
Co-Expression of NEU2 and GBA3 Causes a Drastic Reduction in Cytosolic Sialyl Free N-glycans in Human MKN45 Stomach Cancer Cells—Evidence for the Physical Interaction of NEU2 and GBA3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Involvement of GBA3 in the Catabolic Pathways for Sialyl-FNGs in the Cytosol of MKN45 Cells


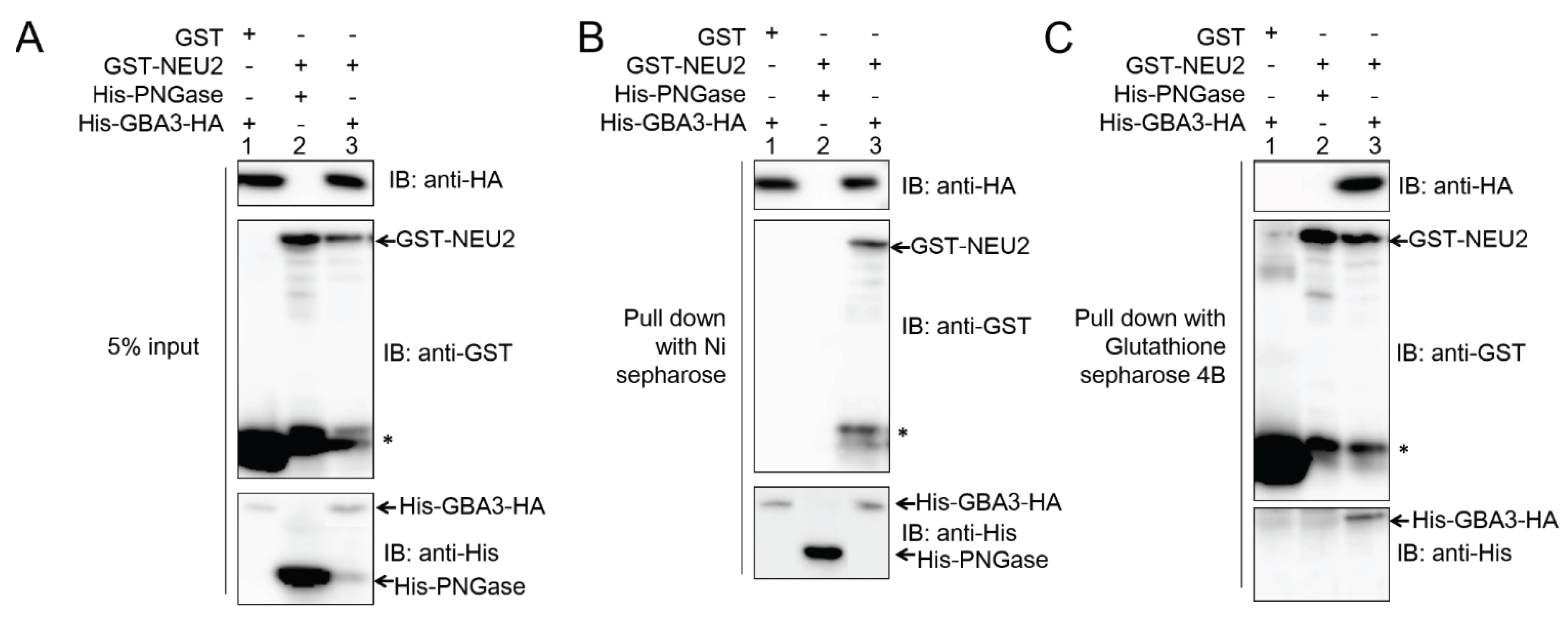
2.2. GBA3 Binds to NEU2 to Form a Complex


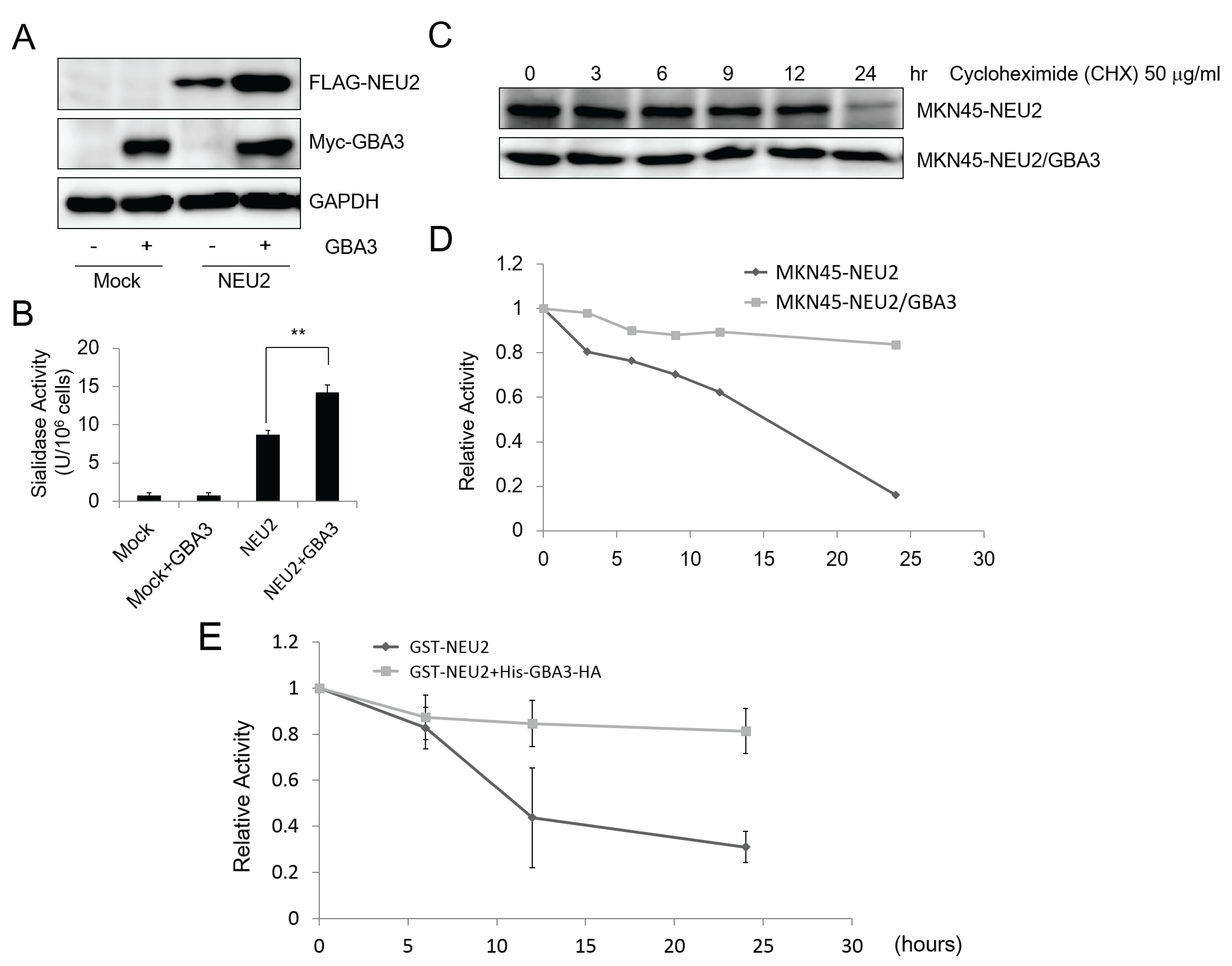
2.3. GBA3 Co-Transfection Stabilized NEU2 Protein and Increased NEU2 Activity both in Cellulo and in Vitro Experiments
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Western Blotting
4.3. RT-PCR
4.4. Extraction and PA-Labeling of Free Oligosaccharides Recovered from Cytosol
4.5. HPLC Analysis
4.6. Plasmid Transfection into Mammalian Cells
4.7. E. coli Expression and Purification of Recombinant GBA3 and NEU2
4.8. Co-Precipitation Assay
4.9. Gel Filtration Assay
4.10. Cycloheximide-Decay Assay
4.11. Sialidase Assay
4.12. GBA3 Activity Assay
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Varki, A. Biological roles of oligosaccharides—All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef] [PubMed]
- Haltiwanger, R.S.; Lowe, J.B. Role of glycosylation indevelopment. Annu. Rev. Biochem. 2004, 73, 491–537. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, Y.; Satoh, T.; Kato, K. Molecular and structural basis for N-glycan-dependent determination of glycoprotein fates in cells. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B. Lysosomal metabolism of glycoproteins. Glycobiology 2005, 15, 1R–15R. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T. Introduction to “Glycometabolome”. Trends Glycosci. Glycotechnol. 2009, 21, 219–227. [Google Scholar] [CrossRef]
- Suzuki, T. Cytoplasmic peptide: N-glycanase and catabolic pathway for free N-glycans in the cytosol. Semin. Cell Dev. Biol. 2007, 18, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Harada, Y. Non-lysosomal degradation pathway for N-linked glycans and dolichol-linked oligosaccharides. Biochem. Biophys. Res. Commun. 2014, 453, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Iwai, K.; Mega, T.; Hase, S. Quantitation and isomeric structure analysis of free oligosaccharides present in the cytosol fraction of mouse liver: Detection of a free disialobiantennary oligosaccharide and glucosylated oligomannosides. J. Biochem. 1999, 126, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, A.; Hashimto, Y.; Naka, R.; Kinoshita, M.; Kakehi, K.; Seino, J.; Funakoshi, Y.; Suzuki, T.; Kameyama, A.; Narimatsu, H. Accumulation of free complex-type N-glycans in MKN7 and MKN45 stomach cancer cells. Biochem. J. 2008, 413, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Seino, J.; Wang, L.; Harada, Y.; Huang, C.C.; Ishii, K.; Mizushima, N.; Suzuki, T. Basal autophagy is required for the efficient catabolism of sialyloligosaccharides. J. Biol. Chem. 2013, 288, 26898–26907. [Google Scholar] [CrossRef] [PubMed]
- Yabu, M.; Korekane, H.; Hatano, K.; Kaneda, Y.; Nonomura, N.; Sato, C.; Kitajima, K.; Miyamoto, Y. Occurrence of free deaminoneuraminic acid (KDN)-containing complex-type N-glycans in human prostate cancers. Glycobiology 2013, 23, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Yabu, M.; Korekane, H.; Takahashi, H.; Ohigashi, H.; Ishikawa, O.; Miyamoto, Y. Accumulation of free Neu5Ac-containing complex-type N-glycans in human pancreatic cancers. Glycoconj. J. 2013, 30, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Hays, W.S.; Jenison, S.A.; Yamada, T.; Pastuszyn, A.; Glew, R.H. Primary structure of the cytosolic beta-glucosidase of guinea pig liver. Biochem. J. 1996, 319, 829–837. [Google Scholar] [PubMed]
- Yahata, K.; Mori, K.; Arai, H.; Koide, S.; Ogawa, Y.; Mukoyama, M.; Sugawara, A.; Ozaki, S.; Tanaka, I.; Nabeshima, Y.; et al. Molecular cloning and expression of a novel klotho-related protein. J. Mol. Med. Jmm 2000, 78, 389–394. [Google Scholar] [CrossRef]
- De Graaf, M.; van Veen, I.C.; van Der Meulen-Muileman, I.H.; Gerritsen, W.R.; Pinedo, H.M.; Haisma, H.J. Cloning and characterization of human liver cytosolic beta-glycosidase. Biochem. J. 2001, 356, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, V.; Pastuszyn, A.; Galey, W.R.; Glew, R.H. Exolytic hydrolysis of toxic plant glucosides by guinea-pig liver cytosolic beta-glucosidase. J. Biol. Chem. 1992, 267, 14027–14032. [Google Scholar] [PubMed]
- Hayashi, Y.; Okino, N.; Kakuta, Y.; Shikanai, T.; Tani, M.; Narimatsu, H.; Ito, M. Klotho-related protein is a novel cytosolic neutral beta-glycosylceramidase. J. Biol. Chem. 2007, 282, 30889–30900. [Google Scholar] [CrossRef] [PubMed]
- Daniels, L.B.; Coyle, P.J.; Chiao, Y.B.; Glew, R.H.; Labow, R.S. Purification and characterization of a cytosolic broad specificity beta-glucosidase from human-liver. J. Biol. Chem. 1981, 256, 3004–3013. [Google Scholar]
- Rossi, S.; Stoppani, E.; Martinet, W.; Bonetto, A.; Costelli, P.; Giuliani, R.; Colombo, F.; Preti, A.; Marchesini, S.; Fanzani, A. The cytosolic sialidase NEU2 is degraded by autophagy during myoblast atrophy. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, F.; Brossmer, R.; Galjaard, H. Purification of acid beta-galactosidase and acid neuraminidase from bovine testis—Evidence for an enzyme complex. Biochem. Biophys. Res. Commun. 1982, 108, 868–875. [Google Scholar] [CrossRef]
- Verheijen, F.W.; Palmeri, S.; Hoogeveen, A.T.; Galjaard, H. Human placental neuraminidase—Activation, stabilization and association with beta-galactosidase and its protective protein. Eur. J. Biochem. 1985, 149, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Nishimura, K. Copurification and separation of beta-galactosidase and sialidase from porcine testis. Int. J. Biochem. 1987, 19, 435–442. [Google Scholar] [CrossRef]
- Potier, M.; Michaud, L.; Tranchemontagne, J.; Thauvette, L. Structure of the lysosomal neuraminidase—Beta-galactosidase-carboxypeptidase multienzymatic complex. Biochem. J. 1990, 267, 197–202. [Google Scholar] [PubMed]
- Pshezhetsky, A.V.; Potier, M. Direct affinity purification and supramolecular organization of human lysosomal cathepsin-A. Arch. Biochem. Biophys. 1994, 313, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Pshezhetsky, A.V.; Potier, M. Association of N-acetylgalactosamine-6-sulfate sulfatase with the multienzyme lysosomal complex of beta-galactosidase, cathepsin A, and neuraminidase—Possible implication for intralysosomal catabolism of keratan sulfate. J. Biol. Chem. 1996, 271, 28359–28365. [Google Scholar] [CrossRef] [PubMed]
- Pshezhetsky, A.V.; Ashmarina, M. Lysosomal multienzyme complex: Biochemistry, genetics, and molecular pathophysiology. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 81–114. [Google Scholar] [PubMed]
- Miyagi, T.; Tsuiki, S. Purification and characterization of cytosolic sialidase from rat-liver. J. Biol. Chem. 1985, 260, 6710–6716. [Google Scholar] [PubMed]
- Koseki, K.; Wada, T.; Hosono, M.; Hata, K.; Yamaguchi, K.; Nitta, K.; Miyagi, T. Human cytosolic sialidase NEU2-low general tissue expression but involvement in PC-3 prostate cancer cell survival. Biochem. Biophys. Res. Commun. 2012, 428, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Lamarco, K.L.; Glew, R.H. Hydrolysis of a naturally-occurring beta-glucoside by a broad-specificity beta-glucosidase from liver. Biochem. J. 1986, 237, 469–476. [Google Scholar] [PubMed]
- Day, A.J.; DuPont, M.S.; Ridley, S.; Rhodes, M.; Rhodes, M.J.C.; Morgan, M.R.A.; Williamson, G. Deglycosylation of flavonoid and isoflavonoid glycosides by human small intestine and liver beta-glucosidase activity. Febs Lett. 1998, 436, 71–75. [Google Scholar] [CrossRef]
- McMahon, L.G.; Nakano, H.; Levy, M.D.; Gregory, J.F. Cytosolic pyridoxine-beta-D-glucoside hydrolase from porcine jejunal mucosa—Purification, properties, and comparison with broad specificity beta-glucosidase. J. Biol. Chem. 1997, 272, 32025–32033. [Google Scholar] [CrossRef] [PubMed]
- Legler, G.; Bieberich, E. Isolation of a cytosolic beta-glucosidase from calf liver and characterization of its active-site with alkyl glucosides and basic glycosyl derivatives. Arch. Biochem. Biophys. 1988, 260, 427–436. [Google Scholar] [CrossRef]
- Wang, L.; Suzuki, T. Dual functions for cytosolic alpha-mannosidase (Man2C1) its down-regulation causes mitochondria-dependent apoptosis independently of its alpha-mannosidase activity. J. Biol. Chem. 2013, 288, 11887–11896. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, H.; Seino, J.; Kitajima, T.; Jigami, Y.; Suzuki, T. Free oligosaccharides to monitor glycoprotein endoplasmic reticulum-associated degradation in saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 12390–12404. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hara, I.; Nakano, M.; Shigeta, M.; Nakagawa, T.; Kondo, A.; Funakoshi, Y.; Taniguchi, N. Man2c1, an alpha-mannosidase, is involved in the trimming of free oligosaccharides in the cytosol. Biochem. J. 2006, 400, 33–41. [Google Scholar] [PubMed]
- Tomiya, N.; Awaya, J.; Kurono, M.; Endo, S.; Arata, Y.; Takahashi, N. Analyses of N-linked oligosaccharides using a two-dimensional mapping technique. Anal. Biochem. 1988, 171, 73–90. [Google Scholar] [CrossRef]
- Suzuki, T.; Matsuo, I.; Totani, K.; Funayama, S.; Seino, J.; Taniguchi, N.; Ito, Y.; Hase, S. Dual-gradient high-performance liquid chromatography for identification of cytosolic high-mannose-type free glycans. Anal. Biochem. 2008, 381, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Chavas, L.M.G.; Tringali, C.; Fusi, P.; Venerando, B.; Tettamanti, G.; Kato, R.; Monti, E.; Wakatsuki, S. Crystal structure of the human cytosolic sialidase NEU2—Evidence for the dynamic nature of substrate recognition. J. Biol. Chem. 2005, 280, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.; Suzuki, T.; Balgobin, B.J.; Lennarz, W.J. Site-directed mutagenesis study of yeast peptide: N-glycanase—Insight into the reaction mechanism of deglycosylation. J. Biol. Chem. 2002, 277, 12953–12959. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Seino, J.; Tomotake, H.; Funakoshi, Y.; Hirayama, H.; Suzuki, T. Co-Expression of NEU2 and GBA3 Causes a Drastic Reduction in Cytosolic Sialyl Free N-glycans in Human MKN45 Stomach Cancer Cells—Evidence for the Physical Interaction of NEU2 and GBA3. Biomolecules 2015, 5, 1499-1514. https://doi.org/10.3390/biom5031499
Wang L, Seino J, Tomotake H, Funakoshi Y, Hirayama H, Suzuki T. Co-Expression of NEU2 and GBA3 Causes a Drastic Reduction in Cytosolic Sialyl Free N-glycans in Human MKN45 Stomach Cancer Cells—Evidence for the Physical Interaction of NEU2 and GBA3. Biomolecules. 2015; 5(3):1499-1514. https://doi.org/10.3390/biom5031499
Chicago/Turabian StyleWang, Li, Junichi Seino, Haruna Tomotake, Yoko Funakoshi, Hiroto Hirayama, and Tadashi Suzuki. 2015. "Co-Expression of NEU2 and GBA3 Causes a Drastic Reduction in Cytosolic Sialyl Free N-glycans in Human MKN45 Stomach Cancer Cells—Evidence for the Physical Interaction of NEU2 and GBA3" Biomolecules 5, no. 3: 1499-1514. https://doi.org/10.3390/biom5031499