Histone Acetylation Promotes Neutrophil Extracellular Trap Formation


Abstract
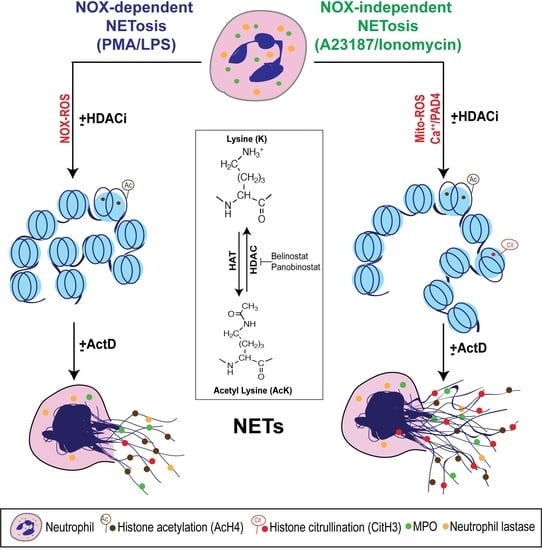
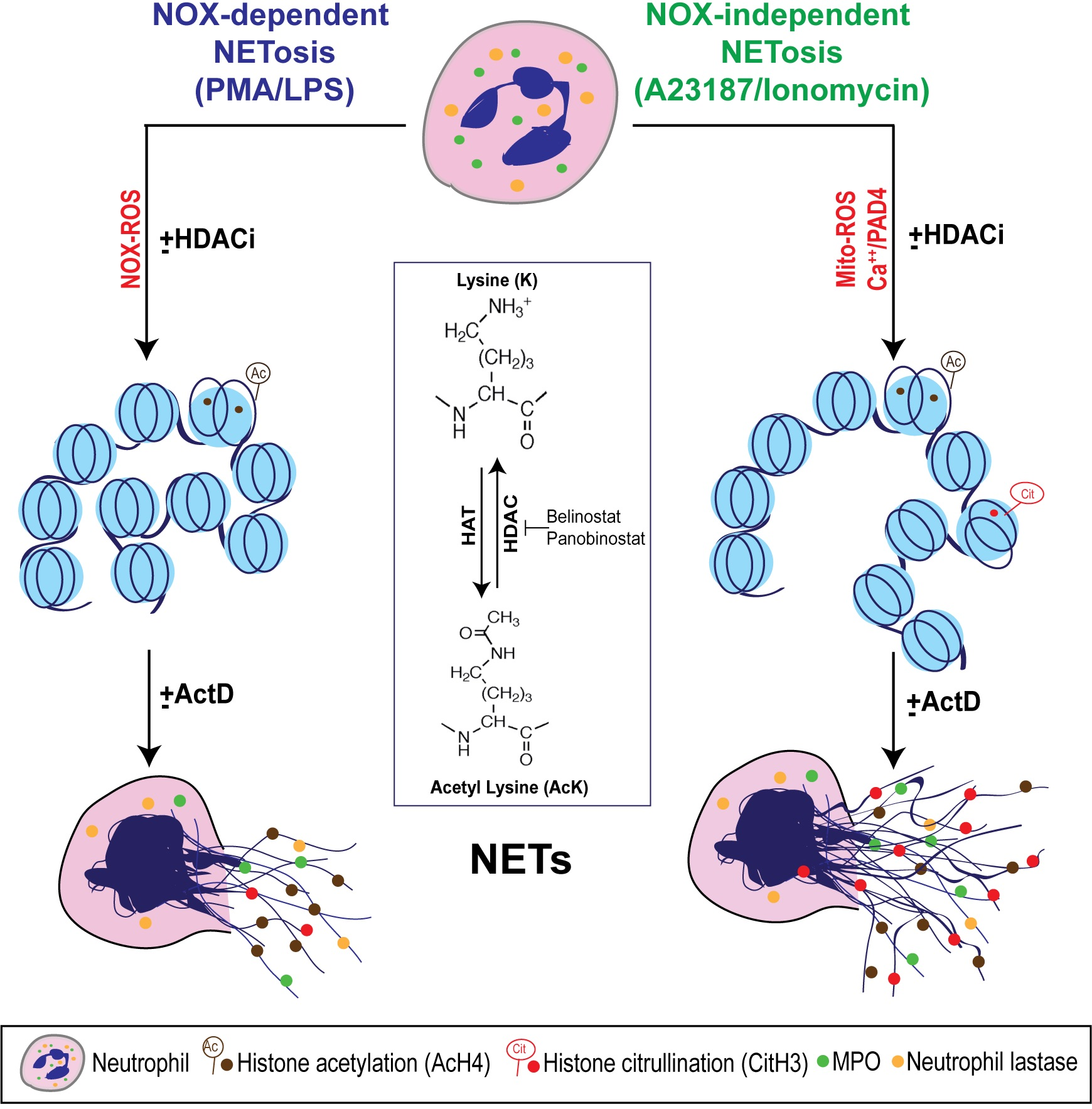
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Research Ethics Board Approval
2.2. Human Peripheral Blood Neutrophil Isolation
2.3. Sytox Green NETosis Assay
2.4. DHR123, DCFDA and MitoSOX Plate Reader Assays (NOX- and Mitochondrial-Mediated ROS Analyses)
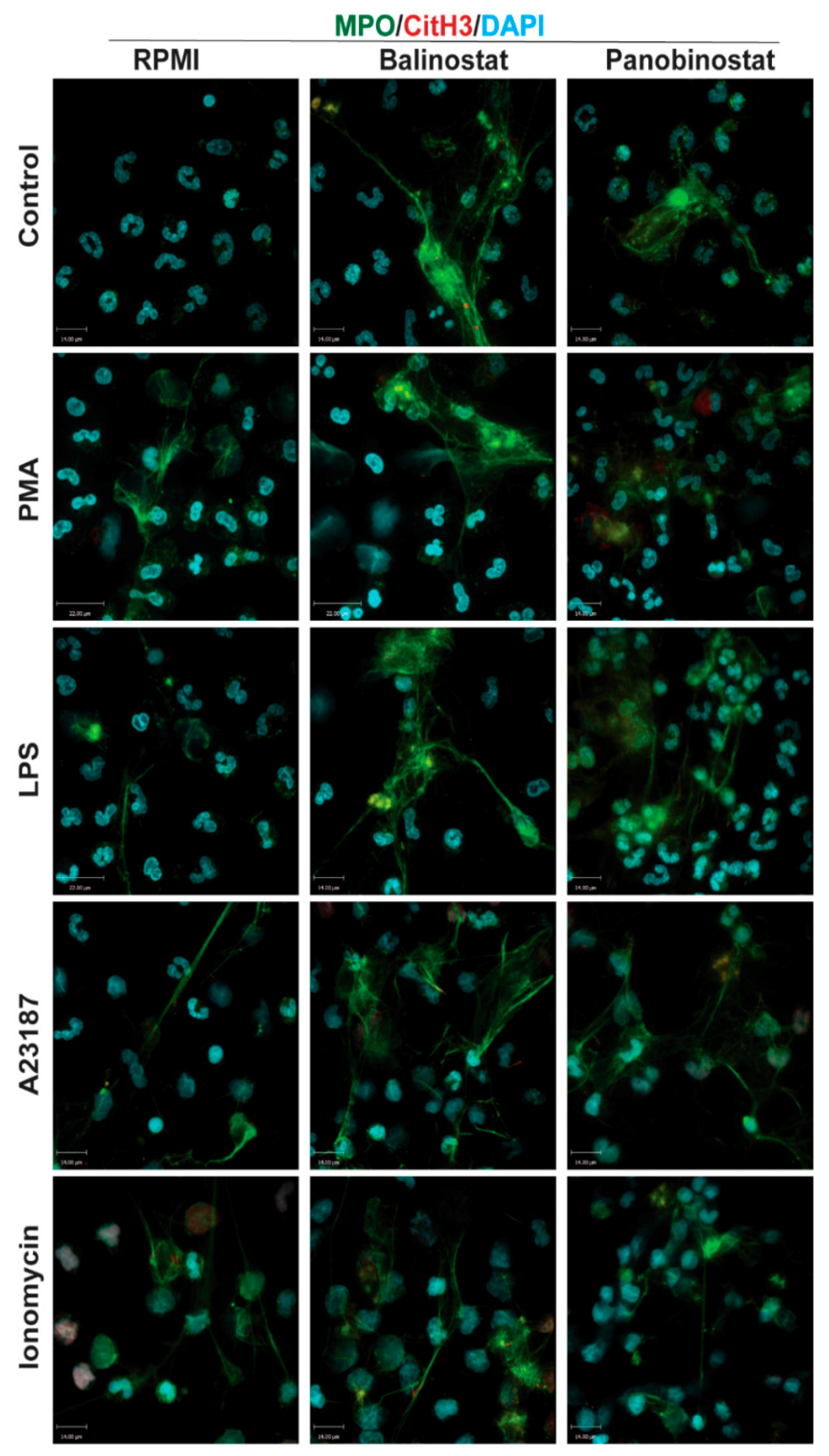
2.5. Fluorescence Confocal Imaging
2.6. Western Blot
2.7. Statistical Analyses
3. Results
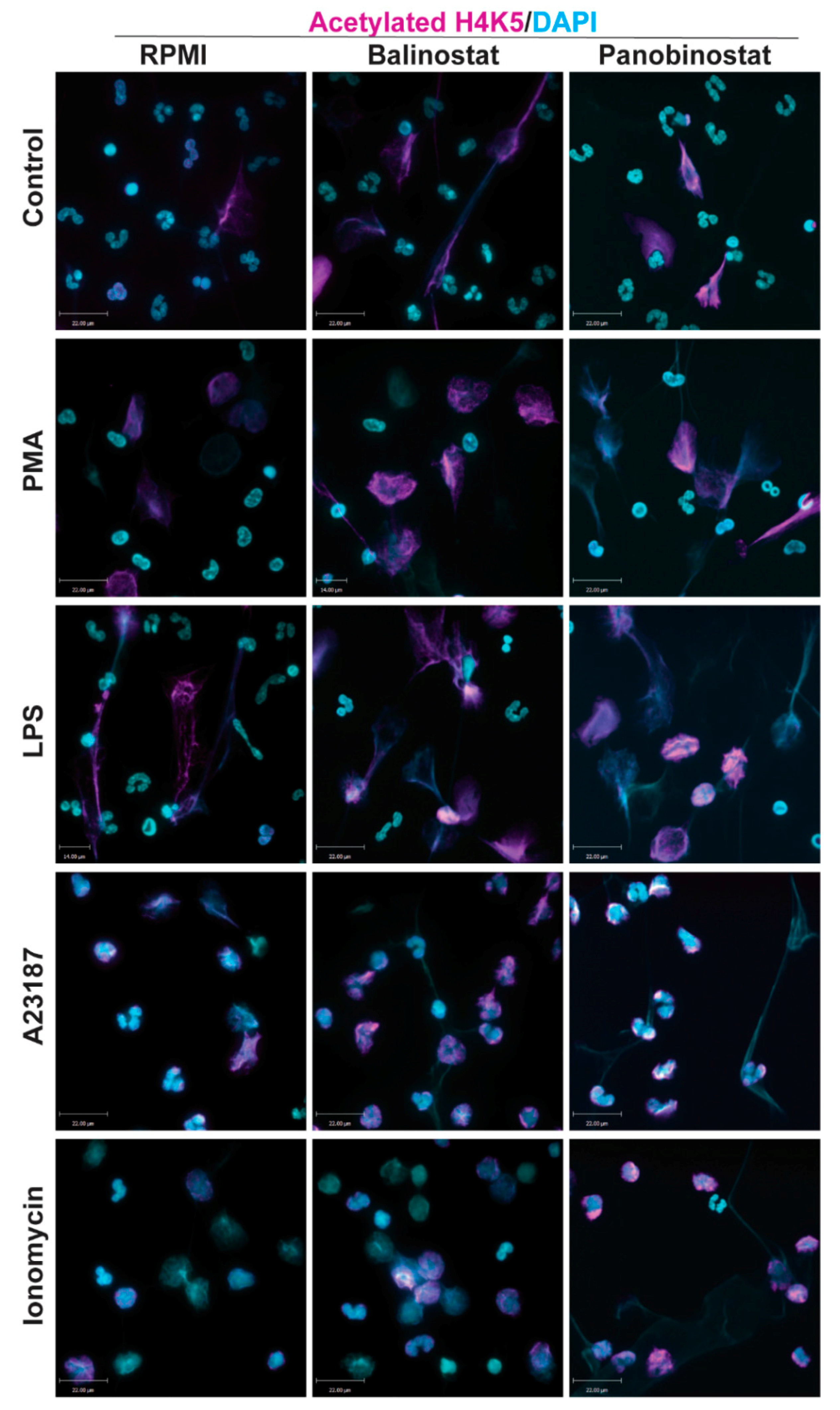
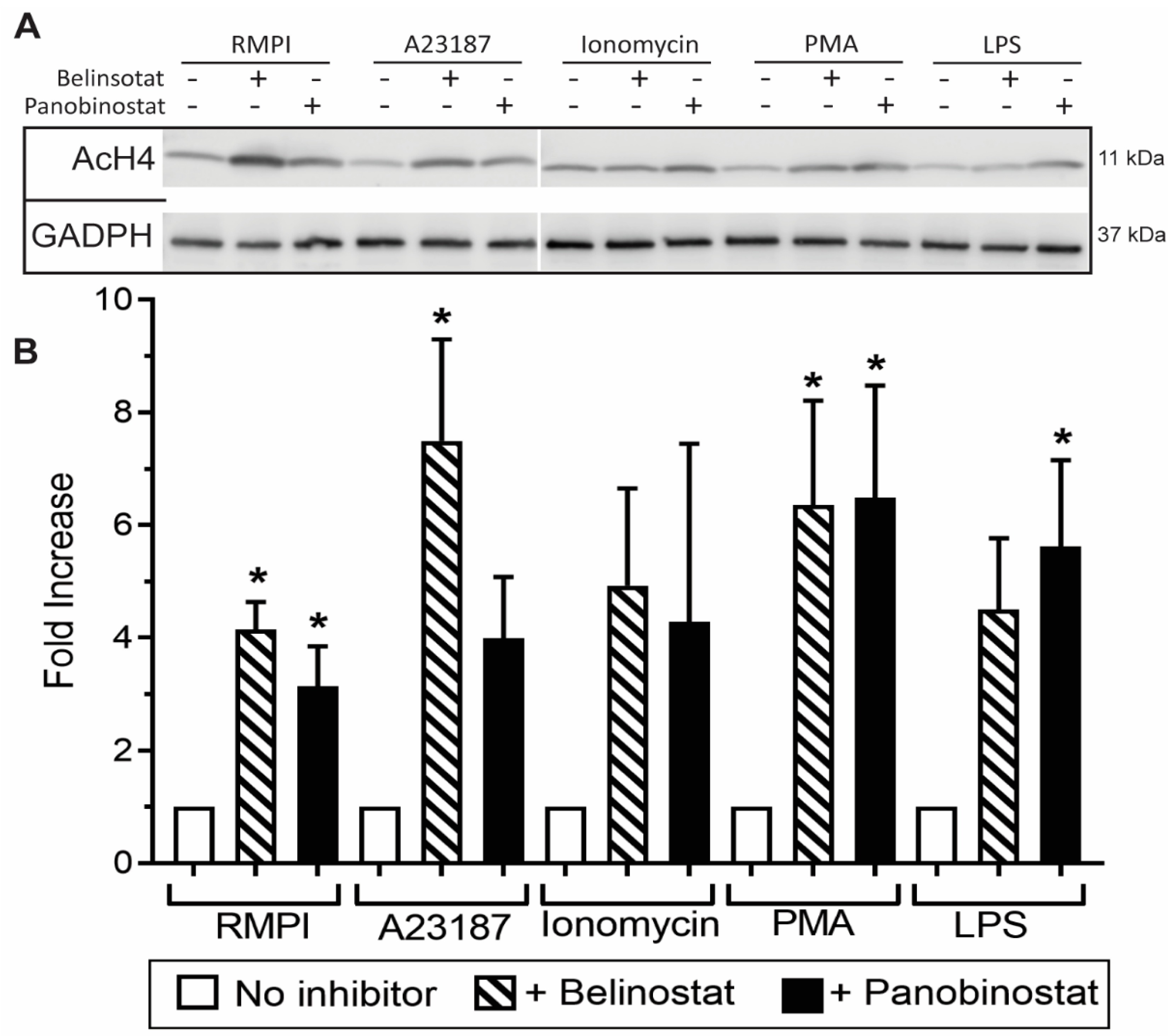
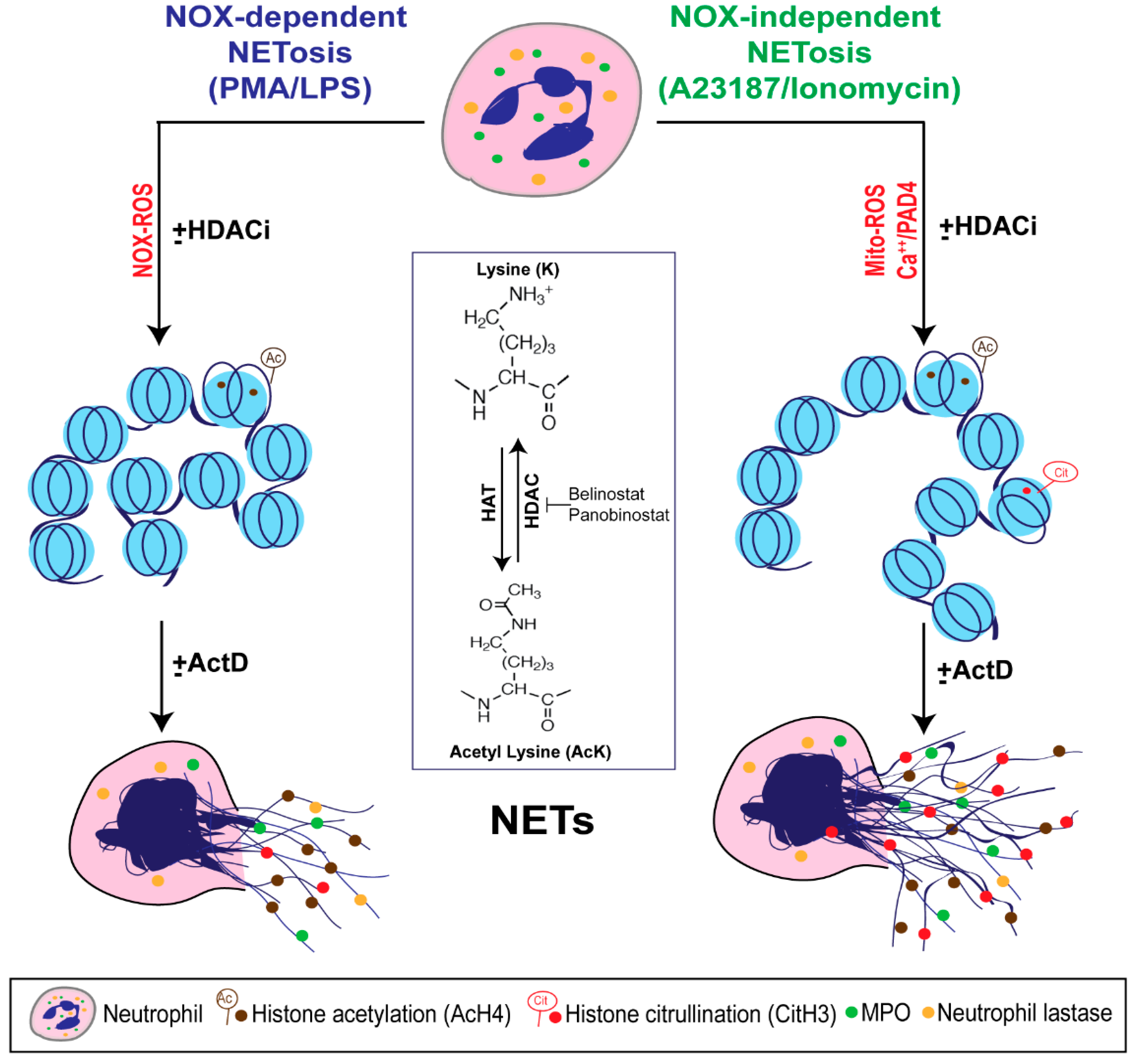
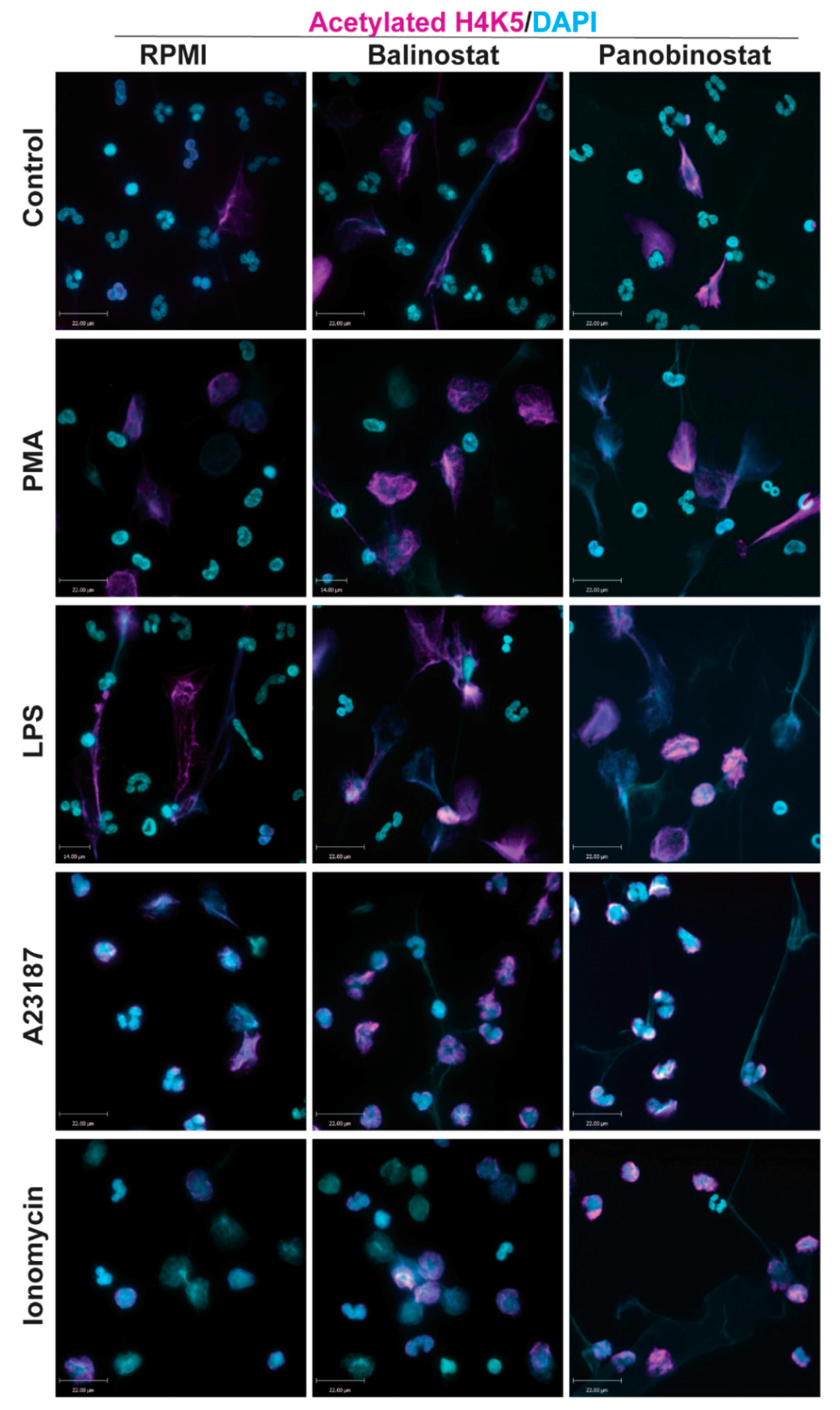
3.1. HDAC Inhibitors Promote Histone Acetylation
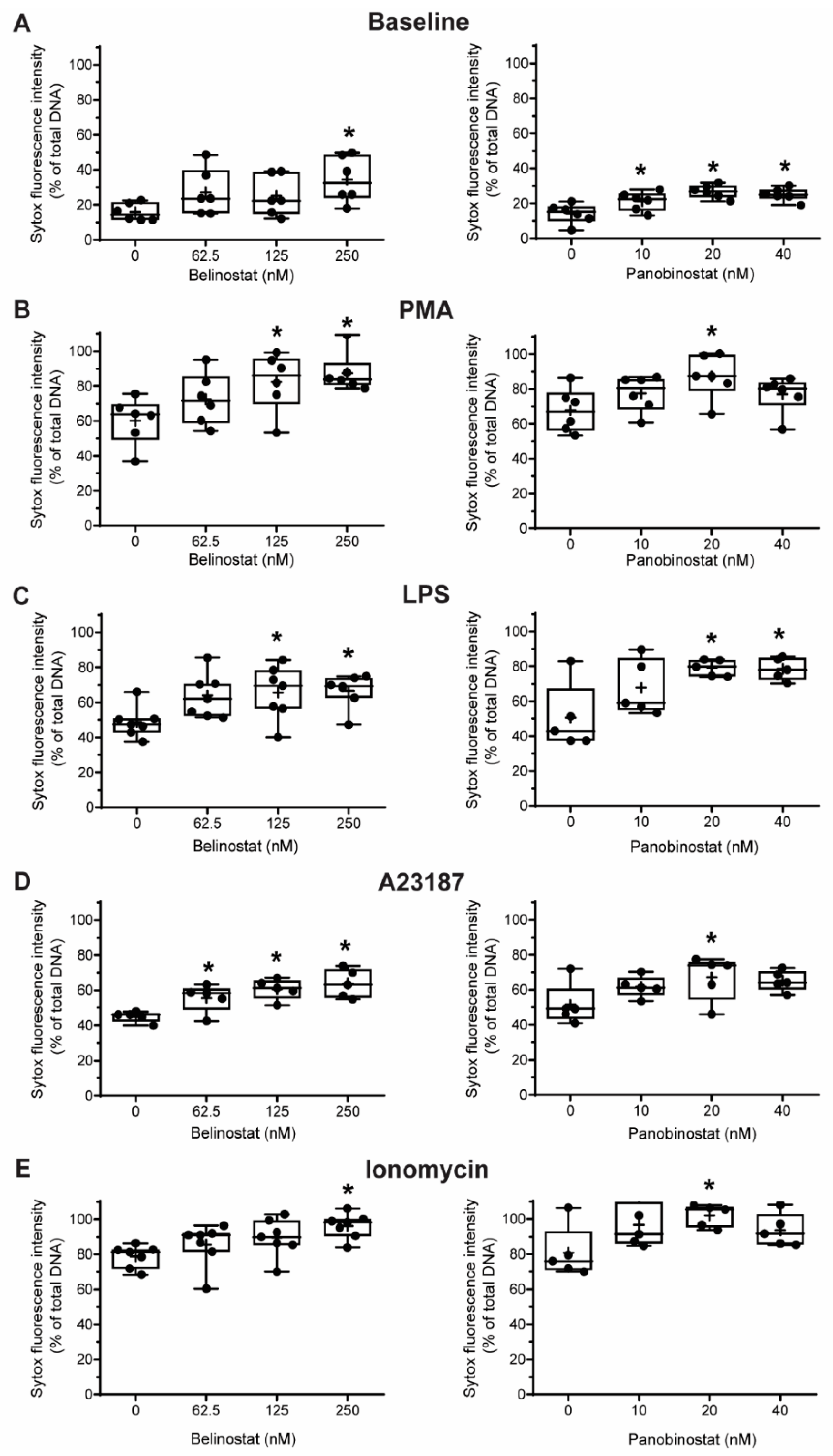
3.2. HDAC Inhibitors Promote Baseline NETosis
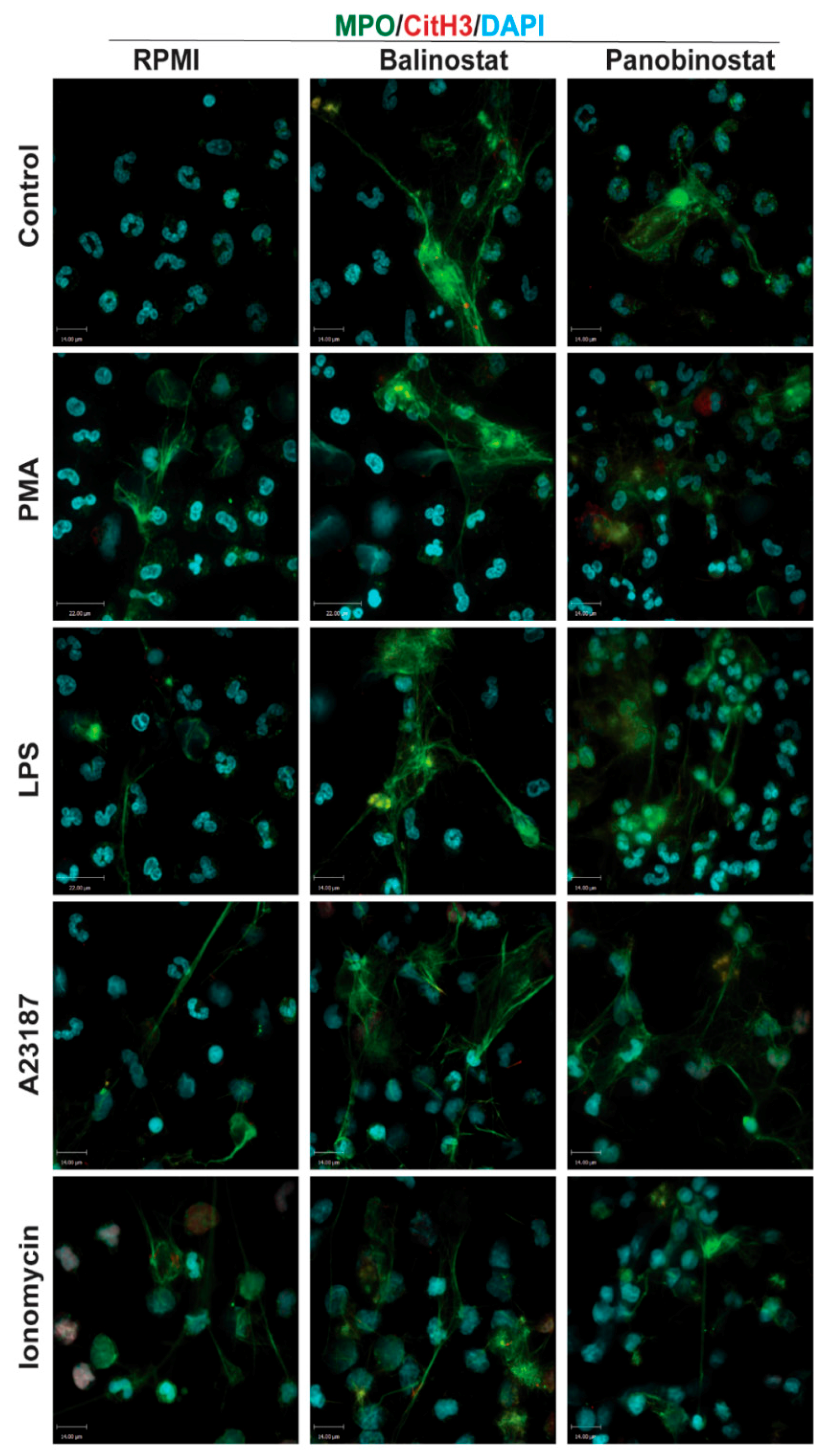
3.3. HDAC Inhibitors Additively Promote NOX-Dependent NETosis
3.4. HDAC Inhibitors Additively Promote NOX-Independent NETosis
3.5. HDAC Inhibitor Do Not Induce Apoptosis
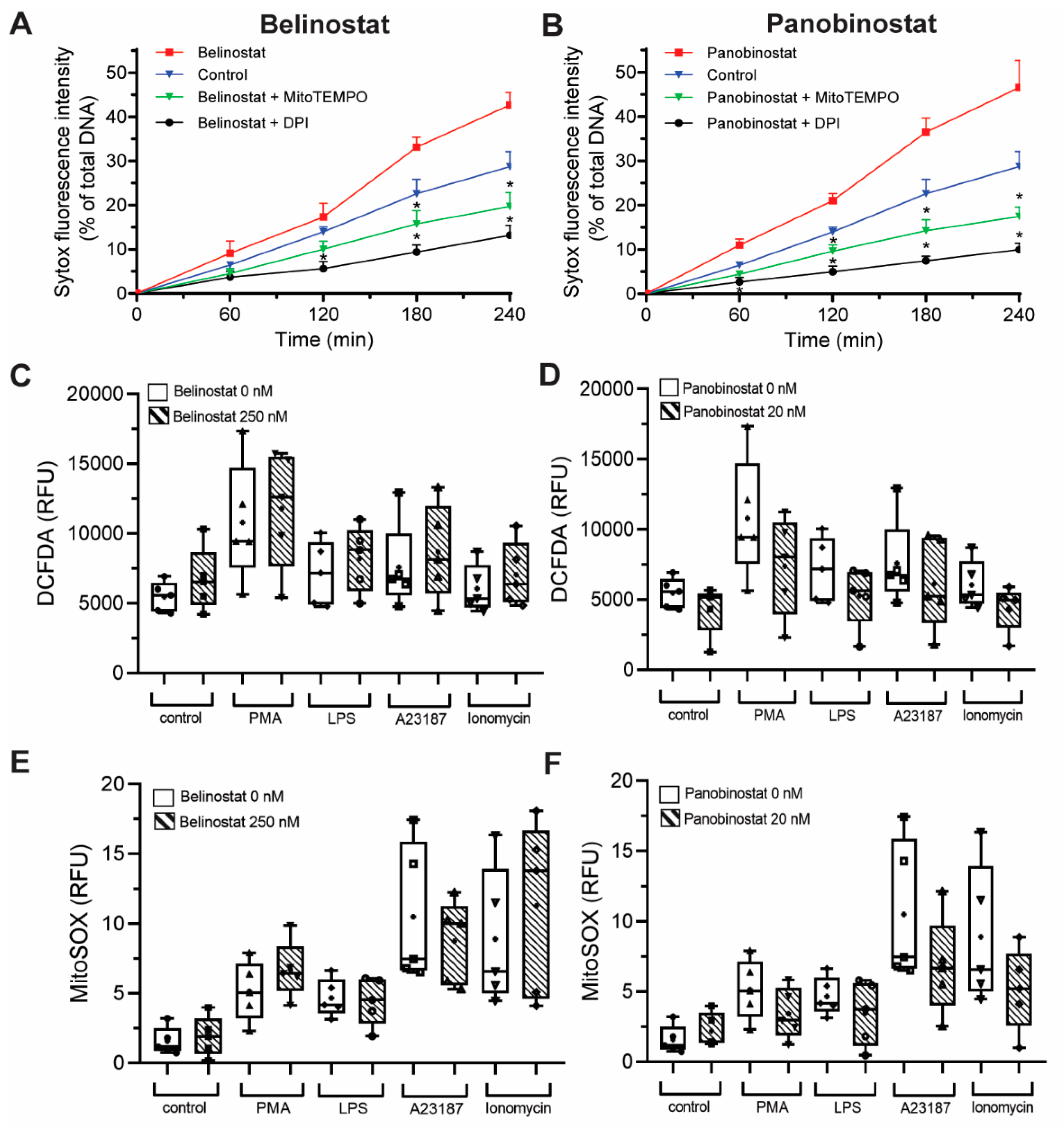
3.6. HDAC Inhibitor-Mediated NETosis Requires Baseline ROS
3.7. HDAC Inhibitors Do Not Promote NOX- and Mitochondrial-Derived ROS Production
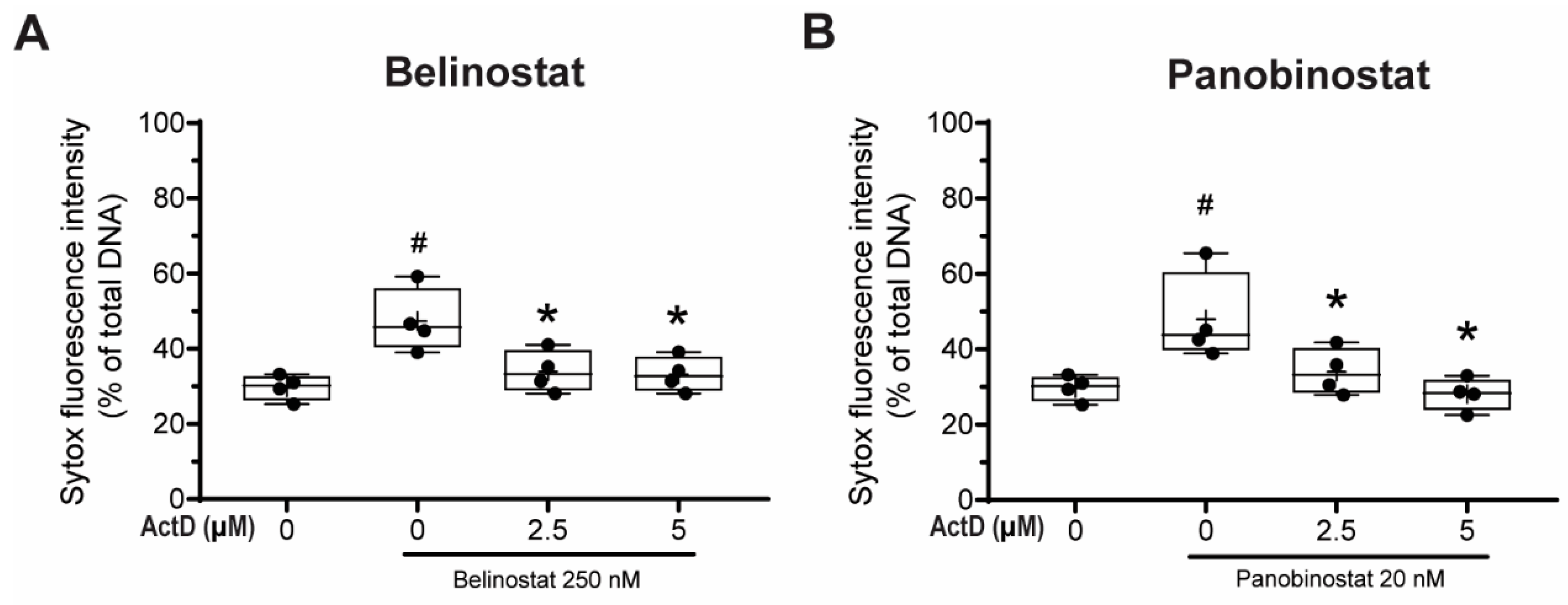
3.8. Transcriptional Firing Is Required for HDAC Inhibitors to Promote NETosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, J.; Pluthero, F.G.; Douda, D.N.; Riedl, M.; Cherry, A.; Ulanova, M.; Kahr, W.H.A.; Palaniyar, N.; Licht, C. NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non-alternative pathways. Front. Immunol. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Palaniyar, N. Transcriptional firing helps to drive NETosis. Sci. Rep. 2017, 7, 41749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. Myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Farahvash, A.; Douda, D.N.; Licht, J.-C.; Grasemann, H.; Sweezey, N.; Palaniyar, N. JNK Activation Turns on LPS- and Gram-Negative Bacteria-Induced NADPH Oxidase-Dependent Suicidal NETosis. Sci. Rep. 2017, 7, 3409. [Google Scholar] [CrossRef]
- Obermayer, A.; Stoiber, W.; Krautgartner, W.-D.; Klappacher, M.; Kofler, B.; Steinbacher, P.; Vitkov, L.; Grabcanovic-Musija, F.; Studnicka, M. New Aspects on the Structure of Neutrophil Extracellular Traps from Chronic Obstructive Pulmonary Disease and In Vitro Generation. PLoS ONE 2014, 9, e97784. [Google Scholar] [CrossRef]
- Sahakian, E.; Chen, J.; Powers, J.J.; Chen, X.; Maharaj, K.; Deng, S.L.; Achille, A.N.; Lienlaf, M.; Wang, H.W.; Cheng, F.; et al. Essential role for histone deacetylase 11 (HDAC11) in neutrophil biology. J. Leukoc. Biol. 2017, 102, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ge, H.; Poulton, C.J.; Hogan, S.L.; Hu, Y.; Jones, B.E.; Henderson, C.D.; McInnis, E.A.; Pendergraft, W.F.; Jennette, J.C.; et al. Histone modification signature at myeloperoxidase and proteinase 3 in patients with anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Clin. Epigenetics 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Kankaanranta, H.; Janka-Junttila, M.; Ilmarinen-Salo, P.; Ito, K.; Jalonen, U.; Ito, M.; Adcock, I.M.; Moilanen, E.; Zhang, X. Histone deacetylase inhibitors induce apoptosis in human eosinophils and neutrophils. J. Inflamm. (Lond.) 2010, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, P.A.; Dokmanovic, M. Histone deacetylase inhibitors: Discovery and development as anticancer agents. Expert Opin. Investig. Drugs 2005, 14, 1497–1511. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, C.N.; Breda, L.C.D.; Khan, M.A.; de Almeida, S.R.; Câmara, N.O.S.; Sweezey, N.; Palaniyar, N. Alkaline pH promotes NADPH oxidase-independent neutrophil extracellular trap formation: A matter of mitochondrial reactive oxygen species generation and citrullination and cleavage of histone. Front. Immunol. 2018, 8. [Google Scholar] [CrossRef]
- Bressenot, A.; Marchal, S.; Bezdetnaya, L.; Garrier, J.; Guillemin, F.; Plénat, F. Assessment of Apoptosis by Immunohistochemistry to Active Caspase-3, Active Caspase-7, or Cleaved PARP in Monolayer Cells and Spheroid and Subcutaneous Xenografts of Human Carcinoma. J. Histochem. Cytochem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Farrell, A.W.; Halliday, G.M.; Lyons, J.G. Chromatin structure following UV-induced DNA damage-repair or death? Int. J. Mol. Sci. 2011, 12, 8063–8065. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, D.; Khan, M.A.; Sweezey, N.; Palaniyar, N. Two-in-one: UV radiation simultaneously induces apoptosis and NETosis. Cell Death Discov. 2018, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.A.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H.; et al. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hollands, A.; Corriden, R.; Gysler, G.; Dahesh, S.; Olson, J.; Ali, S.R.; Kunkel, M.T.; Lin, A.E.; Forli, S.; Newton, A.C.; et al. Natural product anacardic acid from cashew nut shells stimulates neutrophil extracellular trap production and bactericidal activity. J. Biol. Chem. 2016, 291, 13964–13973. [Google Scholar] [CrossRef] [PubMed]
- Mamidyala, S.K.; Ramu, S.; Huang, J.X.; Robertson, A.A.B.; Cooper, M.A. Efficient synthesis of anacardic acid analogues and their antibacterial activities. Bioorganic Med. Chem. Lett. 2013. [Google Scholar] [CrossRef]
- Kubo, J.; Lee, J.R.; Kubo, I. Anti-Helicobacter pylori agents from the cashew apple. J. Agric. Food Chem. 1999. [Google Scholar] [CrossRef]
- Sharma, R.; Kishore, N.; Hussein, A.; Lall, N. Antibacterial and anti-inflammatory effects of Syzygium jambos L. (Alston) and isolated compounds on acne vulgaris. BMC Complement. Altern. Med. 2013. [Google Scholar] [CrossRef]
- Pietrocola, F.; Lachkar, S.; Enot, D.P.; Niso-Santano, M.; Bravo-San Pedro, J.M.; Sica, V.; Izzo, V.; Maiuri, M.C.; Madeo, F.; Mariño, G.; et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015. [Google Scholar] [CrossRef] [PubMed]
- Harsha Raj, M.; Yashaswini, B.; Rössler, J.; Salimath, B.P. Combinatorial treatment with anacardic acid followed by TRAIL augments induction of apoptosis in TRAIL resistant cancer cells by the regulation of p53, MAPK and NFκβ pathways. Apoptosis 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Tangsombatvisit, S.; Rosenberg, J.M.; Mandelbaum, G.; Gillespie, E.C.; Gozani, O.P.; Alizadeh, A.A.; Utz, P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012, 14. [Google Scholar] [CrossRef]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; van der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. https://doi.org/10.3390/biom9010032
Hamam HJ, Khan MA, Palaniyar N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules. 2019; 9(1):32. https://doi.org/10.3390/biom9010032
Chicago/Turabian StyleHamam, Hussein J., Meraj A. Khan, and Nades Palaniyar. 2019. "Histone Acetylation Promotes Neutrophil Extracellular Trap Formation" Biomolecules 9, no. 1: 32. https://doi.org/10.3390/biom9010032
APA StyleHamam, H. J., Khan, M. A., & Palaniyar, N. (2019). Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules, 9(1), 32. https://doi.org/10.3390/biom9010032