
Why are Functional Amyloids Non-Toxic in Humans?


Abstract
:1. Introduction
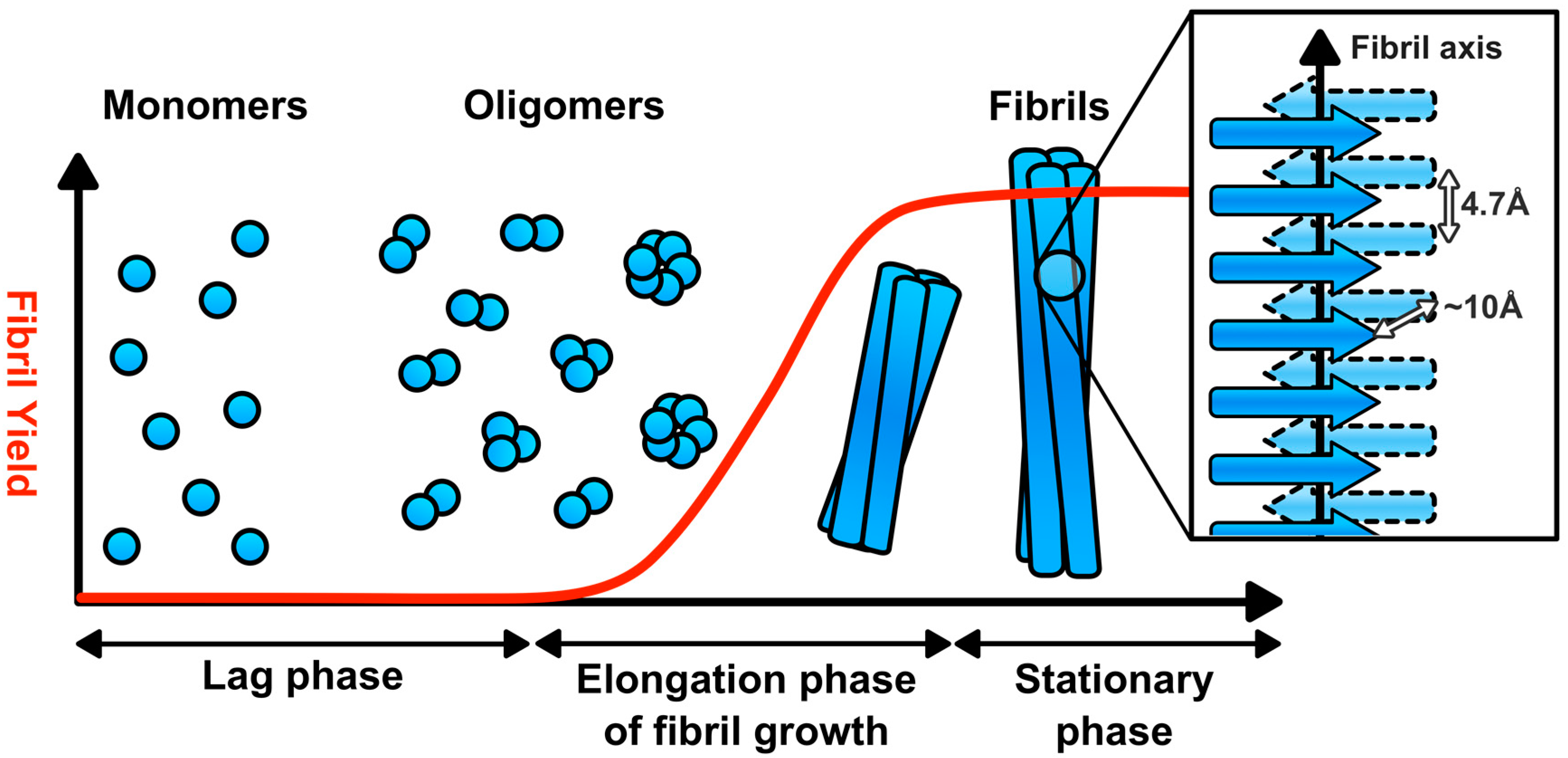
2. Amyloid Fibril Assembly and Structure
3. The Amyloidoses and Mechanisms of Amyloid Toxicity
4. Functional Amyloids and Their Physiological Roles in Humans
5. Does Functional Amyloid Fibril Assembly Generate Toxic Species?
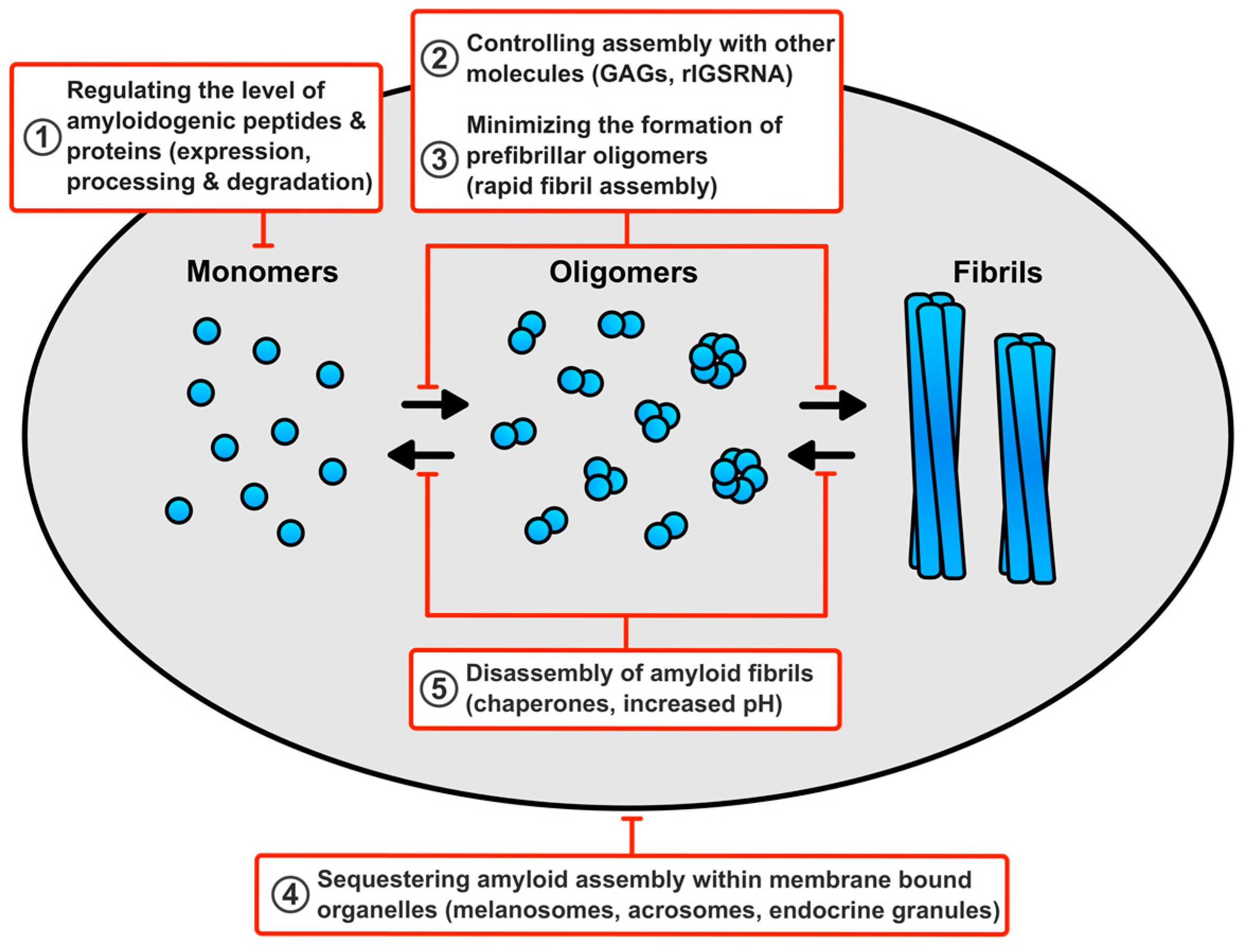
6. How Do Cells Avert Toxicity in Functional Amyloid Assembly?
6.1. Regulating the Level of the Amyloidogenic Peptides and Proteins
6.2. Minimising the Production of Prefibrillar Oligomers
6.3. Controlling Assembly of Functional Amyloid Fibrils with other Molecules
6.4. Sequestering Functional Amyloid Assembly Reactions within Membrane Bound Compartments
6.5. Disassembly of Functional Amyloid Fibrils
7. Summary and Remaining Questions
- Does the dysregulation of functional amyloid production result in disease? Of particular interest is whether Alzheimer’s disease is caused by the overproduction of a functional amyloid.
- Do functional amyloid assembly reactions produce toxic oligomers? Studies suggest that toxic prefibrillar oligomers are a common feature of amyloid assembly, yet surprisingly little is known about the properties of oligomers associated with functional amyloids.
- Are functional amyloids able to assemble more rapidly than disease-associated amyloids, thus limiting the production of any toxic prefibrillar oligomers?
- In addition to promoting the assembly of functional amyloids do rIGSRNA and GAGs also prevent amyloid toxicity?
- How can functional amyloid fibrils be assembled and stored within membrane bound compartments when cellular membranes represent a key target in amyloid toxicity?
Acknowledgments
Conflicts of Interest
References
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Kisilevsky, R.; Raimondi, S.; Bellotti, V. Historical and current concepts of fibrillogenesis and in vivo amyloidogenesis: Implications of amyloid tissue targeting. Front. Mol. Biosci. 2016, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Linke, R.P. A molecular history of the amyloidoses. J. Mol. Biol. 2012, 421, 142–159. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Benson, M.D.; Buxbaum, J.N.; Cohen, A.S.; Frangione, B.; Ikeda, S.I.; Masters, C.L.; Merlini, G.; Saraiva, M.J.; Sipe, J.D. Amyloid: Toward terminology clarification—Report from the nomenclature committee of the international society of amyloidosis. Amyloid 2005, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.I.; Merlini, G.; Saraiva, M.J.; Westermark, P. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification international society of amyloidosis 2016 nomenclature guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.L.L.; Kwan, A.H.; Sunde, M. Functional amyloid: Widespread in nature, diverse in purpose. Essays Biochem. 2014, 56, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Otzen, D. Functional amyloid turning swords into plowshares. Prion 2010, 4, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Audas, T.E.; Audas, D.E.; Jacob, M.D.; Ho, J.J.; Khacho, M.; Wang, M.; Perera, J.K.; Gardiner, C.; Bennett, C.A.; Head, T.; et al. Adaptation to stressors by systemic protein amyloidogenesis. Dev. Cell 2016, 39, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Guyonnet, B.; Egge, N.; Cornwall, G.A. Functional amyloids in the mouse sperm acrosome. Mol. Cell Biol. 2014, 34, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Whelly, S.; Johnson, S.; Powell, J.; Borchardt, C.; Hastert, M.C.; Cornwall, G.A. Nonpathological extracellular amyloid is present during normal epididymal sperm maturation. PLoS ONE 2012, 7, e36394. [Google Scholar] [CrossRef] [PubMed]
- Whelly, S.; Muthusubramanian, A.; Powell, J.; Johnson, S.; Hastert, M.C.; Cornwall, G.A. Cystatin-related epididymal spermatogenic subgroup members are part of an amyloid matrix and associated with extracellular vesicles in the mouse epididymal lumen. Mol. Hum. Reprod. 2016, 22, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Domanov, Y.; Pietiainen, M.; Kontinen, V.P.; Kinnunen, P.K.J. Binding of LL-37 to model biomembranes: Insight into target vs. host cell recognition. Biochem. Biophys. Acta 2008, 1778, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Munch, J.; Rucker, E.; Standker, L.; Adermann, K.; Goffinet, C.; Schindler, M.; Wildum, S.; Chinnadurai, R.; Rajan, D.; Specht, A.; et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell 2007, 131, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Arce, F.T.; Mustata, M.; Ramachandran, S.; Capone, R.; Nussinov, R.; Lal, R. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys. J. 2011, 100, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Roan, N.R.; Muller, J.A.; Liu, H.C.; Chu, S.; Arnold, F.; Sturzel, C.M.; Walther, P.; Dong, M.; Witkowska, H.E.; Kirchhoff, F.; et al. Peptides released by physiological cleavage of semen coagulum proteins form amyloids that enhance HIV infection. Cell Host Microbe 2011, 10, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R.; Wickner, R.B. Molecular structures of amyloid and prion fibrils: Consensus versus controversy. Accounts Chem. Res. 2013, 46, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- O’Nuallain, B.; Wetzel, R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc. Natl. Acad. Sci. USA 2002, 99, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Yau, W.M.; Lu, J.X.; Collinge, J.; Tycko, R. Structural variation in amyloid-β fibrils from alzheimer's disease clinical subtypes. Nature 2017, 541, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010, 277, 4602–4613. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. Ebiomedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 2004, 279, 31374–31382. [Google Scholar] [CrossRef] [PubMed]
- Baglioni, S.; Casamenti, F.; Bucciantini, M.; Luheshi, L.M.; Taddei, N.; Chiti, F.; Dobson, C.M.; Stefani, M. Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J. Neurosci. 2006, 26, 8160–8167. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Tseng, B.P.; Rydel, R.E.; Podlisny, M.B.; Selkoe, D.J. The oligomerization of amyloid β-protein begins intracellularly in cells derived from human brain. Biochemistry 2000, 39, 10831–10839. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Glabe, C.G. Distinct early folding and aggregation properties of alzheimer amyloid-β peptides Aβ40 and Aβ42: Stable trimer or tetramer formation by Aβ42. J. Biol. Chem. 2006, 281, 24414–24422. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Hepler, R.W.; Grimm, K.M.; Nahas, D.D.; Breese, R.; Dodson, E.C.; Acton, P.; Keller, P.M.; Yeager, M.; Wang, H.; Shughrue, P.; et al. Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry 2006, 45, 15157–15167. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Ladiwala, A.R.A.; Lin, J.C.; Bale, S.S.; Marcelino-Cruz, A.M.; Bhattacharya, M.; Dordick, J.S.; Tessier, P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem. 2010, 285, 24228–24237. [Google Scholar] [CrossRef] [PubMed]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.F.; Hellewell, A.L.; Gosal, W.S.; Homans, S.W.; Hewitt, E.W.; Radford, S.E. Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem. 2009, 284, 34272–34282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanesi, L.; Sheynis, T.; Xue, W.F.; Orlova, E.V.; Hellewell, A.L.; Jelinek, R.; Hewitt, E.W.; Radford, S.E.; Saibil, H.R. Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 20455–20460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, M.F.M.; Khemtemourian, L.; Kleijer, C.C.; Meeldijk, H.J.D.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Hoppener, J.W.M. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef] [PubMed]
- Tipping, K.W.; van Oosten-Hawle, P.; Hewitt, E.W.; Radford, S.E. Amyloid fibres: Inert end-stage aggregates or key players in disease? Trends Biochem. Sci. 2015, 40, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Tipping, K.W.; Karamanos, T.K.; Jakhria, T.; Iadanza, M.G.; Goodchild, S.C.; Tuma, R.; Ranson, N.A.; Hewitt, E.W.; Radford, S.E. pH-induced molecular shedding drives the formation of amyloid fibril-derived oligomers. Proc. Natl. Acad. Sci. USA 2015, 112, 5691–5696. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Rochin, L.; van Niel, G. PMEL amyloid fibril formation: The bright steps of pigmentation. Int. J. Mol. Sci. 2016, 17, 1438. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Hassannia, B.; Vandenabeele, P. An outline of necrosome triggers. Cell Mol. Life Sci. 2016, 73, 2137–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, B.L.; Jang, H.; Capone, R.; Teran Arce, F.; Ramachandran, S.; Lal, R.; Nussinov, R. Antimicrobial properties of amyloid peptides. Mol. Pharm. 2012, 9, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Last, N.B.; Miranker, A.D. Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2013, 110, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Hewetson, A.; Do, H.Q.; Myers, C.; Muthusubramanian, A.; Sutton, R.B.; Wylie, B.J.; Cornwall, G.A. Functional amyloids in reproduction. Biomolecules 2017, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Berruti, G.; Paiardi, C. Acrosome biogenesis: Revisiting old questions to yield new insights. Spermatogenesis 2011, 1, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yuan, Q.; Chen, S.H.; Cai, H.; Lu, M.G.; Liu, Y.; Xu, C. Antimicrobial activity and molecular mechanism of the CRES protein. PLoS ONE 2012, 7, e48368. [Google Scholar] [CrossRef] [PubMed]
- Chau, K.M.; Cornwall, G.A. Reduced fertility in vitro in mice lacking the cystatin cres (cystatin-related epididymal spermatogenic): Rescue by exposure of spermatozoa to dibutyryl camp and isobutylmethylxanthine. Biol. Reprod. 2011, 84, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Parent, A.D.; Cornwall, G.A.; Liu, L.Y.; Smith, C.E.; Hermo, L. Alterations in the testis and epididymis associated with loss of function of the cystatin-related epididymal spermatogenic (CRES) protein. J. Androl. 2011, 32, 444–463. [Google Scholar] [CrossRef] [PubMed]
- Roan, N.R.; Sandi-Monroy, N.; Kohgadai, N.; Usmani, S.M.; Hamil, K.G.; Neidleman, J.; Montano, M.; Standker, L.; Rocker, A.; Cavrois, M.; et al. Semen amyloids participate in spermatozoa selection and clearance. Elife 2017, 6, e24888. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.L.; Tickler, A.K.; Squires, A.M.; Devlin, G.L.; Horton, M.A.; Dobson, C.M.; MacPhee, C.E. Functionalised amyloid fibrils for roles in cell adhesion. Biomaterials 2008, 29, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Born, A.K.; Schweizer, T.; Zenobi-Wong, M.; Cerruti, M.; Mezzenga, R. Amyloid-hydroxyapatite bone biomimetic composites. Adv. Mater. 2014, 26, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.K.; Scanlon, D.; Musgrave, I.F.; Carver, J.A. SEVI, the semen enhancer of HIV infection along with fragments from its central region, form amyloid fibrils that are toxic to neuronal cells. Biochim. Biophys. Acta 2014, 1844, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Berson, J.F.; Theos, A.C.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 2003, 161, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, R.M.; Vigneron, N.; Rahner, C.; Cresswell, P. Proprotein convertases process PMEL17 during secretion. J. Biol. Chem. 2011, 286, 9321–9337. [Google Scholar] [CrossRef] [PubMed]
- Rochin, L.; Hurbain, I.; Serneels, L.; Fort, C.; Watt, B.; Leblanc, P.; Marks, M.S.; De Strooper, B.; Raposo, G.; van Niel, G. BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10658–10663. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Hozumi, Y.; Suzuki, T. ADAM protease inhibitors reduce melanogenesis by regulating PMEL17 processing in human melanocytes. J. Dermatol. Sci. 2015, 78, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Maruyama, H.; Huelsmann, C.; Baches, S.; Weggen, S.; Koo, E.H. Formation of PMEL17 amyloid is regulated by juxtamembrane metalloproteinase cleavage, and the resulting C-terminal fragment is a substrate for gamma-secretase. J. Biol. Chem. 2009, 284, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; Tenza, D.; Lemmon, M.A.; Kerje, S.; Raposo, G.; Andersson, L.; Marks, M.S. Mutations in or near the transmembrane domain alter PMEL amyloid formation from functional to pathogenic. PLoS Genet. 2011, 7, e1002286. [Google Scholar] [CrossRef] [PubMed]
- Kerje, S.; Sharma, P.; Gunnarsson, U.; Kim, H.; Bagchi, S.; Fredriksson, R.; Schutz, K.; Jensen, P.; von Heijne, G.; Okimoto, R.; et al. The Dominant white, Dun and Smoky Color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene. Genetics 2004, 168, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, A.R.; Watt, B.; Fard, S.S.; Tenza, D.; Mannstrom, P.; Narfstrom, K.; Ekesten, B.; Ito, S.; Wakamatsu, K.; Larsson, J.; et al. Inactivation of PMEL alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet. 2011, 7, e1002285. [Google Scholar] [CrossRef] [PubMed]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer's disease-associated amyloid β-protein is an antimicrobial peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci. Transl. Med. 2016, 8, 340ra372. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, P.; Condic, M.; Herrmann, M.; Oberstein, T.J.; Scharin-Mehlmann, M.; Gilbert, D.F.; Friedrich, O.; Gromer, T.; Kornhuber, J.; Lang, R.; et al. Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci. Rep. 2016, 6, 32228. [Google Scholar] [CrossRef] [PubMed]
- Sall, J.; Carlsson, M.; Gidlof, O.; Holm, A.; Humlen, J.; Ohman, J.; Svensson, D.; Nilsson, B.O.; Jonsson, D. The antimicrobial peptide LL-37 alters human osteoblast Ca2+ handling and induces Ca2+-independent apoptosis. J. Innate Immun. 2013, 5, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Barlow, P.G.; Beaumont, P.E.; Cosseau, C.; Mackellar, A.; Wilkinson, T.S.; Hancock, R.E.; Haslett, C.; Govan, J.R.; Simpson, A.J.; Davidson, D.J. The human cathelicidin LL-37 preferentially promotes apoptosis of infected airway epithelium. Am. J. Respir. Cell Mol. Biol. 2010, 43, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Li, H.N.; Barlow, P.G.; Bylund, J.; Mackellar, A.; Bjorstad, A.; Conlon, J.; Hiemstra, P.S.; Haslett, C.; Gray, M.; Simpson, A.J.; et al. Secondary necrosis of apoptotic neutrophils induced by the human cathelicidin LL-37 is not proinflammatory to phagocytosing macrophages. J. Leukoc. Biol. 2009, 86, 891–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.F.; Cherryholmes, G.; Shively, J.E. Neutrophil secondary necrosis is induced by LL-37 derived from cathelicidin. J. Leukoc. Biol. 2008, 84, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Ciornei, C.D.; Tapper, H.; Bjartell, A.; Sternby, N.H.; Bodelsson, M. Human antimicrobial peptide LL-37 is present in atherosclerotic plaques and induces death of vascular smooth muscle cells: A laboratory study. BMC Cardiovasc. Disord. 2006, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, M.N.; Gras, S.L. Bioactive TTR105–115-based amyloid fibrils reduce the viability of mammalian cells. Biomaterials 2015, 46, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Wegner, K.W.; Saleh, D.; Degterev, A. Complex pathologic roles of RIPK1 and RIPK3: Moving beyond necroptosis. Trends Pharmacol. Sci. 2017, 38, 202–225. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trend Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.P.; Hewitt, E.W. Cellular proteostasis: Degradation of misfolded proteins by lysosomes. Essays Biochem. 2016, 60, 173–180. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Drueke, T.B.; Massy, Z.A. β2-microglobulin. Semin. Dial. 2009, 22, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Zanini, A.; Giannattasio, G.; Nussdorfer, G.; Margolis, R.K.; Margolis, R.U.; Meldolesi, J. Molecular-organization of prolactin granules. II. Characterization of glycosaminoglycans and glycoproteins of the bovine prolactin matrix. J. Cell Biol. 1980, 86, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Di Domizio, J.; Zhang, R.; Stagg, L.J.; Gagea, M.; Zhuo, M.; Ladbury, J.E.; Cao, W. Binding with nucleic acids or glycosaminoglycans converts soluble protein oligomers to amyloid. J. Biol. Chem. 2012, 287, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Middleton, D.A. Comparison of aggregation enhancement and inhibition as strategies for reducing the cytotoxicity of the aortic amyloid polypeptide medin. Eur. Biophys. J. Biophys. 2010, 39, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Vilasi, S.; Sarcina, R.; Maritato, R.; De Simone, A.; Irace, G.; Sirangelo, I. Heparin induces harmless fibril formation in amyloidogenic W7FW14F apomyoglobin and amyloid aggregation in wild-type protein in vitro. PLoS ONE 2011, 6, e22076. [Google Scholar] [CrossRef] [PubMed]
- Sheynis, T.; Friediger, A.; Xue, W.F.; Hellewell, A.L.; Tipping, K.W.; Hewitt, E.W.; Radford, S.E.; Jelinek, R. Aggregation modulators interfere with membrane interactions of β2-microglobulin fibrils. Biophys. J. 2013, 105, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, S.C.; Sheynis, T.; Thompson, R.; Tipping, K.W.; Xue, W.F.; Ranson, N.A.; Beales, P.A.; Hewitt, E.W.; Radford, S.E. β2-microglobulin amyloid fibril-induced membrane disruption is enhanced by endosomal lipids and acidic pH. PLoS ONE 2014, 9, e104492. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, E.; Cecchi, C.; Cascella, R.; Sgromo, C.; Becatti, M.; Dobson, C.M.; Chiti, F.; Stefani, M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci. 2012, 125, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Shewmaker, F.; McPhie, P.; Monterroso, B.; Thurber, K.; Wickner, R.B. The repeat domain of the melanosome fibril protein PMEL17 forms the amyloid core promoting melanin synthesis. Proc. Natl. Acad. Sci. USA 2009, 106, 13731–13736. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Gruschus, J.M.; Nagy, A.; Lee, J.C. Probing fibril dissolution of the repeat domain of a functional amyloid, PMEL17, on the microscopic and residue level. Biochemistry 2011, 50, 10567–10569. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein, Peptide or Cellular Structure | Proposed Functions | Experimental Evidence for Amyloid Fibrils |
|---|---|---|
| Amyloid-bodies (A-bodies) | Stores of proteins in stressed cells | A-bodies are stained by Congo red and thioflavin-S. Proteins that accumulate in A-bodies can form fibrils with a cross-β X-ray fiber diffraction pattern [11]. |
| Acrosomes | The acrosome reaction during fertilisation of oocytes. | Acrosomes in sperm are stained by thioflavin-S and are recognised by amyloid-specific antibodies. Purified acrosomal matrix has a cross-β X-ray fiber diffraction pattern [12]. |
| Cystatin-related epididymal spermatogenicis (CRES) subgroup proteins | Antimicrobial activity, acrosome reaction and lysosomal function | Material from the epididymis has a cross cross-β X-ray fiber diffraction pattern, is recognised by amyloid-specific antibodies and binds thioflavin-S and Congo red. CRES proteins co-localises with thioflavin-S. Fibrils of CRES proteins bind thioflavin-T and are recognised by amyloid-specific antibodies [13,14]. |
| LL-37 | Antimicrobial | Fibrils exhibit green birefringence with Congo red [15]. |
| Peptide hormones | Storage of the hormone in secretory granules | Purified granules from endocrine cells have a cross-β X-ray fiber diffraction pattern and exhibit green birefringence with Congo red. The fibrils bind Congo red and have a cross-β X-ray fiber diffraction pattern [16]. |
| Pigment cell-specific pre-melanosomal protein (PMEL) | Pigmentation | Purified melansomes are stained by thioflavin-S and Congo red. The fibrils have a cross-β X-ray fiber diffraction pattern, bind Congo red and thioflavin-T and have a far ultraviolet circular dichroism spectrum consistent with β-sheet content [17]. |
| Prostatic acid phosphatase peptides | Removal of damaged sperm | The fibrils have a cross-β X-ray fiber diffraction pattern, bind thioflavin-T and exhibit green birefringence with Congo red [18]. |
| Protegrin-1 | Antimicrobial | The fibrils bind thioflavin-T [19]. |
| Receptor-interacting protein 1 (RIP1)/RIP3 | Regulated necrosis | The fibrils have a cross-β X-ray fiber diffraction pattern, a solid state NMR spectra consistent with a β-sheet core and bind thioflavin T and Congo red [20]. |
| Semenogelin proteins (SEM1 and SEM2) | Removal of damaged sperm | The fibrils bind thioflavin-T and Congo red and an amyloid-specific antibody pulls down SEM 1 and SEM 2 from seminal fluid [21]. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, M.P.; Hewitt, E.W. Why are Functional Amyloids Non-Toxic in Humans? Biomolecules 2017, 7, 71. https://doi.org/10.3390/biom7040071
Jackson MP, Hewitt EW. Why are Functional Amyloids Non-Toxic in Humans? Biomolecules. 2017; 7(4):71. https://doi.org/10.3390/biom7040071
Chicago/Turabian StyleJackson, Matthew P., and Eric W. Hewitt. 2017. "Why are Functional Amyloids Non-Toxic in Humans?" Biomolecules 7, no. 4: 71. https://doi.org/10.3390/biom7040071
APA StyleJackson, M. P., & Hewitt, E. W. (2017). Why are Functional Amyloids Non-Toxic in Humans? Biomolecules, 7(4), 71. https://doi.org/10.3390/biom7040071