
Arsenic Disruption of DNA Damage Responses—Potential Role in Carcinogenesis and Chemotherapy

Abstract
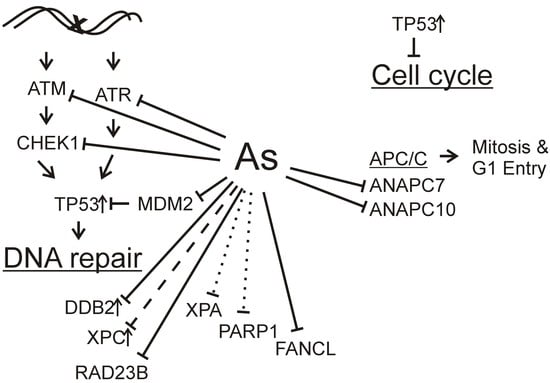
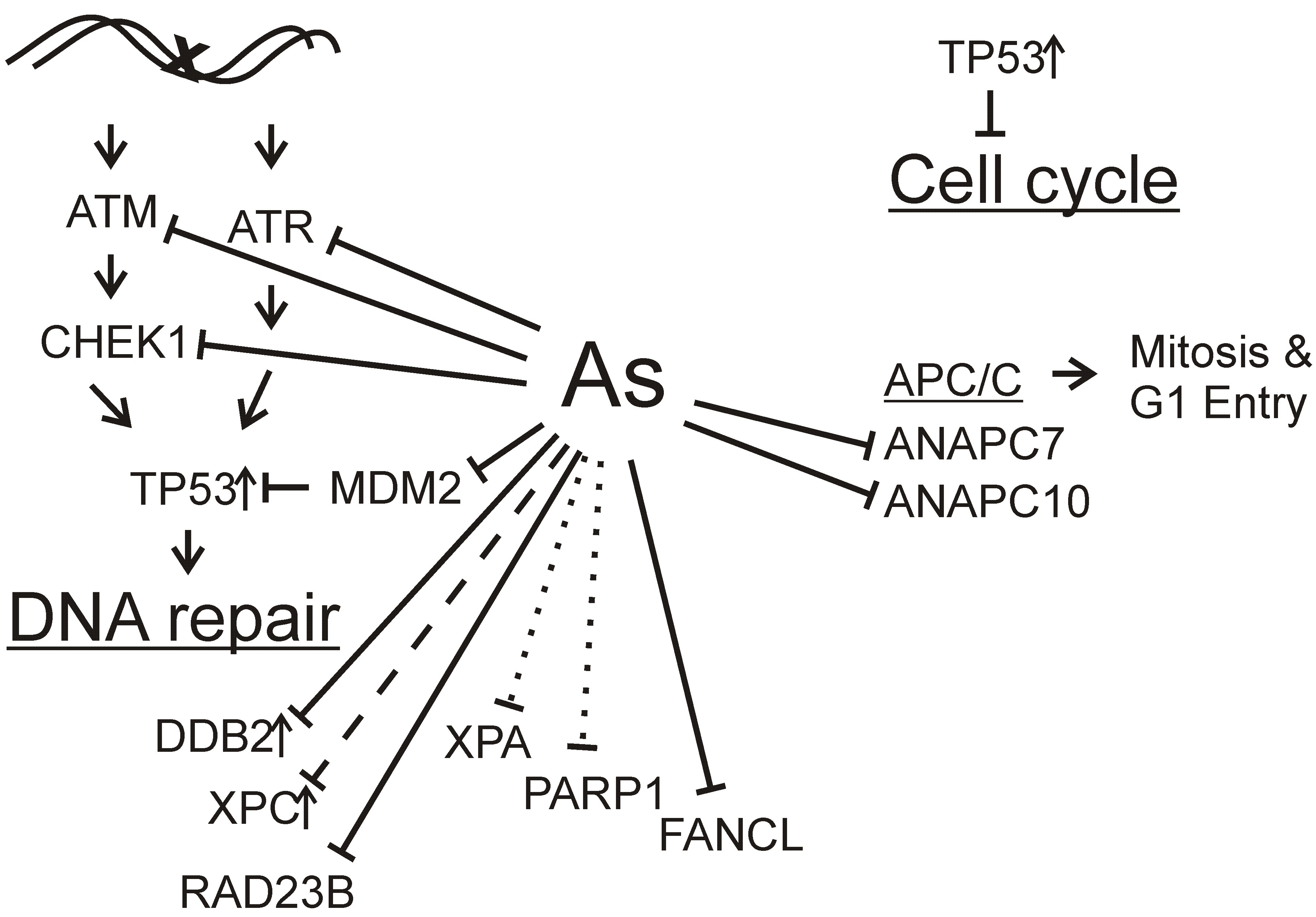
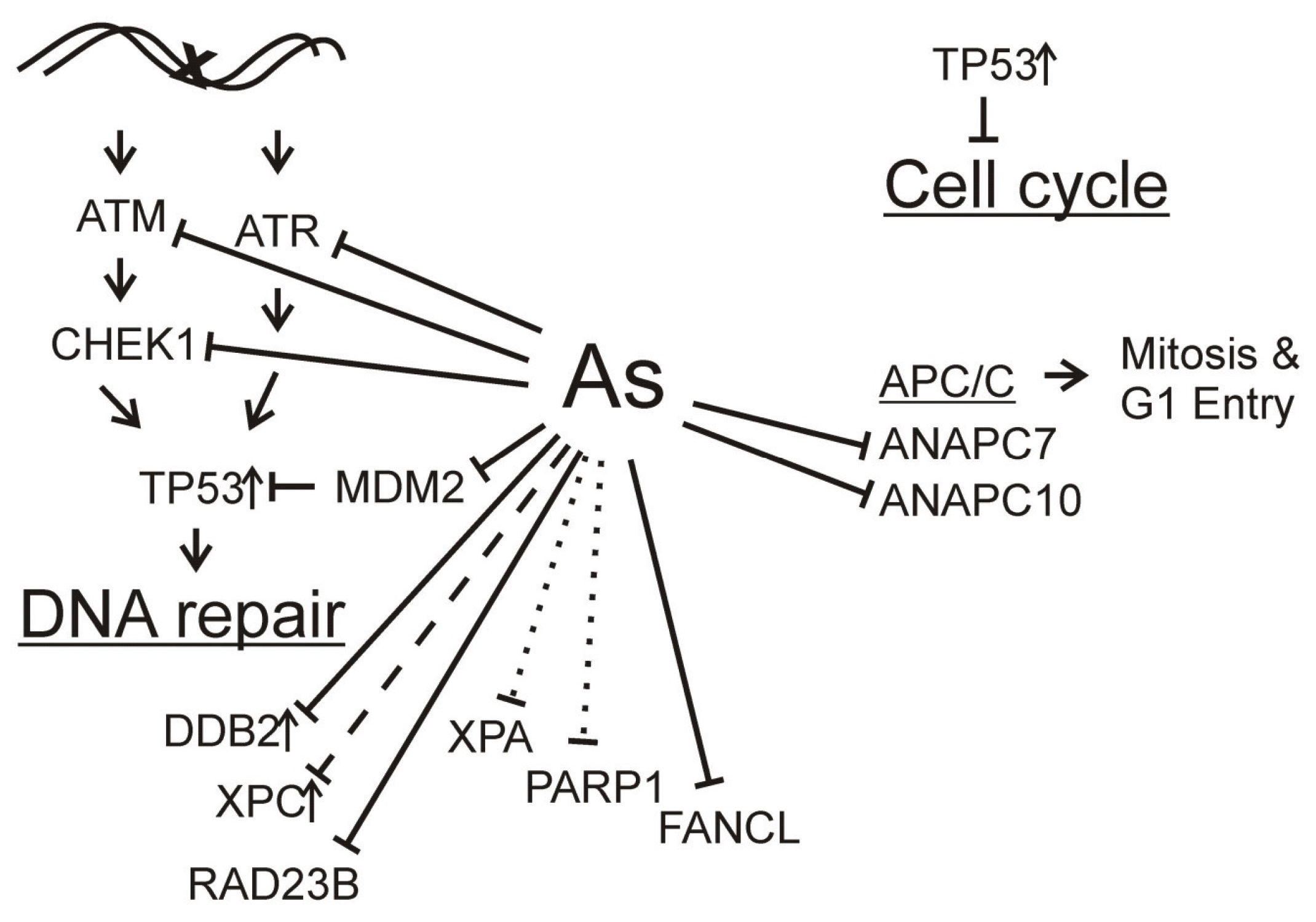
:
{kind=link}
{kind=link}

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cui, X.; Kobayashi, Y.; Akashi, M.; Okayasu, R. Metabolism and the paradoxical effects of arsenic: Carcinogenesis and anticancer. Curr. Med. Chem. 2008, 15, 2293–2304. [Google Scholar] [CrossRef] [PubMed]
- Desoize, B. Metals and metal compounds in cancer treatment. Anticancer Res. 2004, 24, 1529–1544. [Google Scholar] [PubMed]
- Gibb, H.; Haver, C.; Gaylor, D.; Ramasamy, S.; Lee, J.S.; Lobdell, D.; Wade, T.; Chen, C.; White, P.; Sams, R. Utility of recent studies to assess the national research council 2001 estimates of cancer risk from ingested arsenic. Environ. Health Perspect. 2011, 119, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Banerjee, M.; Kundu, M.; Banerjee, N.; Bhattacharya, U.; Giri, A.K.; Ganguli, B.; Roy, S.S.; Polya, D.A. Comparison of drinking water, raw rice and cooking of rice as arsenic exposure routes in three contrasting areas of West Bengal, India. Environ. Geochem. Health 2010, 32, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Saint-Jacques, N.; Parker, L.; Brown, P.; Dummer, T.J. Arsenic in drinking water and urinary tract cancers: A systematic review of 30 years of epidemiological evidence. Environ. Health 2014. [Google Scholar] [CrossRef]
- Steinmaus, C.; Ferreccio, C.; Yuan, Y.; Acevedo, J.; Gonzalez, F.; Perez, L.; Cortes, S.; Balmes, J.R.; Liaw, J.; Smith, A.H. Elevated lung cancer in younger adults and low concentrations of arsenic in water. Am. J. Epidemiol. 2014, 180, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, S.; Zhang, D. Association of inorganic arsenic exposure with liver cancer mortality: A meta-analysis. Environ. Res. 2014, 135, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Tokar, E.J.; Qu, W.; Liu, J.; Liu, W.; Webber, M.M.; Phang, J.M.; Waalkes, M.P. Arsenic-specific stem cell selection during malignant transformation. J. Nat. Cancer Inst. 2010, 102, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Salazar, A.M.; Miller, H.L.; McNeely, S.C.; Sordo, M.; Ostrosky-Wegman, P.; States, J.C. Suppression of p53 and p21CIP1/WAF1 reduces arsenite-induced aneuploidy. Chem. Res. Toxicol. 2010, 23, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Vega, L.; Gonsebatt, M.E.; Ostrosky-Wegman, P. Aneugenic effect of sodium arsenite on human lymphocytes in vitro: An individual susceptibility effect detected. Mutat. Res. 1995, 334, 365–373. [Google Scholar] [CrossRef]
- Waalkes, M.P.; Qu, W.; Tokar, E.J.; Kissling, G.E.; Dixon, D. Lung tumors in mice induced by “whole-life” inorganic arsenic exposure at human-relevant doses. Arch. Toxicol. 2014, 88, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Burns, F.J.; Uddin, A.N.; Wu, F.; Nadas, A.; Rossman, T.G. Arsenic-induced enhancement of ultraviolet radiation carcinogenesis in mouse skin: A dose-response study. Environ. Health Perspect. 2004, 112, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Rossman, T.G.; Uddin, A.N.; Burns, F.J. Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol. Appl. Pharmacol. 2004, 198, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Rossman, T.G.; Uddin, A.N.; Burns, F.J.; Bosland, M.C. Arsenite is a cocarcinogen with solar ultraviolet radiation for mouse skin: An animal model for arsenic carcinogenesis. Toxicol. Appl. Pharmacol. 2001, 176, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Rossman, T.G.; Uddin, A.N.; Burns, F.J.; Bosland, M.C. Arsenite cocarcinogenesis: An animal model derived from genetic toxicology studies. Environ. Health Perspect. 2002, 110, S749–S752. [Google Scholar] [CrossRef]
- Schwarz, M.; Munzel, P.A.; Braeuning, A. Non-melanoma skin cancer in mouse and man. Arch. Toxicol. 2013, 87, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Waalkes, M.P.; Liu, J.; Germolec, D.R.; Trempus, C.S.; Cannon, R.E.; Tokar, E.J.; Tennant, R.W.; Ward, J.M.; Diwan, B.A. Arsenic exposure in utero exacerbates skin cancer response in adulthood with contemporaneous distortion of tumor stem cell dynamics. Cancer Res. 2008, 68, 8278–8285. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.S.; Karagas, M.R.; Hamilton, J.W. Decreased DNA repair gene expression among individuals exposed to arsenic in united states drinking water. Int. J. Cancer 2003, 104, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.S.; Burgess, J.L.; Meza, M.M.; Demidenko, E.; Waugh, M.G.; Hamilton, J.W.; Karagas, M.R. Arsenic exposure is associated with decreased DNA repair in vitro and in individuals exposed to drinking water arsenic. Environ. Health Perspect. 2006, 114, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, K.T.; Hirao, T.; Hirao, S.; Devi-Ashok, T.; Nelson, H.H.; Andrew, A.; Colt, J.; Baris, D.; Morris, J.S.; Schned, A.; et al. Tp53 alterations and patterns of carcinogen exposure in a US Population-based study of bladder cancer. Int. J. Cancer 2005, 117, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Ohneseit, P.F.; Tsai, Y.C.; Spruck, C.H., 3rd; Nichols, P.W.; Chiang, H.S.; Lai, M.K.; Jones, P.A. Mutational spectrum in the p53 gene in bladder tumors from the endemic area of black foot disease in taiwan. Carcinogenesis 1994, 15, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. Roles of the transcription factor p53 in keratinocyte carcinomas. Br. J. Dermatol. 2006, 154, S8–S10. [Google Scholar] [CrossRef] [PubMed]
- Castren, K.; Ranki, A.; Welsh, J.A.; Vahakangas, K.H. Infrequent p53 mutations in arsenic-related skin lesions. Oncol. Res. 1998, 10, 475–482. [Google Scholar] [PubMed]
- Hsieh, L.L.; Chen, H.J.; Hsieh, J.T.; Jee, S.H.; Chen, G.S.; Chen, C.J. Arsenic-related bowen’s disease and paraquat-related skin cancerous lesions show no detectable ras and p53 gene alterations. Cancer Lett. 1994, 86, 59–65. [Google Scholar] [CrossRef]
- Hsu, C.H.; Yang, S.A.; Wang, J.Y.; Yu, H.S.; Lin, S.R. Mutational spectrum of p53 gene in arsenic-related skin cancers from the blackfoot disease endemic area of Taiwan. Br. J. Cancer 1999, 80, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Komissarova, E.V.; Rossman, T.G. Arsenite induced poly(ADP-ribosyl)ation of tumor suppressor p53 in human skin keratinocytes as a possible mechanism for carcinogenesis associated with arsenic exposure. Toxicol. Appl. Pharmacol. 2010, 243, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Liu, W.; Cooper, K.L.; Qin, X.J.; de Souza Bergo, P.L.; Hudson, L.G.; Liu, K.J. Inhibition of poly (ADP-ribose) polymerase-1 by arsenite interferes with repair of oxidative DNA damage. J. Biol. Chem. 2009, 284, 6809–6817. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhou, X.; Du, L.; Liu, W.; Liu, Y.; Hudson, L.G.; Liu, K.J. Arsenite binding-induced zinc loss from parp-1 is equivalent to zinc deficiency in reducing parp-1 activity, leading to inhibition of DNA repair. Toxicol. Appl. Pharmacol. 2014, 274, 313–318. [Google Scholar] [CrossRef] [PubMed]
- McNeely, S.C.; Belshoff, A.C.; Taylor, B.F.; Fan, T.W.; McCabe, M.J., Jr.; Pinhas, A.R.; States, J.C. Sensitivity to sodium arsenite in human melanoma cells depends upon susceptibility to arsenite-induced mitotic arrest. Toxicol. Appl. Pharmacol. 2008, 229, 252–261. [Google Scholar] [CrossRef] [PubMed]
- McNeely, S.C.; Taylor, B.F.; States, J.C. Mitotic arrest-associated apoptosis induced by sodium arsenite in A375 melanoma cells is BUBR1-dependent. Toxicol. Appl. Pharmacol. 2008, 231, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.F.; McNeely, S.C.; Miller, H.L.; Lehmann, G.M.; McCabe, M.J., Jr.; States, J.C. p53 Suppression of arsenite-induced mitotic catastrophe is mediated by p21CIP1/WAF1. J. Pharmacol. Exp. Ther. 2006, 318, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Bau, D.T.; Wang, T.S.; Chung, C.H.; Wang, A.S.; Wang, A.S.; Jan, K.Y. Oxidative DNA adducts and DNA-protein cross-links are the major DNA lesions induced by arsenite. Environ. Health Perspect. 2002, 110, S753–S756. [Google Scholar] [CrossRef]
- Salnikow, K.; Zhitkovich, A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: Nickel, arsenic, and chromium. Chem. Res. Toxicol. 2008, 21, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hudson, L.G.; Ding, W.; Wang, S.; Cooper, K.L.; Liu, S.; Chen, Y.; Shi, X.; Liu, K.J. Arsenite causes DNA damage in keratinocytes via generation of hydroxyl radicals. Chem. Res. Toxicol. 2004, 17, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hudson, L.G.; Liu, K.J. Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free. Radic. Biol. Med. 2004, 37, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Shi, X.; Liu, K.J. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem. 2004, 255, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Ebert, F.; Weiss, A.; Bultemeyer, M.; Hamann, I.; Hartwig, A.; Schwerdtle, T. Arsenicals affect base excision repair by several mechanisms. Mutat. Res. 2011, 715, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Osmond, M.J.; Kunz, B.A.; Snow, E.T. Age and exposure to arsenic alter base excision repair transcript levels in mice. Mutagenesis 2010, 25, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, A.; Pelzer, A.; Asmuss, M.; Burkle, A. Very low concentrations of arsenite suppress poly (ADP-ribosyl)ation in mammalian cells. Int. J. Cancer 2003, 104, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Schwerdtle, T.; Walter, I.; Hartwig, A. Arsenite and its biomethylated metabolites interfere with the formation and repair of stable BPDE-induced DNA adducts in human cells and impair XPAzf and Fpg. DNA Repair 2003, 2, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, B. Arsenic trioxide (As2O3) inhibits peritoneal invasion of ovarian carcinoma cells in vitro and in vivo. Gynecol. Oncol. 2006, 103, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Hirschfeld, S.; Honig, S.F.; Ibrahim, A.; Johnson, J.R.; O’Leary, J.J.; White, R.M.; Williams, G.A.; Pazdur, R. Drug approval summaries: Arsenic trioxide, tamoxifen citrate, anastrazole, paclitaxel, bexarotene. Oncologist 2001, 6, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Akao, Y.; Morikawa, H.; Hirata, I.; Katsu, K.; Naoe, T.; Ohishi, N.; Yagi, K. Arsenic trioxide-induced apoptosis through oxidative stress in cells of colon cancer cell lines. Life Sci. 2002, 70, 2253–2269. [Google Scholar] [CrossRef]
- Uslu, R.; Sanli, U.A.; Sezgin, C.; Karabulut, B.; Terzioglu, E.; Omay, S.B.; Goker, E. Arsenic trioxide-mediated cytotoxicity and apoptosis in prostate and ovarian carcinoma cell lines. Clin. Cancer Res. 2000, 6, 4957–4964. [Google Scholar] [PubMed]
- Zhang, T.C.; Cao, E.H.; Li, J.F.; Ma, W.; Qin, J.F. Induction of apoptosis and inhibition of human gastric cancer MGC-803 cell growth by arsenic trioxide. Eur. J. Cancer 1999, 35, 1258–1263. [Google Scholar] [CrossRef]
- Maeda, H.; Hori, S.; Nishitoh, H.; Ichijo, H.; Ogawa, O.; Kakehi, Y.; Kakizuka, A. Tumor growth inhibition by arsenic trioxide (As2O3) in the orthotopic metastasis model of androgen-independent prostate cancer. Cancer Res. 2001, 61, 5432–5440. [Google Scholar] [PubMed]
- Chun, Y.J.; Park, I.C.; Park, M.J.; Woo, S.H.; Hong, S.I.; Chung, H.Y.; Kim, T.H.; Lee, Y.S.; Rhee, C.H.; Lee, S.J. Enhancement of radiation response in human cervical cancer cells in vitro and in vivo by arsenic trioxide (As2O3). FEBS Lett. 2002, 519, 195–200. [Google Scholar] [CrossRef]
- Wang, W.; Qin, S.K.; Chen, B.A.; Chen, H.Y. Experimental study on antitumor effect of arsenic trioxide in combination with cisplatin or doxorubicin on hepatocellular carcinoma. World J. Gastroenterol. 2001, 7, 702–705. [Google Scholar] [PubMed]
- Muenyi, C.S.; States, V.A.; Masters, J.H.; Fan, T.W.; Helm, C.W.; States, J.C. Sodium arsenite and hyperthermia modulate cisplatin-DNA damage responses and enhance platinum accumulation in murine metastatic ovarian cancer xenograft after hyperthermic intraperitoneal chemotherapy (HIPEC). J. Ovarian Res. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Neher, T.M.; Rechkunova, N.I.; Lavrik, O.I.; Turchi, J.J. Photo-cross-linking of XPC-Rad23B to cisplatin-damaged DNA reveals contacts with both strands of the DNA duplex and spans the DNA adduct. Biochemistry 2010, 49, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Sameer, A.S.; Nissar, S.; Fatima, K. Mismatch repair pathway: Molecules, functions, and role in colorectal carcinogenesis. Eur. J. Cancer Prev. 2014, 23, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Melton, D.W.; Gourley, C. Mismatch repair deficiency in ovarian cancer—Molecular characteristics and clinical implications. Gynecol. Oncol. 2014, 132, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Adimoolam, S.; Ford, J.M. P53 and regulation of DNA damage recognition during nucleotide excision repair. DNA Repair 2003, 2, 947–954. [Google Scholar] [CrossRef]
- Ford, J.M. Regulation of DNA damage recognition and nucleotide excision repair: Another role for p53. Mutat. Res. 2005, 577, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.M.; Hanawalt, P.C. Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc. Natl. Acad. Sci. USA 1995, 92, 8876–8880. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.M.; Hanawalt, P.C. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J. Biol. Chem. 1997, 272, 28073–28080. [Google Scholar] [CrossRef] [PubMed]
- Nollen, M.; Ebert, F.; Moser, J.; Mullenders, L.H.; Hartwig, A.; Schwerdtle, T. Impact of arsenic on nucleotide excision repair: XPC function, protein level, and gene expression. Mol. Nutr. Food Res. 2009, 53, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Muenyi, C.S.; Pinhas, A.R.; Fan, T.W.; Brock, G.N.; Helm, C.W.; States, J.C. Sodium arsenite ± hyperthermia sensitizes p53-expressing human ovarian cancer cells to cisplatin by modulating platinum-DNA damage responses. Toxicol. Sci. 2012, 127, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Muenyi, C.S.; Trivedi, A.P.; Helm, C.W.; States, J.C. Cisplatin plus sodium arsenite and hyperthermia induces pseudo-G1 associated apoptotic cell death in ovarian cancer cells. Toxicol. Sci. 2014, 139, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, M.T.; Veloso, A.; Prasad, J.; Bedi, K.; Ljungman, E.A.; Magnuson, B.; Wilson, T.E.; Ljungman, M. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 2014, 67, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Reichard, J.F.; Puga, A. Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics 2010, 2, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Chervona, Y.; Arita, A.; Costa, M. Carcinogenic metals and the epigenome: Understanding the effect of nickel, arsenic, and chromium. Metallomics 2012, 4, 619–627. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muenyi, C.S.; Ljungman, M.; States, J.C. Arsenic Disruption of DNA Damage Responses—Potential Role in Carcinogenesis and Chemotherapy. Biomolecules 2015, 5, 2184-2193. https://doi.org/10.3390/biom5042184
Muenyi CS, Ljungman M, States JC. Arsenic Disruption of DNA Damage Responses—Potential Role in Carcinogenesis and Chemotherapy. Biomolecules. 2015; 5(4):2184-2193. https://doi.org/10.3390/biom5042184
Chicago/Turabian StyleMuenyi, Clarisse S., Mats Ljungman, and J. Christopher States. 2015. "Arsenic Disruption of DNA Damage Responses—Potential Role in Carcinogenesis and Chemotherapy" Biomolecules 5, no. 4: 2184-2193. https://doi.org/10.3390/biom5042184
APA StyleMuenyi, C. S., Ljungman, M., & States, J. C. (2015). Arsenic Disruption of DNA Damage Responses—Potential Role in Carcinogenesis and Chemotherapy. Biomolecules, 5(4), 2184-2193. https://doi.org/10.3390/biom5042184