Ryffel, B.; et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Rabolli, V.; Badissi, A.A.; Devosse, R.; Uwambayinema, F.; Yakoub, Y.; Palmai-Pallag, M.; Lebrun, A.; de Gussem, V.; Couillin, I.; Ryffel, B.; et al. The alarmin IL-1alpha is a master cytokine in acute lung inflammation induced by silica micro- and nanoparticles. Part. Fibre Toxicol. 2014. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Øvrevik, J.; Refsnes, M.; Låg, M.; Holme, J.A.; Schwarze, P.E. Activation of Proinflammatory Responses in Cells of the Airway Mucosa by Particulate Matter: Oxidant- and Non-Oxidant-Mediated Triggering Mechanisms. Biomolecules 2015, 5, 1399-1440. https://doi.org/10.3390/biom5031399
Øvrevik J, Refsnes M, Låg M, Holme JA, Schwarze PE. Activation of Proinflammatory Responses in Cells of the Airway Mucosa by Particulate Matter: Oxidant- and Non-Oxidant-Mediated Triggering Mechanisms. Biomolecules. 2015; 5(3):1399-1440. https://doi.org/10.3390/biom5031399
Chicago/Turabian StyleØvrevik, Johan, Magne Refsnes, Marit Låg, Jørn A. Holme, and Per E. Schwarze. 2015. "Activation of Proinflammatory Responses in Cells of the Airway Mucosa by Particulate Matter: Oxidant- and Non-Oxidant-Mediated Triggering Mechanisms" Biomolecules 5, no. 3: 1399-1440. https://doi.org/10.3390/biom5031399