
The TORC2‐Dependent Signaling Network in the Yeast Saccharomyces cerevisiae

 and
and 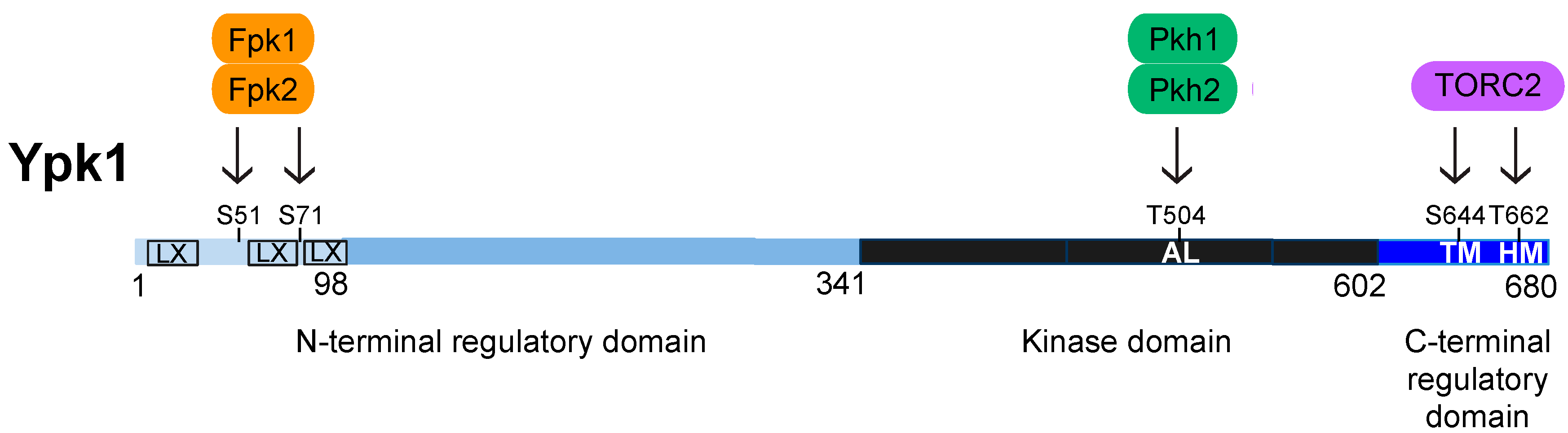{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. TORC2 Structure and Function
3. TORC2 Effectors
4. Structure, Function, and Regulation of Ypk1
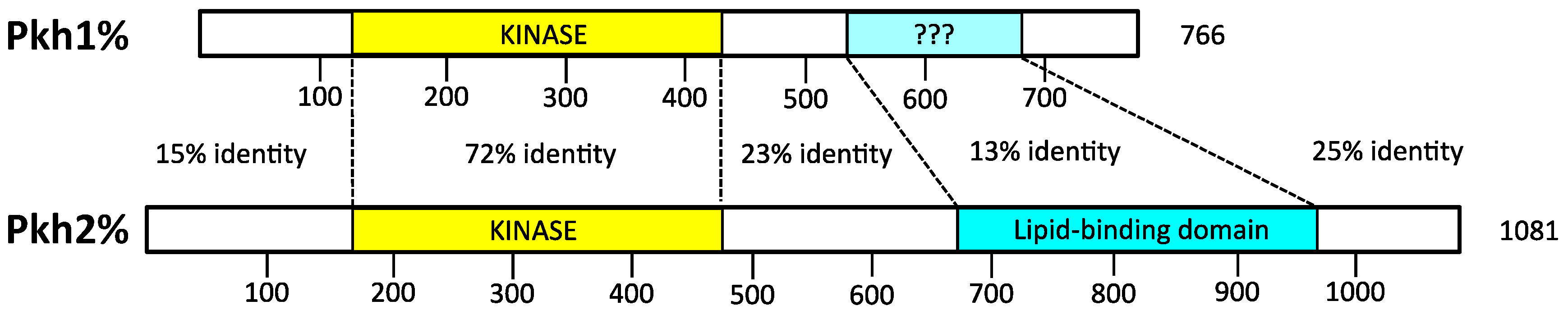
4.1. Activation Loop Phosphorylation by Pkh1 and Pkh2
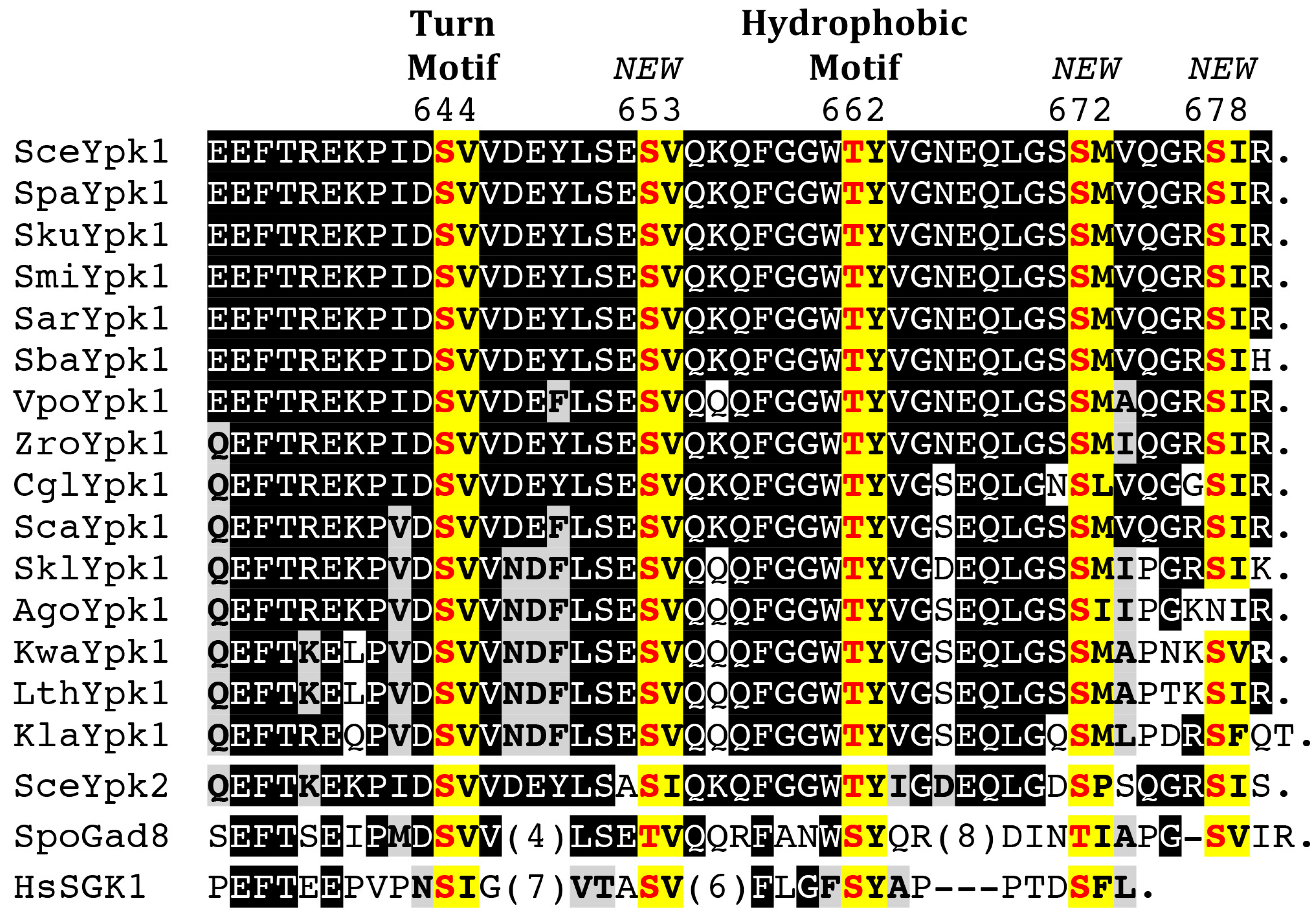
4.2. C-Terminal Phosphorylation by TORC2
4.3. N-Terminal Phosphorylation by Fpk1 and Fpk2
4.4. Ypk1 Activity Is Not Controlled by Sterol
5. Substrates of Ypk1
5.1. Control of Aminoglycerophospholipid Asymmetry in the Plasma Membrane Bilayer
5.2. Control of Sphingolipid Biosynthesis
5.3. Control of Intracellular Glycerol Concentration
5.4. Control of Integral Plasma Membrane Protein Endocytosis
5.5. Other Potential Targets
6. Structure, Function and Regulation of Pkc1
6.1. Activation Loop Phosphorylation by Pkh1 and Pkh2
6.2. C-Terminal Phosphorylation by TORC2
6.3. Interaction with Rho1-GTP
6.4. Modulation by PtdSer and DAG
7. Substrates of Pkc1
7.1. Cell Wall Integrity Pathway
7.2. Nuclear Targets
7.3. Modulation of Triacylglycerol Synthesis
7.4. Other Potential Targets
8. Cross-Talk between Ypk1- and Pkc1-Dependent Signaling
9. Prospectus
Acknowledgments
Conflicts of Interest
References
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing Streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Martel, R.R.; Klicius, J.; Galet, S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can. J. Physiol. Pharmacol. 1977, 55, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.P.; Sehgal, S.N.; Vézina, C. Activity of rapamycin (AY-22,989) against transplanted tumors. J. Antibiot. 1984, 37, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Hall, M.N. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 2011, 189, 1177–1201. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.R.; Hall, M.N. Target of rapamycin in yeast, Tor2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Helliwell, S.B.; Wagner, P.; Kunz, J.; Deuter-Reinhard, M.; Henriquez, R.; Hall, M.N. Tor1 and Tor2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol. Biol. Cell 1994, 5, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two Tor complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Wedaman, K.P.; Reinke, A.; Anderson, S.; Yates, J., 3rd; McCaffery, J.M.; Powers, T. Tor kinases are in distinct membrane-associated protein complexes in Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 1204–1220. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. A complex interplay between AKT, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Oliveira, T.M.; Prouteau, M.; Leitner, A.; Karuppasamy, M.; Konstantinidou, G.; Rispal, D.; Eltschinger, S.; Robinson, G.C.; Thore, S.; et al. Molecular basis of the rapamycin insensitivity of Target of Rapamycin complex 2. Mol. Cell 2015, 58, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Ikai, N.; Nakazawa, N.; Hayashi, T.; Mitsuhiro, Y. The reverse, but coordinated, roles of Tor2 (TORC1) and TOR1 (TORC2) kinases for growth, cell cycle and separase-mediated mitosis in Schizosaccharomyces pombe. Open Biol. 2011, 1, 110007. [Google Scholar] [CrossRef] [PubMed]
- Eltschinger, S.; Loewith, R. TOR complexes and the maintenance of cellular homeostasis. Trends Cell Biol. 2016, 26, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Aylett, C.H.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mTOR complex 1. Science 2015, 351, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Baretić, D.; Berndt, A.; Ohashi, Y.; Johnson, C.M.; Williams, R.L. TOR forms a dimer through an N-terminal helical solenoid with a complex topology. Nat. Commun. 2016, 7, 11016. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Prouteau, M.; Kusmider, B.; Loewith, R. TORC2 structure and function. Trends Biochem. Sci. 2016, 41, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Breslow, D.K.; Muir, A.; Weissman, J.S.; Thorner, J. Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2011, 108, 19222–19227. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.; Ramachandran, S.; Roelants, F.M.; Timmons, G.; Thorner, J. TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. Elife 2014, 3, e03779. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.; Roelants, F.M.; Timmons, G.; Leskoske, K.L.; Thorner, J. Down-regulation of torc2-ypk1 signaling promotes mapk-independent survival under hyperosmotic stress. Elife 2015, 4, 09336. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, C.G.; Aindow, A.; Thorner, J. Differential phosphorylation provides a switch to control how α-arrestin Rod1 down-regulates mating pheromone response in Saccharomyces cerevisiae. Genetics 2016, 203, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Leskoske, K.L.; Pedersen, R.T.; Muir, A.; Liu, J.M.; Finnigan, G.C.; Thorner, J. TOR complex 2-regulated protein kinase Fpk1 stimulates endocytosis via inhibition of Ark1/Prk1-related protein kinase Akl1 in Saccharomyces cerevisiae. Mol. Cell. Biol. 2017, 37, e00627-16. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Walther, T.C. TORC2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol. Biol. Cell 2009, 20, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Piccolis, M.; Chiaruttini, N.; Riezman, I.; Riezman, H.; Roux, A.; Walther, T.C.; Loewith, R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat. Cell Biol. 2012, 14, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Niles, B.J.; Mogri, H.; Hill, A.; Vlahakis, A.; Powers, T. Plasma membrane recruitment and activation of the AGC kinase Ypk1 is mediated by Target of Rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2. Proc. Natl. Acad. Sci. USA 2012, 109, 1536–1541. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Miao, Y.; Yamane, Y.; Zhang, C.; Shokat, K.M.; Takematsu, H.; Kozutsumi, Y.; Drubin, D.G. Orm protein phosphoregulation mediates transient sphingolipid biosynthesis response to heat stress via the Pkh-Ypk and Cdc55 PP2A pathways. Mol. Biol. Cell 2012, 23, 2388–2398. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, F.; Olson, D.K.; Christiano, R.; Farese, R.V.J.; Walther, T.C. Proteomic and phosphoproteomic analyses of yeast reveal the global cellular response to sphingolipid depletion. Proteomics 2016, 16, 2759–2763. [Google Scholar] [CrossRef] [PubMed]
- deHart, A.K.A.; Schnell, J.D.; Allen, D.A.; Hicke, L. The conserved Pkh-Ypk kinase cascade is required for endocytosis in yeast. J. Cell Biol. 2002, 156, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, K.; Kim, K. Insight into Tor2, a budding yeast microdomain protein. Eur. J. Cell Biol. 2014, 93, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Rispal, D.; Eltschinger, S.; Stahl, M.; Vaga, S.; Bodenmiller, B.; Abraham, Y.; Filipuzzi, I.; Movva, N.R.; Aebersold, R.; Helliwell, S.B.; et al. Target of Rapamycin complex 2 regulates actin polarization and endocytosis via multiple pathways. J. Biol. Chem. 2015, 290, 14963–14978. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.; Schneider, U.; Howald, I.; Schmidt, A.; Hall, M.N. Heat repeats mediate plasma membrane localization of Tor2p in yeast. J. Biol. Chem. 2000, 275, 37011–37020. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, T.W.; Cohen, S.; Diefenbacher, M.; Trautwein, M.; Martin, D.E.; Hall, M.N. Tor1 and Tor2 have distinct locations in live cells. Eukaryot. Cell 2008, 7, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Lee, K.S.; Levin, D.E. A pair of putative protein kinase genes (YPK1 and YPK2) is required for cell growth in Saccharomyces cerevisiae. Mol. Gen. Genet. 1993, 236, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.E.; Fields, F.O.; Kunisawa, R.; Bishop, J.M.; Thorner, J. A candidate protein kinase C gene, Pkc1, is required for the S. cerevisiae cell cycle. Cell 1990, 62, 213–224. [Google Scholar] [CrossRef]
- Leskoske, K.L.; Roelants, F.M.; Martinez Marshall, M.N.; Hill, J.M.; Thorner, J. The stress-sensing TORC2 complex activates yeast AGC-family protein kinase Ypk1 at multiple novel sites. Genetics 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.E.; Schulze, J.O.; Biondi, R.M. AGC kinases, mechanisms of regulation and innovative drug development. Semin. Cancer Biol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Plowman, G.D. The protein kinases of budding yeast: Six score and more. Trends Biochem. Sci. 1997, 22, 18–22. [Google Scholar] [CrossRef]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]
- Rubenstein, E.M.; Schmidt, M.C. Mechanisms regulating the protein kinases of Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Casamayor, A.; Torrance, P.D.; Kobayashi, T.; Thorner, J.; Alessi, D.R. Functional counterparts of mammalian protein kinases PDK1 and SGK in budding yeast. Curr. Biol. 1999, 9, 186–197. [Google Scholar] [CrossRef]
- Roelants, F.M.; Torrance, P.D.; Bezman, N.; Thorner, J. Pkh1 and Pkh2 differentially phosphorylate and activate Ypk1 and Ykr2 and define protein kinase modules required for maintenance of cell wall integrity. Mol. Biol. Cell 2002, 13, 3005–3028. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Torrance, P.D.; Thorner, J. Differential roles of PDK1- and PDK2-phosphorylation sites in the yeast AGC kinases Ypk1, Pkc1 and Sch9. Microbiology 2004, 150, 3289–3304. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Komander, D.; van Aalten, D.M.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell. Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Baltz, A.G.; Trott, A.E.; Fereres, S.; Thorner, J. A protein kinase network regulates the function of aminophospholipid flippases. Proc. Natl. Acad. Sci. USA 2010, 107, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Biondi, R.M.; Kieloch, A.; Currie, R.A.; Deak, M.; Alessi, D.R. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001, 20, 4380–4390. [Google Scholar] [CrossRef] [PubMed]
- Biondi, R.M.; Komander, D.; Thomas, C.C.; Lizcano, J.M.; Deak, M.; Alessi, D.R.; van Aalten, D.M. High resolution crystal structure of the human PDK1 catalytic domain defines the regulatory phosphopeptide docking site. EMBO J. 2002, 21, 4219–4228. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Thompson, V.; Good, V.M.; Hess, D.; Hemmings, B.A.; Barford, D. Molecular mechanism for the regulation of protein kinase B/AKT by hydrophobic motif phosphorylation. Mol. Cell 2002, 9, 1227–1240. [Google Scholar] [CrossRef]
- Voordeckers, K.; Kimpe, M.; Haesendonckx, S.; Louwet, W.; Versele, M.; Thevelein, J.M. Yeast 3-phosphoinositide-dependent protein kinase-1 (PDK1) orthologs Pkh1-3 differentially regulate phosphorylation of protein kinase A (PKA) and the protein kinase B (PKB)/S6K ortholog Sch9. J. Biol. Chem. 2011, 286, 22017–22027. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Qadota, H.; Python, C.P.; Anraku, Y.; Ohya, Y.; Levin, D.E. Activation of yeast protein kinase C by Rho1 GTPase. J. Biol. Chem. 1996, 271, 9193–9196. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.; Settleman, J. The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol. Cell. Biol. 1997, 17, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Durgan, J.; Magalhaes, A.; Hall, A. Rho GTPases regulate PRK2/PKN2 to control entry into mitosis and exit from cytokinesis. EMBO J. 2007, 26, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.W.; Magalhaes, A.; Hall, A. The Rho target PRK2 regulates apical junction formation in human bronchial epithelial cells. Mol. Cell. Biol. 2011, 31, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Maurer, R.A. Isolation of a yeast protein kinase gene by screening with a mammalian protein kinase cDNA. DNA 1988, 7, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.; Ohno, S.; Matsumoto, S.; Yahara, I.; Suzuki, K. A novel yeast gene coding for a putative protein kinase. Gene 1989, 76, 177–180. [Google Scholar] [PubMed]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 546–563. [Google Scholar] [CrossRef] [PubMed]
- Mewes, H.W.; Albermann, K.; Bähr, M.; Frishman, D.; Gleissner, A.; Hani, J.; Heumann, K.; Kleine, K.; Maierl, A.; Oliver, S.G.; et al. Overview of the yeast genome. Nature 1997, 387, 7–65. [Google Scholar] [CrossRef] [PubMed]
- Hittinger, C.T.; Rokas, A.; Bai, F.Y.; Boekhout, T.; Gonçalves, P.; Jeffries, T.W.; Kominek, J.; Lachance, M.A.; Libkind, D.; Rosa, C.A.; et al. Genomics and the making of yeast biodiversity. Curr. Opin. Genet. Dev. 2015, 35, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H.; Armisén, D.; Proux-Wera, E.; ÓhÉigeartaigh, S.S.; Azam, H.; Gordon, J.L.; Byrne, K.P. Clade- and species-specific features of genome evolution in the Saccharomycetaceae. FEMS Yeast Res. 2015, 15, fov035. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Fujioka, Y.; Suzuki, N.N.; Inagaki, F.; Wullschleger, S.; Loewith, R.; Hall, M.N.; Ohsumi, Y. Tor2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol. Cell. Biol. 2005, 25, 7239–7248. [Google Scholar] [CrossRef] [PubMed]
- Aronova, S.; Wedaman, K.; Aronov, P.A.; Fontes, K.; Ramos, K.; Hammock, B.D.; Powers, T. Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab 2008, 7, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, M.; Schmelzle, T.; Yamaguchi, K.; Irie, K.; Hall, M.N.; Matsumoto, K. PDK1 homologs activate the Pkc11-mitogen-activated protein kinase pathway in yeast. Mol. Cell. Biol. 1999, 19, 8344–8352. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O.; Betts, M.J.; Gvozdenovic-Jeremic, J.; Maeda, K.; Matetzki, C.; Aguilar-Gurrieri, C.; Beltran-Alvarez, P.; Bonn, S.; Fernández-Tornero, C.; Jensen, L.J.; et al. A systematic screen for protein–lipid interactions in Saccharomyces cerevisiae. Mol. Syst. Biol. 2010, 6, 430. [Google Scholar] [CrossRef] [PubMed]
- Olivera-Couto, A.; Graña, M.; Harispe, L.; Aguilar, P.S. The eisosome core is composed of BAR domain proteins. Mol. Biol. Cell 2011, 22, 2360–2372. [Google Scholar] [CrossRef] [PubMed]
- Gavin, A.C.; European Molecular Biology Laboratory (EMBL), Heidelberg, Germany. Personal communication, 2014.
- McDonald, N.A.; Gould, K.L. Linking up at the BAR: Oligomerization and F-BAR protein function. Cell Cycle 2016, 15, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Karotki, L.; Huiskonen, J.T.; Stefan, C.J.; Ziółkowska, N.E.; Roth, R.; Surma, M.A.; Krogan, N.J.; Emr, S.D.; Heuser, J.; Grünewald, K.; et al. Eisosome proteins assemble into a membrane scaffold. J. Cell Biol. 2011, 195, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Brickner, J.H.; Aguilar, P.S.; Bernales, S.; Pantoja, C.; Walter, P. Eisosomes mark static sites of endocytosis. Nature 2006, 439, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.M.; Konopka, J.B. Fungal membrane organization: The eisosome concept. Annu. Rev. Microbiol. 2014, 68, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Moreira, K.E.; Schuck, S.; Schrul, B.; Fröhlich, F.; Moseley, J.B.; Walther, T.C.; Walter, P. Seg1 controls eisosome assembly and shape. J. Cell Biol. 2012, 198, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Tashiro, K.; Muta, S.; Ozawa, R.; Chiba, T.; Nishizawa, M.; Yamamoto, K.; Kuhara, S.; Sakaki, Y. Toward a protein–protein interaction map of the budding yeast: A comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar] [PubMed]
- Gavin, A.C.; Bösche, M.; Krause, R.; Grandi, P.; Marzioch, M.; Bauer, A.; Schultz, J.; Rick, J.M.; Michon, A.M.; Cruciat, C.M.; et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.; Gruhler, A.; Heilbut, A.; Bader, G.D.; Moore, L.; Adams, S.L.; Millar, A.; Taylor, P.; Bennett, K.; Boutilier, K.; et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 415, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Thorner, J. Deletion analysis of Pkh1 sequence elements required for association with eisosome component Pil1. Unpublished work. 2017. [Google Scholar]
- Lu, R.; Drubin, D.G.; Sun, Y. Clathrin-mediated endocytosis in budding yeast at a glance. J. Cell Sci. 2016, 129, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Brach, T.; Specht, T.; Kaksonen, M. Reassessment of the role of plasma membrane domains in the regulation of vesicular traffic in yeast. J. Cell Sci. 2011, 124, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lester, R.L.; Dickson, R.C. Pil1p and lsp1p negatively regulate the 3-phosphoinositide-dependent protein kinase-like kinase Pkh1p and downstream signaling pathways Pkc1p and Ypk1p. J. Biol. Chem. 2004, 279, 22030–22038. [Google Scholar] [CrossRef] [PubMed]
- Kabeche, R.; Howard, L.; Moseley, J.B. Eisosomes provide membrane reservoirs for rapid expansion of the yeast plasma membrane. J. Cell Sci. 2015, 128, 4057–4062. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Seaman, M.; Nemoto, Y.; Daniell, L.; Suchy, S.F.; Emr, S.; De Camilli, P.; Nussbaum, R. Disruption of three phosphatidylinositol-polyphosphate 5-phosphatase genes from Saccharomyces cerevisiae results in pleiotropic abnormalities of vacuole morphology, cell shape, and osmohomeostasis. Eur. J. Cell Biol. 1997, 74, 350–360. [Google Scholar] [PubMed]
- Stolz, L.E.; Huynh, C.V.; Thorner, J.; York, J.D. Identification and characterization of an essential family of inositol polyphosphate 5-phosphatases (Inp51, Inp52 and Inp53 gene products) in the yeast Saccharomyces cerevisiae. Genetics 1998, 148, 1715–1729. [Google Scholar] [PubMed]
- Fröhlich, F.; Christiano, R.; Olson, D.K.; Alcazar-Roman, A.; DeCamilli, P.; Walther, T.C. A role for eisosomes in maintenance of plasma membrane phosphoinositide levels. Mol. Biol. Cell 2014, 25, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Morales-Johansson, H.; Jenoe, P.; Cooke, F.T.; Hall, M.N. Negative regulation of phosphatidylinositol 4,5-bisphosphate levels by the Inp51-associated proteins Tax4 and Irs4. J. Biol. Chem. 2004, 279, 39604–39610. [Google Scholar] [CrossRef] [PubMed]
- Kabeche, R.; Roguev, A.; Krogan, N.J.; Moseley, J.B. A Pil1-Sle1-Syj1-Tax4 functional pathway links eisosomes with PI(4,5)P2 regulation. J. Cell Sci. 2014, 127, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Kabeche, R.; Madrid, M.; Cansado, J.; Moseley, J.B. Eisosomes regulate phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) cortical clusters and mitogen-activated protein (MAP) kinase signaling upon osmotic stress. J. Biol. Chem. 2015, 290, 25960–25973. [Google Scholar] [CrossRef] [PubMed]
- Vonkova, I.; Saliba, A.E.; Deghou, S.; Annand, K.; Ceschia, S.; Doerks, T.; Galih, A.; Kugler, K.G.; Maeda, K.; Rybin, V.; et al. Lipid cooperativity as a general membrane-recruitment principle for PH domains. Cell Rep. 2015, 12, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Ohya, Y.; Nakano, A.; Anraku, Y. Genetic interactions among genes involved in the Stt4-Pkc1 pathway of Saccharomyces cerevisiae. Mol. Gen. Genet. 1994, 242, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Saba, J.D.; Obeid, L.M. The dihydrosphingosine-1-phosphate phosphatases of Saccharomyces cerevisiae are important regulators of cell proliferation and heat stress responses. Biochem J. 1999, 342, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Skrzypek, M.S.; Nagiec, M.M.; Lester, R.L.; Dickson, R.C. Analysis of phosphorylated sphingolipid long-chain bases reveals potential roles in heat stress and growth control in Saccharomyces. J. Bacteriol. 1999, 181, 1134–1140. [Google Scholar] [PubMed]
- Friant, S.; Lombardi, R.; Schmelzle, T.; Hall, M.N.; Riezman, H. Sphingoid base signaling via Pkh kinases is required for endocytosis in yeast. EMBO J. 2001, 20, 6783–6792. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, X.; Lester, R.L.; Dickson, R.C. The sphingoid long chain base phytosphingosine activates AGC-type protein kinases in Saccharomyces cerevisiae including Ypk1, Ypk2, and Sch9. J. Biol. Chem. 2005, 280, 22679–22687. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Gruhler, A.; Liu, Y.; Jensen, O.; Dickson, R. The sphingolipid long-chain base-Pkh1/2-Ypk1/2 signaling pathway regulates eisosome assembly and turnover. J. Biol. Chem. 2008, 283, 10433–10444. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Okamoto, Y.; Mao, C. Yeast sphingolipids: Metabolism and biology. Biochim Biophys Acta 2002, 1585, 163–171. [Google Scholar] [CrossRef]
- Dickson, R.C.; Sumanasekera, C.; Lester, R.L. Functions and metabolism of sphingolipids in Saccharomyces cerevisiae. Prog. Lipid Res. 2006, 45, 447–465. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.C. Roles for sphingolipids in Saccharomyces cerevisiae. Adv. Exp. Med. Biol. 2010, 688, 217–231. [Google Scholar] [PubMed]
- Fröhlich, F.; Moreira, K.; Aguilar, P.S.; Hubner, N.C.; Mann, M.; Walter, P.; Walther, T.C. A genome-wide screen for genes affecting eisosomes reveals Nce102 function in sphingolipid signaling. J Cell Biol. 2009, 185, 1227–1242. [Google Scholar]
- García-Marqués, S.; Randez-Gil, F.; Dupont, S.; Garre, E.; Prieto, J.A. Sng1 associates with Nce102 to regulate the yeast Pkh-Ypk signalling module in response to sphingolipid status. Biochim. Biophys. Acta 2016, 1863, 1319–1333. [Google Scholar] [CrossRef] [PubMed]
- Haesendonckx, S.; Tudisca, V.; Voordeckers, K.; Moreno, S.; Thevelein, J.M.; Portela, P. The activation loop of PKA catalytic isoforms is differentially phosphorylated by Pkh protein kinases in Saccharomyces cerevisiae. Biochem J. 2012, 448, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Aguilar, P.S.; Fröhlich, F.; Chu, F.; Moreira, K.; Burlingame, A.L.; Walter, P. Pkh-kinases control eisosome assembly and organization. EMBO J. 2007, 26, 4946–4955. [Google Scholar] [CrossRef] [PubMed]
- Kimpe, M.; Voordeckers, K.; Thevelein, J.M.; Van Zeebroeck, G. Pkh1 interacts with and phosphorylates components of the yeast Gcn2/eIF2α system. Biochem. Biophys. Res. Commun. 2012, 419, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Morvan, J.; Rinaldi, B.; Friant, S. Pkh1/2-dependent phosphorylation of Vps27 regulates ESCRT-I recruitment to endosomes. Mol. Biol. Cell 2012, 23, 4054–4064. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Stenmark, H.; Emr, S.D. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb. Perspect. Biol. 2013, 5, a016766. [Google Scholar] [CrossRef] [PubMed]
- Schöneberg, J.; Lee, I.H.; Iwasa, J.H.; Hurley, J.H. Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 2017, 18, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, D.; Kobayashi, T.; Sun, Y.; Fujita, T.; Takematsu, H.; Kozutsumi, Y. The requirement for the hydrophobic motif phosphorylation of Ypk1 in yeast differs depending on the downstream events, including endocytosis, cell growth, and resistance to a sphingolipid biosynthesis inhibitor, ISP-1. Arch. Biochem. Biophys. 2005, 437, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, J.F.; Muir, A.; Ramachandran, S.; Thorner, J.; Sá-Correia, I. Sphingolipid biosynthesis upregulation by TOR complex 2-Ypk1 signaling during yeast adaptive response to acetic acid stress. Biochem. J. 2016, 473, 4311–4325. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Good, V.M.; Thompson, V.; Hemmings, B.A.; Barford, D. Crystal structure of an activated AKT/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 2002, 9, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Grodsky, N.; Li, Y.; Bouzida, D.; Love, R.; Jensen, J.; Nodes, B.; Nonomiya, J.; Grant, S. Structure of the catalytic domain of human protein kinase C β II complexed with a bisindolylmaleimide inhibitor. Biochemistry 2006, 45, 13970–13981. [Google Scholar] [CrossRef] [PubMed]
- Hauge, C.; Antal, T.L.; Hirschberg, D.; Doehn, U.; Thorup, K.; Idrissova, L.; Hansen, K.; Jensen, O.N.; Jørgensen, T.J.; Biondi, R.M.; et al. Mechanism for activation of the growth factor-activated AGC kinases by turn motif phosphorylation. EMBO J. 2007, 26, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of AKT kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, V.; Ouyang, W.; Wei, H.; Soto, N.; Lazorchak, A.; Gould, C.; Lowry, C.; Newton, A.C.; Mao, Y.; Miao, R.Q.; et al. The mammalian target of rapamycin complex 2 controls folding and stability of AKT and protein kinase C. EMBO J. 2008, 27, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and AKT turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Fadri, M.; Daquinag, A.; Wang, S.; Xue, T.; Kunz, J. The pleckstrin homology domain proteins Slm1 and Slm2 are required for actin cytoskeleton organization in yeast and bind phosphatidylinositol-4,5-bisphosphate and TORC2. Mol. Biol. Cell 2005, 16, 1883–1900. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, M.; Audhya, A.; Parsons, A.B.; Boone, C.; Emr, S.D. The phosphatidylinositol 4,5-biphosphate and TORC2 binding proteins Slm1 and Slm2 function in sphingolipid regulation. Mol. Cell. Biol. 2006, 26, 5861–5875. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wang, J.; Ives, H.E.; Pearce, D. mSIN1 protein mediates SGK1 protein interaction with mTORC2 protein complex and is required for selective activation of the epithelial sodium channel. J. Biol. Chem. 2011, 286, 30647–30654. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.L. Identification of SIN1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Morigasaki, S.; Tatebe, H.; Tamanoi, F.; Shiozaki, K. Fission yeast TOR complex 2 activates the AGC-family Gad8 kinase essential for stress resistance and cell cycle control. Cell Cycle 2008, 7, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, K.; Tatebe, H.; Murayama, S.; Kojima, C. Mechanisms that determine the substrate specificity of TOR kinase. In Cell Biology of Yeasts; Cyert, M., Lew, D.J., Sawin, K., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2013; p. 68. [Google Scholar]
- Liao, H.C.; Chen, M.Y. Target of rapamycin complex 2 signals to downstream effector yeast protein kinase 2 (Ypk2) through Adheres-Voraciously-to-Target-of-Rapamycin-2 protein 1 (Avo1) in Saccharomyces cerevisiae. J. Biol. Chem. 2012, 287, 6089–6099. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Oppliger, W.; Hall, M.N. Molecular organization of target of rapamycin complex 2. J. Biol. Chem. 2005, 280, 30697–30704. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Murayama, S.; Yonekura, T.; Hatano, T.; Richter, D.; Furuya, T.; Kataoka, S.; Furuita, K.; Kojima, C.; Shiozaki, K. Substrate specificity of TOR complex 2 is determined by a ubiquitin-fold domain of the Sin1 subunit. Elife 2017, 6, e19594. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Mendrola, J.M.; Audhya, A.; Singh, S.; Keleti, D.; DeWald, D.B.; Murray, D.; Emr, S.D.; Lemmon, M.A. Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol. Cell 2004, 13, 677–688. [Google Scholar] [CrossRef]
- Daquinag, A.; Fadri, M.; Jung, S.Y.; Qin, J.; Kunz, J. The yeast PH domain proteins Slm1 and Slm2 are targets of sphingolipid signaling during the response to heat stress. Mol. Cell. Biol. 2007, 27, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Omnus, D.J.; Manford, A.G.; Bader, J.M.; Emr, S.D.; Stefan, C.J. Phosphoinositide kinase signaling controls ER-PM cross-talk. Mol. Biol. Cell 2016, 27, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Bultynck, G.; Heath, V.L.; Majeed, A.P.; Galan, J.M.; Haguenauer-Tsapis, R.; Cyert, M.S. Slm1 and Slm2 are novel substrates of the calcineurin phosphatase required for heat stress-induced endocytosis of the yeast uracil permease. Mol. Cell. Biol. 2006, 26, 4729–4745. [Google Scholar] [CrossRef] [PubMed]
- Mulet, J.M.; Martin, D.E.; Loewith, R.; Hall, M.N. Mutual antagonism of Target of Rapamycin and calcineurin signaling. J. Biol. Chem. 2006, 281, 33000–33007. [Google Scholar] [CrossRef] [PubMed]
- Audhya, A.; Loewith, R.; Parsons, A.B.; Gao, L.; Tabuchi, M.; Zhou, H.; Boone, C.; Hall, M.N.; Emr, S.D. Genome-wide lethality screen identifies new PI4,5P2 effectors that regulate the actin cytoskeleton. EMBO J. 2004, 23, 3747–3757. [Google Scholar] [CrossRef] [PubMed]
- Shioda, R. Functional analysis of TOR complex 2 and its control of sphingolipid biosynthesis in Saccharomyces cerevisiae. Ph.D. Thesis, University of Basel, Basel, Switzerland, 2006; p. 108. [Google Scholar]
- Pearce, L.R.; Sommer, E.M.; Sakamoto, K.; Wullschleger, S.; Alessi, D.R. Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. Biochem. J. 2011, 436, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Mok, J.; Kim, P.M.; Lam, H.Y.; Piccirillo, S.; Zhou, X.; Jeschke, G.R.; Sheridan, D.L.; Parker, S.A.; Desai, V.; Jwa, M.; et al. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 2010, 3, ra12. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Jeschke, G.R.; Roelants, F.M.; Thorner, J.; Turk, B.E. Reciprocal phosphorylation of yeast glycerol-3-phosphate dehydrogenases in adaptation to distinct types of stress. Mol. Cell. Biol. 2012, 32, 4705–4717. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Forte, G.M.; Smith, D.; Petersen, J. Phosphorylation of the amino-terminus of the AGC kinase Gad8 prevents its interaction with TORC2. Open Biol. 2016, 6, 150189. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Chauhan, N.; Muir, A.; Davis, J.; Menon, A.K.; Levine, T.P.; Thorner, J. Tor complex 2-regulated protein kinase Ypk1 controls sterol distribution by inhibiting StART domain-containing proteins located at plasma membrane-endoplasmic reticulum contact sites. 2017; manuscript in preparation. [Google Scholar]
- Basson, M.E.; Thorsness, M.; Rine, J. Saccharomyces cerevisiae contains two functional genes encoding 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proc. Natl. Acad. Sci. USA 1986, 83, 5563–5567. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y.; Rine, J. Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J. Cell Biol. 1994, 125, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Kuranda, K.; François, J.; Palamarczyk, G. The isoprenoid pathway and transcriptional response to its inhibitors in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2010, 190, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gianoulis, T.A.; Yip, K.Y.; Gerstein, M.; Snyder, M. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell 2010, 143, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Yamamoto, T.; Kishimoto, T.; Noji, T.; Tanaka, K. Protein kinases Fpk1p and Fpk2p are novel regulators of phospholipid asymmetry. Mol. Biol. Cell 2008, 19, 1783–1797. [Google Scholar] [CrossRef] [PubMed]
- Roland, B.P.; Graham, T.R. Decoding P4-ATPase substrate interactions. Crit.Rev. Biochem. Mol. Biol. 2016, 51, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, T.T.; Baldridge, R.D.; Xu, P.; Graham, T.R. Phospholipid flippases: Building asymmetric membranes and transport vesicles. Biochim. Biophys. Acta 2012, 1821, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, B.P.; Raychaudhuri, S.; Natarajan, P.; Abe, F.; Liu, K.; Prinz, W.A.; Graham, T.R. Control of protein and sterol trafficking by antagonistic activities of a type IV P-type ATPase and oxysterol binding protein homologue. Mol. Biol. Cell 2009, 20, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Fujimura-Kamada, K.; Hanamatsu, H.; Kato, U.; Umeda, M.; Kozminski, K.G.; Tanaka, K. Transbilayer phospholipid flipping regulates Cdc42p signaling during polarized cell growth via Rga GTPase-activating proteins. Dev. Cell 2007, 13, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Slaughter, B.D.; Unruh, J.R.; Bradford, W.D.; Alexander, R.; Rubinstein, B.; Li, R. Flippase-mediated phospholipid asymmetry promotes fast Cdc42 recycling in dynamic maintenance of cell polarity. Nat. Cell Biol. 2012, 14, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, M.E.; Couso, I.; Crespo, J.L. The TOR signaling network in the model unicellular green alga Chlamydomonas reinhardtii. Biomolecules 2017, 7. in press. [Google Scholar] [CrossRef] [PubMed]
- van der Mark, V.A.; Elferink, R.P.; Paulusma, C.C. P4 ATPases: Flippases in health and disease. Int. J. Mol. Sci. 2013, 14, 7897–7922. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Lemke, G. TAM receptor signaling and autoimmune disease. Curr. Opin. Immunol. 2010, 22, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Tufail, Y.; Cook, D.; Fourgeaud, L.; Powers, C.J.; Merten, K.; Clark, C.L.; Hoffman, E.; Ngo, A.; Sekiguchi, K.J.; O’Shea, C.C.; et al. Phosphatidylserine exposure controls viral innate immune responses by microglia. Neuron 2017, 93, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Taniguchi, R.; Tanoue, D.; Yamaji, T.; Takematsu, H.; Mori, K.; Fujita, T.; Kawasaki, T.; Kozutsumi, Y. Sli2 (Ypk1), a homologue of mammalian protein kinase SGK, is a downstream kinase in the sphingolipid-mediated signaling pathway of yeast. Mol. Cell. Biol. 2000, 20, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Kozutsumi, Y.; Nakamura, S.; Fujita, T.; Kawasaki, T. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem. Biophys. Res. Commun. 1995, 211, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Ikushiro, H.; Hayashi, H.; Kagamiyama, H. Reactions of serine palmitoyltransferase with serine and molecular mechanisms of the actions of serine derivatives as inhibitors. Biochemistry 2004, 43, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Yeung, B.K. Natural product drug discovery: The successful optimization of ISP-1 and halichondrin b. Curr. Opin. Chem. Biol. 2011, 15, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Dunn, T.M.; Lynch, D.V.; Michaelson, L.V.; Napier, J.A. A post-genomic approach to understanding sphingolipid metabolism in Arabidopsis thaliana. Ann. Bot. 2004, 93, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Megyeri, M.; Riezman, H.; Schuldiner, M.; Futerman, A.H. Making sense of the yeast sphingolipid pathway. J. Mol. Biol. 2016, 428, 4765–4775. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.K.; Fröhlich, F.; Farese, R.V.J.; Walther, T.C. Taming the Sphinx: Mechanisms of cellular sphingolipid homeostasis. Biochim. Biophys. Acta 2016, 1861, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Collins, S.R.; Bodenmiller, B.; Aebersold, R.; Simons, K.; Shevchenko, A.; Ejsing, C.S.; Weissman, J.S. Orm family proteins mediate sphingolipid homeostasis. Nature 2010, 463, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.; Santos, A.; Gable, K.; Epstein, S.; Gururaj, C.; Chymkowitch, P.; Pultz, D.; Rødkær, S.V.; Clay, L.; Bjørås, M.; et al. TORC1 inhibits Gsk3-mediated Elo2 phosphorylation to regulate very long chain fatty acid synthesis and autophagy. Cell Rep. 2013, 5, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Carman, G.M. CTP synthetase and its role in phospholipid synthesis in the yeast Saccharomyces cerevisiae. Prog. Lipid Res. 2008, 47, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, F.; Smith, T.K. The Kennedy pathway—de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Fei, W.; Shui, G.; Zhang, Y.; Krahmer, N.; Ferguson, C.; Kapterian, T.S.; Lin, R.C.; Dawes, I.W.; Brown, A.J.; Li, P.; et al. A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLoS Genet. 2011, 7, e1002201. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, M.; Knittelfelder, O.; Cristobal-Sarramian, A.; Kolb, D.; Wolinski, H.; Kohlwein, S.D. The emergence of lipid droplets in yeast: Current status and experimental approaches. Curr. Genet. 2013, 59, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Goodman, J.M. The lipid droplet—a well-connected organelle. Front. Cell Dev. Biol. 2015, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Pahlman, A.K.; Granath, K.; Ansell, R.; Hohmann, S.; Adler, L. The yeast glycerol 3-phosphatases Gpp1p and Gpp2p are required for glycerol biosynthesis and differentially involved in the cellular responses to osmotic, anaerobic, and oxidative stress. J. Biol. Chem. 2001, 276, 3555–3563. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, S. An integrated view on a eukaryotic osmoregulation system. Curr. Genet. 2015, 61, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Posas, F. Response to hyperosmotic stress. Genetics 2012, 192, 289–318. [Google Scholar] [CrossRef] [PubMed]
- Ansell, R.; Granath, K.; Hohmann, S.; Thevelein, J.M.; Adler, L. The two isoenzymes for yeast NAD+-dependent glycerol 3-phosphate dehydrogenase encoded by Gpd1 and Gpd2 have distinct roles in osmoadaptation and redox regulation. EMBO J. 1997, 16, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Albertyn, J.; Hohmann, S.; Thevelein, J.M.; Prior, B.A. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in Saccharomyces cerevisiae, and its expression is regulated by the high-osmolarity glycerol response pathway. Mol. Cell. Biol. 1994, 14, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Reiter, W.; Dohnal, I.; Gregori, C.; Beese-Sims, S.; Kuchler, K.; Ammerer, G.; Levin, D.E. MAPK Hog1 closes the S. cerevisiae glycerol channel Fps1 by phosphorylating and displacing its positive regulators. Genes Dev. 2013, 27, 2590–2601. [Google Scholar] [CrossRef] [PubMed]
- Tamás, M.J.; Luyten, K.; Sutherland, F.C.; Hernandez, A.; Albertyn, J.; Valadi, H.; Li, H.; Prior, B.A.; Kilian, S.G.; Ramos, J.; et al. Fps1p controls the accumulation and release of the compatible solute glycerol in yeast osmoregulation. Mol. Microbiol. 1999, 31, 1087–1104. [Google Scholar] [CrossRef] [PubMed]
- Babazadeh, R.; Furukawa, T.; Hohmann, S.; Furukawa, K. Rewiring yeast osmostress signalling through the MAPK network reveals essential and non-essential roles of Hog1 in osmoadaptation. Sci. Rep. 2014, 4, 4697. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, C.G.; O’Donnell, A.F.; Prosser, D.C.; Augustine, A.A.; Goldman, A.; Brodsky, J.L.; Cyert, M.S.; Wendland, B.; Thorner, J. Specific α-arrestins negatively regulate Saccharomyces cerevisiae pheromone response by down-modulating the G-protein coupled receptor Ste2. Mol. Cell. Biol. 2014, 34, 2660–2681. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; MacGurn, J.A.; Chu, T.; Stefan, C.J.; Emr, S.D. Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell 2008, 135, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Nikko, E.; Pelham, H.R. Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic 2009, 10, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Becuwe, M.; Herrador, A.; Haguenauer-Tsapis, R.; Vincent, O.; Léon, S. Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem. Res. Int. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Cope, M.J.; Yang, S.; Shang, C.; Drubin, D.G. Novel protein kinases Ark1p and Prk1p associate with and regulate the cortical actin cytoskeleton in budding yeast. J. Cell Biol. 1999, 144, 1203–1218. [Google Scholar] [CrossRef] [PubMed]
- Dodou, E.; Treisman, R. The Saccharomyces cerevisiae MADS-box transcription factor Rlm1 is a target for the Mpk1 mitogen-activated protein kinase pathway. Mol. Cell. Biol. 1997, 17, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- De Nadal, E.; Casadomé, L.; Posas, F. Targeting the Mef2-like transcription factor Smp1 by the stress-activated Hog1 mitogen-activated protein kinase. Mol. Cell. Biol. 2003, 23, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Muir, A. Systematic identification of proteins regulated by the TOR complex2-dependent kinase Ypk1 in Saccharomyces cerevisiae. Ph.D. thesis, University of California, Berkeley, Berkeley, CA, USA, 2015; p. 123. [Google Scholar]
- Ruiz-Roig, C.; Noriega, N.; Duch, A.; Posas, F.; de Nadal, E. The Hog1 SAPK controls the Rtg1/Rtg3 transcriptional complex activity by multiple regulatory mechanisms. Mol. Biol. Cell 2012, 23, 4286–4296. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Huang, X.; Kropat, J.; Henras, A.; Merchant, S.S.; Dickson, R.C.; Chanfreau, G.F. Sphingolipid signaling mediates iron toxicity. Cell Metab. 2012, 16, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, S.; Roelants, F.M.; Thorner, J. Microarray analysis of the Smp1-regulated transcriptional program. unpublished work. 2017. [Google Scholar]
- Yerlikaya, S.; Meusburger, M.; Kumari, R.; Huber, A.; Anrather, D.; Costanzo, M.; Boone, C.; Ammerer, G.; Baranov, P.V.; Loewith, R. TORC1 and TORC2 work together to regulate ribosomal protein S6 phosphorylation in Saccharomyces cerevisiae. Mol. Biol. Cell 2016, 27, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Gelperin, D.; Horton, L.; DeChant, A.; Hensold, J.; Lemmon, S.K. Loss of Ypk1 function causes rapamycin sensitivity, inhibition of translation initiation and synthetic lethality in 14-3-3-deficient yeast. Genetics 2002, 161, 1453–1464. [Google Scholar] [PubMed]
- Clarkson, B.K.; Gilbert, W.V.; Doudna, J.A. Functional overlap between eIF4G isoforms in Saccharomyces cerevisiae. PLoS ONE 2010, 5, e9114. [Google Scholar] [CrossRef] [PubMed]
- Niles, B.J.; Joslin, A.C.; Fresques, T.; Powers, T. TOR complex 2-Ypk1 signaling maintains sphingolipid homeostasis by sensing and regulating ROS accumulation. Cell Rep. 2014, 6, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Niles, B.J.; Powers, T. TOR complex 2-Ypk1 signaling regulates actin polarization via reactive oxygen species. Mol. Biol. Cell 2014, 25, 3962–3972. [Google Scholar] [CrossRef] [PubMed]
- Vlahakis, A.; Graef, M.; Nunnari, J.; Powers, T. TOR complex 2-Ypk1 signaling is an essential positive regulator of the general amino acid control response and autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 10586–10591. [Google Scholar] [CrossRef] [PubMed]
- Vlahakis, A.; Powers, T. A role for TOR complex 2 signaling in promoting autophagy. Autophagy 2014, 10, 2085–2086. [Google Scholar] [CrossRef] [PubMed]
- Grosshans, B.L.; Grötsch, H.; Mukhopadhyay, D.; Fernández, I.M.; Pfannstiel, J.; Idrissi, F.; Lechner, J.; Riezman, H.; Geli, M. TEDS site phosphorylation of the yeast myosins I is required for ligand-induced but not for constitutive endocytosis of the G protein-coupled receptor Ste2p. J. Biol. Chem. 2006, 281, 11104–11114. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.L.; Lee, L.; Pierce, B.D.; Maldonado-Báez, L.; Drubin, D.G.; Wendland, B. Interaction of the endocytic scaffold protein Pan1 with the type I myosins contributes to the late stages of endocytosis. Mol. Biol. Cell 2007, 18, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lytvyn, V.; Thomas, D.Y.; Leberer, E. The phosphorylation site for Ste20p-like protein kinases is essential for the function of myosin-I in yeast. J. Biol. Chem. 1997, 272, 30623–30626. [Google Scholar] [CrossRef] [PubMed]
- Sakchaisri, K.; Asano, S.; Yu, L.R.; Shulewitz, M.J.; Park, C.J.; Park, J.E.; Cho, Y.W.; Veenstra, T.D.; Thorner, J.; Lee, K.S. Coupling morphogenesis to mitotic entry. Proc. Natl. Acad. Sci. USA 2004, 101, 4124–4129. [Google Scholar] [CrossRef] [PubMed]
- Versele, M.; Thorner, J. Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. J. Cell Biol. 2004, 164, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Specht, K.M.; Shokat, K.M. The emerging power of chemical genetics. Curr. Opin. Cell Biol. 2002, 14, 155–159. [Google Scholar] [CrossRef]
- Kliegman, J.I.; Fiedler, D.; Ryan, C.J.; Xu, Y.F.; Su, X.Y.; Thomas, D.; Caccese, M.C.; Cheng, A.; Shales, M.; Rabinowitz, J.D.; et al. Chemical genetics of rapamycin-insensitive TORC2 in S. cerevisiae. Cell Rep. 2013, 5, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Filipuzzi, I.; Stahl, M.; Helliwell, S.B.; Studer, C.; Hoepfner, D.; Seeber, A.; Loewith, R.; Movva, N.R.; Gasser, S.M. TORC2 signaling pathway guarantees genome stability in the face of DNA strand breaks. Mol. Cell 2013, 51, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, A.M.; Cyert, M.S. Calcineurin acts through the Crz1/Tcn1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 1997, 11, 3432–3444. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.S.; Kliegman, J.I.; Shokat, K.M. The logic and design of analog-sensitive kinases and their small molecule inhibitors. Methods Enzymol. 2014, 548, 189–213. [Google Scholar] [PubMed]
- Vlahakis, A.; Lopez Muniozguren, N.; Powers, T. Calcium channel regulator Mid1 links TORC2-mediated changes in mitochondrial respiration to autophagy. J. Cell Biol. 2016, 215, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Nomura, W.; Inoue, Y. Methylglyoxal activates the Target of Rapamycin complex 2-protein kinase C signaling pathway in Saccharomyces cerevisiae. Mol. Cell. Biol. 2015, 35, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Ikeda, E.; Uno, I.; Mitsuzawa, H. Characterization of a staurosporine- and temperature-sensitive mutant, stt1, of Saccharomyces cerevisiae: STT1 is allelic to PKC1. Mol. Gen. Genet. 1992, 231, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Rüegg, U.T.; Burgess, G.M. Staurosporine, K-252 and UCN-01: Potent but non-specific inhibitors of protein kinases. Trends Pharmacol. Sci. 1989, 10, 218–220. [Google Scholar] [CrossRef]
- Paravicini, G.; Cooper, M.; Friedli, L.; Smith, D.J.; Carpentier, J.L.; .Klig, L.S.; Payton, M.A. The osmotic integrity of the yeast cell requires a functional PKC1 gene product. Mol. Cell. Biol. 1992, 12, 4896–4905. [Google Scholar] [CrossRef] [PubMed]
- Baymiller, J.; McCullough, J.E. Saccharomyces cerevisiae cell lysis mutations cly5 and cly7 define temperature-sensitive alleles of PKC1, the gene encoding yeast protein kinase C. Yeast 1997, 13, 305–312. [Google Scholar] [CrossRef]
- Shimizu, J.; Yoda, K.; Yamasaki, M. The hypo-osmolarity-sensitive phenotype of the Saccharomyces cerevisiae hpo2 mutant is due to a mutation in PKC1, which regulates expression of β-glucanase. Mol. Gen. Genet. 1994, 242, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.E.; Bartlett-Heubusch, E. Mutants in the S. cerevisiae PKC1 gene display a cell cycle-specific osmotic stability defect. J. Cell Biol. 1992, 116, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Schmelzle, T.; Helliwell, S.B.; Hall, M.N. Yeast protein kinases and the Rho1 exchange factor Tus1 are novel components of the cell integrity pathway in yeast. Mol. Cell. Biol. 2002, 22, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.E.; Bowers, B.; Chen, C.Y.; Kamada, Y.; Watanabe, M. Dissecting the protein kinase C/MAP kinase signalling pathway of Saccharomyces cerevisiae. Cell. Mol. Biol. Res. 1994, 40, 229–239. [Google Scholar] [PubMed]
- Fischer, R.; Zekert, N.; Takeshita, N. Polarized growth in fungi—interplay between the cytoskeleton, positional markers and membrane domains. Mol. Microbiol. 2008, 68, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.L.; Shiau, Y.S.; Chen, M.Y. Saccharomyces cerevisiae Tsc11/Avo3 participates in regulating cell integrity and functionally interacts with components of the Tor2 complex. Curr. Genet. 2005, 47, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Madaule, P.; Axel, R.; Myers, A.M. Characterization of two members of the rho gene family from the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1987, 84, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Pruyne, D.; Legesse-Miller, A.; Gao, L.; Dong, Y.; Bretscher, A. Mechanisms of polarized growth and organelle segregation in yeast. Annu. Rev. Cell Dev. Biol. 2004, 20, 559–591. [Google Scholar] [CrossRef] [PubMed]
- Ohya, Y.; Qadota, H.; Anraku, Y.; Pringle, J.R.; Botstein, D. Suppression of yeast geranylgeranyl transferase I defect by alternative prenylation of two target gtpases, Rho1p and Cdc42p. Mol. Biol. Cell 1993, 4, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Bartolini, S.; Pellman, D. Mechanisms for concentrating Rho1 during cytokinesis. Genes Dev. 2009, 23, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Yamochi, W.; Tanaka, K.; Nonaka, H.; Maeda, A.; Musha, T.; Takai, Y. Growth site localization of Rho1 small GTP-binding protein and its involvement in bud formation in Saccharomyces cerevisiae. J. Cell Biol. 1994, 125, 1077–1093. [Google Scholar] [CrossRef] [PubMed]
- Meitinger, F.; Richter, H.; Heisel, S.; Hub, B.; Seufert, W.; Pereira, G. A safeguard mechanism regulates Rho GTOPases to coordinate cytokinesis with the establishment of cell polarity. PLoS Biol. 2013, 11, e1001495. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Tanaka, K.; Imamura, H.; Hihara, T.; Kameyama, T.; Nonaka, H.; Hirano, H.; Matsuura, Y.; Takai, Y. Rom1p and Rom2p are GDP/GTP exchange proteins (GEPs) for the Rho1p small GTP binding protein in Saccharomyces cerevisiae. EMBO J. 1996, 15, 2196–2207. [Google Scholar] [PubMed]
- Philip, B.; Levin, D.E. Wsc1 and Mid2 are cell surface sensors for cell wall integrity signaling that act through Rom2, a guanine nucleotide exchange factor for Rho1. Mol. Cell. Biol. 2001, 21, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Qadota, H.; Python, C.P.; Inoue, S.B.; Arisawa, M.; Anraku, Y.; Zheng, Y.; Watanabe, T.; Levin, D.E.; Ohya, Y. Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-β-glucan synthase. Science 1996, 272, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.; Baginsky, W. In vitro activity of 1,3-β-d-glucan synthase requires the GTP-binding protein Rho1. J. Biol. Chem. 1996, 271, 14604–14609. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, E.M.; Rossio, V.; Hatakeyama, R.; Abe, M.; Ohya, Y.; Yoshida, S. Zds1/Zds2-PP2A-Cdc55 complex specifies signaling output from Rho1 GTPase. J. Cell Biol. 2016, 212, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Maesaki, R.; Ihara, K.; Shimizu, T.; Kuroda, S.; Kaibuchi, K.; Hakoshima, T. The structural basis of Rho effector recognition revealed by the crystal structure of human RhoA complexed with the effector domain of PKN/PRK1. Mol. Cell 1999, 4, 793–803. [Google Scholar] [CrossRef]
- Hutchinson, C.L.; Lowe, P.N.; McLaughlin, S.H.; Mott, H.R.; Owen, D. Differential binding of RhoA, RhoB, and RhoC to protein kinase C-related kinase (PRK) isoforms PRK1, PRK2, and PRK3: PRKs have the highest affinity for RhoB. Biochemistry 2013, 52, 7999–8011. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, H.P.; Lorberg, A.; Heinisch, J.J. Regulation of yeast protein kinase C activity by interaction with the small GTPase Rho1p through its amino-terminal Hr1 domain. Mol. Microbiol. 2002, 44, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Fields, F.O. Biochemical and molecular genetic analysis of protein kinase C function in the yeast Saccharomyces cerevisiae. Ph.D. thesis, University of California, Berkeley, CA, USA, 1991; p. 257. [Google Scholar]
- Dey, P.; Su, W.M.; Han, G.S.; Carman, G.M. Phosphorylation of lipid metabolic enzymes by yeast protein kinase C requires phosphatidylserine and diacylglycerol. J. Lipid Res. 2017, 58, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Nalefski, E.A.; Falke, J.J. The C2 domain calcium-binding motif: Structural and functional diversity. Protein Sci. 1996, 5, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.A.; Evans, J.H.; Landgraf, K.E.; Falke, J.J. Mechanism of specific membrane targeting by C2 domains: Localized pools of target lipids enhance Ca2+ affinity. Biochemistry 2007, 46, 4322–4336. [Google Scholar] [CrossRef] [PubMed]
- Corbalan-Garcia, S.; Gómez-Fernández, J.C. Signaling through C2 domains: More than one lipid target. Biochim. Biophys. Acta 2014, 1838, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Chapa-Y-Lazo, B.; Ayscough, K.R. Apm4, the mu subunit of yeast AP-2 interacts with Pkc1, and mutation of the Pkc1 consensus phosphorylation site Thr176 inhibits AP-2 recruitment to endocytic sites. Commun. Integr. Biol. 2014, 7, e28522. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.E. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2005, 69, 262–291. [Google Scholar] [CrossRef] [PubMed]
- Thai, V.; Dephoure, N.; Weiss, A.; Ferguson, J.; Leitao, R.; Gygi, S.P.; Kellogg, D.R. Protein kinase C controls binding of Igo/ENSA proteins to protein phosphatase 2A in budding yeast. J. Biol. Chem. 2017, 292, 4925–4941. [Google Scholar] [CrossRef] [PubMed]
- Darieva, Z.; Webber, A.; Warwood, S.; Sharrocks, A.D. Protein kinase C coordinates histone H3 phosphorylation and acetylation. Elife 2015, 4, e09886. [Google Scholar] [CrossRef] [PubMed]
- Darieva, Z.; Han, N.; Warwood, S.; Doris, K.S.; Morgan, B.A.; Sharrocks, A.D. Protein kinase C regulates late cell cycle-dependent gene expression. Mol. Cell. Biol. 2012, 32, 4651–4661. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Bolognesi, A.; Juanes, M.A.; Vandermoere, F.; Courtellemont, T.; Pascolutti, R.; Seveno, M.; Barral, Y.; Piatti, S. Rho1- and Pkc1-dependent phosphorylation of the F-bar protein Syp1 contributes to septin ring assembly. Mol. Biol. Cell. 2015, 26, 3245–3262. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; O’Brien, D.J.; Carman, G.M. Phosphorylation of CTP synthetase on Ser36, Ser330, Ser354, and Ser454 regulates the levels of CTP and phosphatidylcholine synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 20785–20794. [Google Scholar] [CrossRef] [PubMed]
- Colón-González, F.; Kazanietz, M.G. C1 domains exposed: From diacylglycerol binding to protein–protein interactions. Biochim. Biophys. Acta 2006, 1761, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Rahman, G.M. C1 domains: Structure and ligand-binding properties. Chem. Rev. 2014, 114, 12108–12131. [Google Scholar] [CrossRef] [PubMed]
- Nomura, W.; Ito, Y.; Inoue, Y. Role of phosphatidylserine in the activation of Rho1-related Pkc1 signaling in Saccharomyces cerevisiae. Cellul. Signal. 2017, 31, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Mukai, H. The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J. Biochem. 2003, 133, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Newton, A.C. Tuning the signalling output of protein kinase C. Biochem. Soc. Trans. 2014, 42, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Denis, V.; Cyert, M.S. Molecular analysis reveals localization of Saccharomyces cerevisiae protein kinase C to sites of polarized growth and Pkc1p targeting to the nucleus and mitotic spindle. Eukaryot. Cell. 2005, 4, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
- Güttler, T.; Madl, T.; Neumann, P.; Deichsel, D.; Corsini, L.; Monecke, T.; Ficner, R.; Sattler, M.; Görlich, D. NES consensus redefined by structures of PKI-type and REV-type nuclear export signals bound to Crm1. Nat. Struct. Mol. Biol. 2010, 17, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Ahn, N.G.; Seger, R.; Krebs, E.G. The mitogen-activated protein kinase activator. Curr. Opin. Cell Biol. 1992, 4, 992–999. [Google Scholar] [CrossRef]
- Cairns, B.R.; Ramer, S.W.; Kornberg, R.D. Order of action of components in the yeast pheromone response pathway revealed with a dominant allele of the Ste11 kinase and the multiple phosphorylation of the Ste7 kinase. Genes Dev. 1992, 6, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Neiman, A.M.; Stevenson, B.J.; Xu, H.P.; Sprague, G.F.J.; Herskowitz, I.; Wigler, M.; Marcus, S. Functional homology of protein kinases required for sexual differentiation in Schizosaccharomyces pombe and Saccharomyces cerevisiae suggests a conserved signal transduction module in eukaryotic organisms. Mol. Biol. Cell 1993, 4, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Gartner, A.; Cade, R.; Ammerer, G.; Errede, B. Pheromone-induced signal transduction in Saccharomyces cerevisiae requires the sequential function of three protein kinases. Mol. Cell. Biol. 1993, 13, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.E.; Thorner, J. Function and regulation in MAPK signaling pathways: Lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, C.G.; Thorner, J. Heterotrimeric G protein-coupled receptor signaling in yeast mating pheromone response. J. Biol. Chem. 2016, 291, 7788–7795. [Google Scholar] [CrossRef] [PubMed]
- Westfall, P.J.; Ballon, D.R.; Thorner, J. When the stress of your environment makes you go HOG wild. Science 2004, 306, 1511–1512. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.C.; Klimenko, E.S.; Thorner, J. Single-cell analysis reveals that insulation maintains signaling specificity between two yeast MAPK pathways with common components. Sci. Signal. 2010, 3, ra75. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175. [Google Scholar] [CrossRef] [PubMed]
- Kock, C.; Dufrêne, Y.F.; Heinisch, J.J. Up against the wall: Is yeast cell wall integrity ensured by mechanosensing in plasma membrane microdomains? Appl. Environ. Microbiol. 2015, 81, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, B.; Montessuit, S.; Friedli, L.; Payton, M.A.; Paravicini, G. Protein kinase C in yeast. Characteristics of the Saccharomyces cerevisiae PKC1 gene product. J. Biol. Chem. 1994, 269, 16821–16828. [Google Scholar] [PubMed]
- Watanabe, M.; Chen, C.Y.; Levin, D.E. Saccharomyces cerevisiae PKC1 encodes a protein kinase C (PKC) homolog with a substrate specificity similar to that of mammalian PKC. J. Biol. Chem. 1994, 269, 16829–16836. [Google Scholar] [PubMed]
- Lee, K.S.; Irie, K.; Gotoh, Y.; Watanabe, Y.; Araki, H.; Nishida, E.; Matsumoto, K.; Levin, D.E. A yeast mitogen-activated protein kinase homolog (Mpk1p) mediates signalling by protein kinase C. Mol. Cell. Biol. 1993, 13, 3067–3075. [Google Scholar] [CrossRef] [PubMed]
- Mascaraque, V.; Hernaez, M.L.; Jimenez-Sanchez, M.; Hansen, R.; Gil, C.; Martin, H.; Cid, V.J.; Molina, M. Phosphoproteomic analysis of protein kinase C signaling in Saccharomyces cerevisiae reveals Slt2 mitogen-activated protein kinase (MAPK)-dependent phosphorylation of eisosome core components. Mol. Cell. Proteomics 2013, 12, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Garrett-Engele, P.; Moilanen, B.; Cyert, M.S. Calcineurin, the Ca2+/calmodulin-dependent protein phosphatase, is essential in yeast mutants with cell integrity defects and in mutants that lack a functional vacuolar H+-ATPase. Mol. Cell. Biol. 1995, 15, 4103–4114. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Jung, U.S.; Garrett-Engele, P.; Roe, T.; Cyert, M.S.; Levin, D.E. Temperature-induced expression of yeast FKS2 is under the dual control of protein kinase C and calcineurin. Mol. Cell. Biol. 1998, 18, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.L.; Bruno, M.E.C.; Carman, G.M. Regulation of yeast CTP synthetase activity by protein kinase C. J. Biol. Chem. 1996, 271, 11113–11119. [Google Scholar] [CrossRef] [PubMed]
- Pascual, F.; Carman, G.M. Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim. Biophys. Acta 2013, 1831, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Su, W.M.; Morgan, J.M.; Han, G.S.; Xu, Z.; Karanasios, S.; Siniossoglou, S.; Carman, G.M. Phosphorylation of phosphatidate phosphatase regulates its membrane association and physiological functions in Saccharomyces cerevisiae: Identification of Ser(602), Thr(723), and Ser(744) as the sites phosphorylated by Cdc28 (Cdk1)-encoded cyclin-dependent kinase. J. Biol. Chem. 2011, 286, 1486–1498. [Google Scholar] [PubMed]
- Hsieh, L.S.; Su, W.M.; Han, G.S.; Carman, G.M. Phosphorylation regulates the ubiquitin-independent degradation of yeast Pah1 phosphatidate phosphatase by the 20S proteasome. J. Biol. Chem. 2015, 290, 11467–11478. [Google Scholar] [CrossRef] [PubMed]
- Su, W.M.; Han, G.S.; Carman, G.M. Yeast Nem1-Spo7 protein phosphatase activity on Pah1 phosphatidate phosphatase is specific for the Pho85-Pho80 protein kinase phosphorylation sites. J. Biol. Chem. 2014, 289, 34699–34708. [Google Scholar] [CrossRef] [PubMed]
- Su, W.M.; Han, G.S.; Carman, G.M. Cross-talk phosphorylations by protein kinase C and Pho85p-Pho80p protein kinase regulate Pah1p phosphatidate phosphatase abundance in Saccharomyces cerevisiae. J. Biol. Chem. 2014, 289, 18818–18830. [Google Scholar] [CrossRef] [PubMed]
- Dubots, E.; Cottier, S.; Péli-Gulli, M.P.; Jaquenoud, M.; Bontron, S.; Schneiter, R.; De Virgilio, C. TORC1 regulates Pah1 phosphatidate phosphatase activity via the Nem1/Spo7 protein phosphatase complex. PLoS ONE 2014, 9, e104194. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T. On the regulation of protein phosphatase 2A and its role in controlling entry into and exit from mitosis. Adv. Biol. Regul. 2013, 53, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Voets, E.; Wolthuis, R.M. Stable government of mitosis by Greatwall: The emperor’s best servant. Mol. Cell. Biol. 2012, 32, 1334–1336. [Google Scholar] [CrossRef] [PubMed]
- Bontron, S.; Jaquenoud, M.; Vaga, S.; Talarek, N.; Bodenmiller, B.; Aebersold, R.; De Virgilio, C. Yeast endosulfines control entry into quiescence and chronological life span by inhibiting protein phosphatase 2A. Cell Rep. 2013, 3, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.C.; Filter, J.J.; Blake-Hodek, K.A.; Wadzinski, B.E.; Fuda, N.J.; Shalloway, D.; Goldberg, M.L. Greatwall-phosphorylated endosulfine is both an inhibitor and a substrate of PP2A-B55 heterotrimers. Elife 2014, 3, e01695. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Neo, S.P.; Yu, X.; Cai, M. A novel septin-associated protein, Syp1p, is required for normal cell cycle-dependent septin cytoskeleton dynamics in yeast. Genetics 2008, 180, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, C.P.; Smolka, M.B.; Payne, S.H.; Bafna, V.; Eng, J.; Zhou, H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics 2008, 7, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Soulard, A.; Cremonesi, A.; Moes, S.; Schütz, F.; Jenö, P.; Hall, M.N. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol. Biol. Cell 2010, 21, 3475–3486. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villén, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Annan, R.S.; Huddleston, M.J.; Carr, S.A.; Reynard, G.; Deshaies, R.J. Phosphorylation of Sic1p by G1 CDK required for its degradation and entry into S phase. Science 1997, 278, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Su, B.M.; von Wulffen, J.; Ramachandran, S.; Sartorel, E.; Trott, A.E.; Thorner, J. Protein kinase Gin4 negatively regulates flippase function and controls plasma membrane asymmetry. J. Cell. Biol. 2015, 208, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Boettner, D.R.; D’Agostino, J.L.; Torres, O.T.; Daugherty-Clarke, K.; Uygur, A.; Reider, A.; Wendland, B.; Lemmon, S.K.; Goode, B.L. The F-bar protein Syp1 negatively regulates WASP-Arp2/3 complex activity during endocytic patch formation. Curr. Biol. 2009, 19, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Reider, A.; Barker, S.L.; Mishra, S.K.; Im, Y.J.; Maldonado-Báez, L.; Hurley, J.H.; Traub, L.M.; Wendland, B. Syp1 is a conserved endocytic adaptor that contains domains involved in cargo selection and membrane tubulation. EMBO J. 2009, 28, 3103–3116. [Google Scholar] [CrossRef] [PubMed]
- Stimpson, H.E.; Toret, C.P.; Cheng, A.T.; Pauly, B.S.; Drubin, D.G. Early-arriving Syp1p and Ede1p function in endocytic site placement and formation in budding yeast. Mol. Biol. Cell 2009, 20, 4640–4651. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Costanzo, M.; Boone, C.; Dickson, R.C. Nutrients and the Pkh1/2 and Pkc1 protein kinases control mRNA decay and P-body assembly in yeast. J. Biol. Chem. 2011, 286, 8759–8770. [Google Scholar] [CrossRef] [PubMed]
- Pujol, N.; Bonet, C.; Vilella, F.; Petkova, M.I.; Mozo-Villarías, A.; de la Torre-Ruiz, M.A. Two proteins from Saccharomyces cerevisiae: Pfy1 and Pkc1, play a dual role in activating actin polymerization and in increasing cell viability in the adaptive response to oxidative stress. FEMS Yeast Res. 2009, 9, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, S.B.; Howald, I.; Barbet, N.; Hall, M.N. Tor2 is part of two related signaling pathways coordinating cell growth in Saccharomyces cerevisiae. Genetics 1998, 148, 99–112. [Google Scholar] [PubMed]
- Pracheil, T.; Thornton, J.; Liu, Z. TORC2 signaling is antagonized by protein phosphatase 2A and the FAR complex in Saccharomyces cerevisiae. Genetics 2012, 190, 1325–1339. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, R.; Kono, K.; Yoshida, S. Ypk1 and Ypk2 kinases maintain Rho1 at the plasma membrane by flippase-dependent lipid remodeling after membrane stresses. J. Cell Sci. 2017, 130, 1169–1178. [Google Scholar] [PubMed]
- Nomura, W.; Inoue, Y. Contribution of phosphatidylserine to Rho1- and Pkc1-related repolarization of the actin cytoskeleton under stressed conditions in Saccharomyces cerevisiae. Small GTPases 2017, in press. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roelants, F.M.; Leskoske, K.L.; Martinez Marshall, M.N.; Locke, M.N.; Thorner, J. The TORC2‐Dependent Signaling Network in the Yeast Saccharomyces cerevisiae. Biomolecules 2017, 7, 66. https://doi.org/10.3390/biom7030066
Roelants FM, Leskoske KL, Martinez Marshall MN, Locke MN, Thorner J. The TORC2‐Dependent Signaling Network in the Yeast Saccharomyces cerevisiae. Biomolecules. 2017; 7(3):66. https://doi.org/10.3390/biom7030066
Chicago/Turabian StyleRoelants, Françoise M., Kristin L. Leskoske, Maria Nieves Martinez Marshall, Melissa N. Locke, and Jeremy Thorner. 2017. "The TORC2‐Dependent Signaling Network in the Yeast Saccharomyces cerevisiae" Biomolecules 7, no. 3: 66. https://doi.org/10.3390/biom7030066
APA StyleRoelants, F. M., Leskoske, K. L., Martinez Marshall, M. N., Locke, M. N., & Thorner, J. (2017). The TORC2‐Dependent Signaling Network in the Yeast Saccharomyces cerevisiae. Biomolecules, 7(3), 66. https://doi.org/10.3390/biom7030066