Translation from the Ribosome to the Clinic: Implication in Neurological Disorders and New Perspectives from Recent Advances


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Translational Dysregulation in Neurological Disorders
2.1. Autism Spectrum Disorders and Other Neurodevelopmental Deficits
2.2. Neuropsychiatric and Mood Disorders
2.3. Neurodegenerative Disorders
3. Translational Stalling and Neurodegeneration
3.1. tRNA-Induced Ribosome Stalling
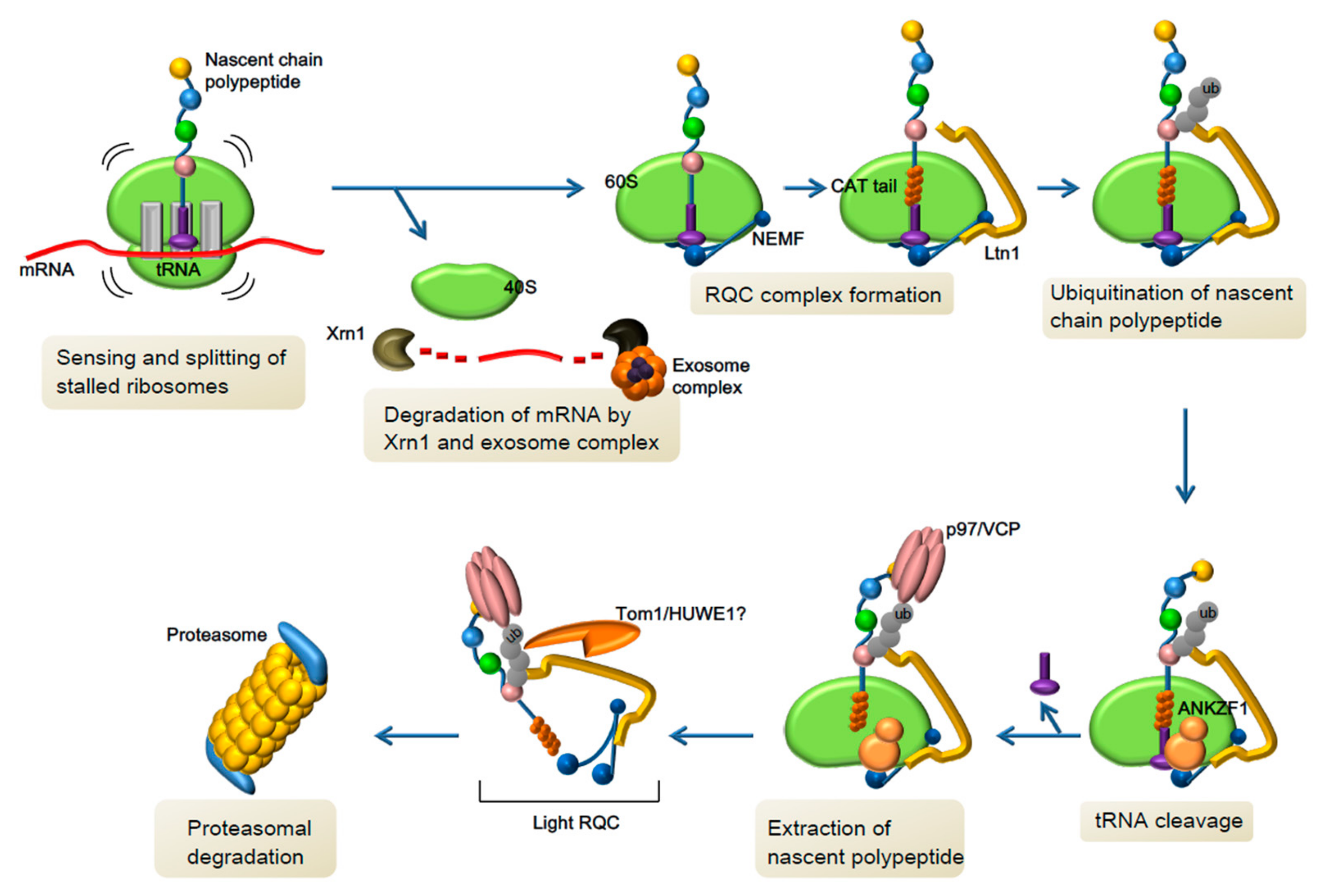
3.2. Novel Pathway of Co-Translational Quality Control and Neurological Disorders
4. Toxic RAN Translation Products in Neurodegeneration
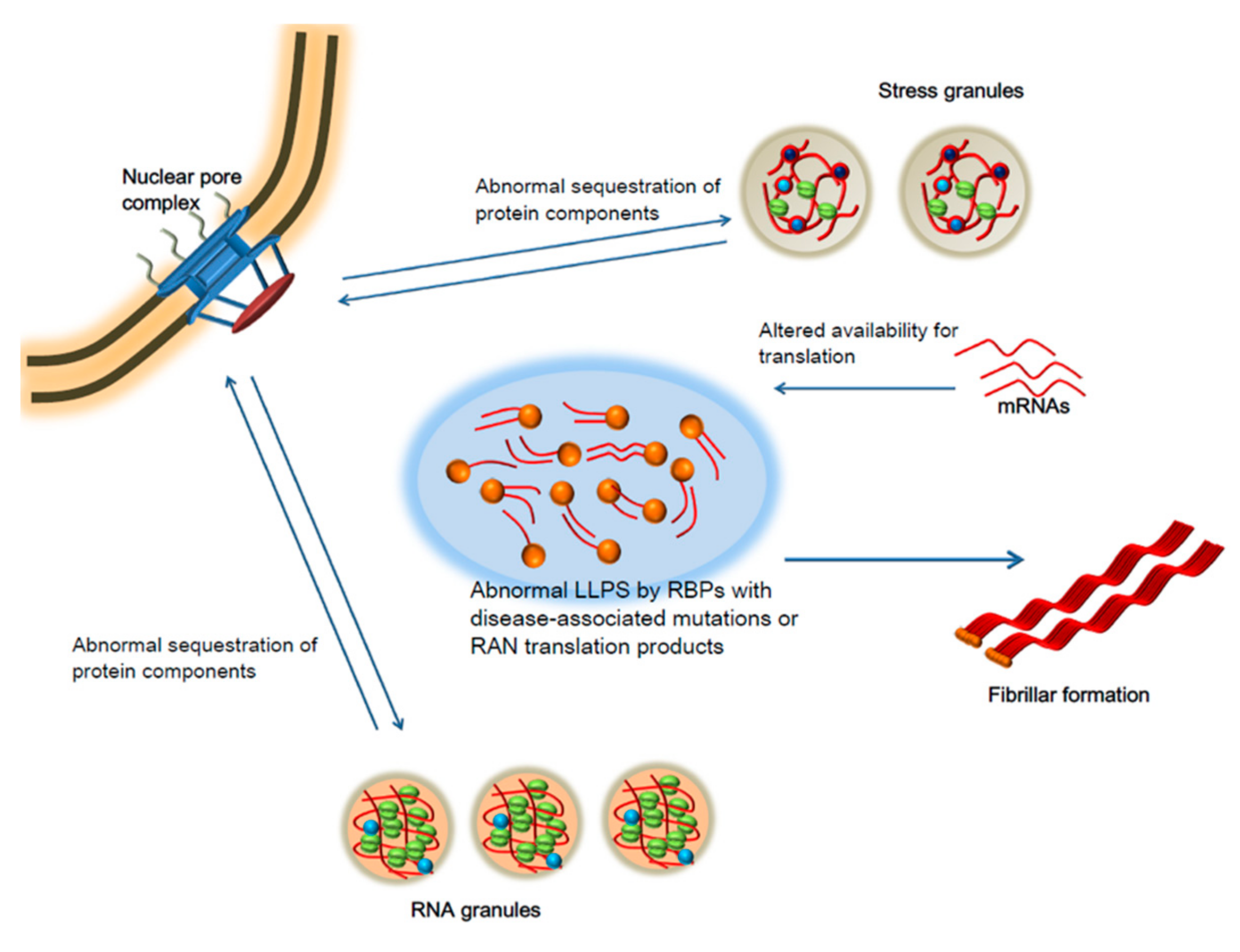
5. Liquid-Liquid Phase Separation and Neurodegeneration
6. Future Perspectives
Promises and Cautions for Novel Therapeutic Strategies
Acknowledgments
Conflicts of Interest
References
- Hui, K.; Katayama, Y.; Nakayama, K.I.; Nomura, J.; Sakurai, T. Characterizing vulnerable brain areas and circuits in mouse models of autism: Towards understanding pathogenesis and new therapeutic approaches. Neurosci. Biobehav. Rev. 2018. [Google Scholar] [CrossRef]
- Auerbach, B.D.; Osterweil, E.K.; Bear, M.F. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011, 480, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.G.; Khoutorsky, A.; Cao, R.; Jafarnejad, S.M.; Prager-Khoutorsky, M.; Giannakas, N.; Kaminari, A.; Fragkouli, A.; Nader, K.; Price, T.J.; et al. Pharmacogenetic Inhibition of eIF4E-Dependent Mmp9 mRNA Translation Reverses Fragile X Syndrome-like Phenotypes. Cell Rep. 2014, 9, 1742–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterweil, E.K.; Krueger, D.D.; Reinhold, K.; Bear, M.F. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 2010, 30, 15616–15627. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Huynh, T.N.; MacAskill, A.F.; Carter, A.G.; Pierre, P.; Ruggero, D.; Kaphzan, H.; Klann, E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013, 493, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Huynh, T.N.; Longo, F.; Koo, S.Y.; Mojica, E.; D’Andrea, L.; Bagni, C.; Klann, E. Reducing eIF4E-eIF4G interactions restores the balance between protein synthesis and actin dynamics in fragile X syndrome model mice. Sci. Signal. 2017, 10, eaan0665. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Kaphzan, H.; Alvarez-Dieppa, A.C.; Murphy, J.P.; Pierre, P.; Klann, E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 2012, 76, 325–337. [Google Scholar] [CrossRef]
- Ascano, M.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef]
- Greenblatt, E.J.; Spradling, A.C. Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 2018, 361, 709–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, S.R.; Seo, S.S.; Barnes, S.A.; Louros, S.R.; Muscas, M.; Dando, O.; Kirby, C.; Wyllie, D.J.A.; Hardingham, G.E.; Kind, P.C.; et al. Cell-Type-Specific Translation Profiling Reveals a Novel Strategy for Treating Fragile X Syndrome. Neuron 2017, 95, 550–563.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxbaum, J.D.; Silverman, J.M.; Smith, C.J.; Greenberg, D.A.; Kilifarski, M.; Reichert, J.; Cook, E.H., Jr.; Fang, Y.; Song, C.-Y.; Vitale, R. Association between a GABRB3 polymorphism and autism. Mol. Psychiatr. 2002, 7, 311. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Heinrich, J.; Delorme, R.; Proepper, C.; Betancur, C.; Huguet, G.; Konyukh, M.; Chaste, P.; Ey, E.; Rastam, M.; et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012, 8, e1002521. [Google Scholar] [CrossRef]
- Neves-Pereira, M.; Müller, B.; Massie, D.; Williams, J.H.G.; O’Brien, P.C.M.; Hughes, A.; Shen, S.-B.; Clair, D.S.; Miedzybrodzka, Z. Deregulation of EIF4E: A novel mechanism for autism. J. Med. Genet. 2009, 46, 759–765. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [Green Version]
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.M.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef]
- Nakajima, J.; Okamoto, N.; Tohyama, J.; Kato, M.; Arai, H.; Funahashi, O.; Tsurusaki, Y.; Nakashima, M.; Kawashima, H.; Saitsu, H.; et al. De novo EEF1A2 mutations in patients with characteristic facial features, intellectual disability, autistic behaviors and epilepsy. Clin. Genet. 2015, 87, 356–361. [Google Scholar] [CrossRef]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 2015, 4, e05033. [Google Scholar] [CrossRef]
- Tărlungeanu, D.C.; Deliu, E.; Dotter, C.P.; Kara, M.; Janiesch, P.C.; Scalise, M.; Galluccio, M.; Tesulov, M.; Morelli, E.; Sonmez, F.M.; et al. Impaired Amino Acid Transport at the Blood Brain Barrier Is a Cause of Autism Spectrum Disorder. Cell 2016, 167, 1481–1494.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novarino, G.; El-Fishawy, P.; Kayserili, H.; Meguid, N.A.; Scott, E.M.; Schroth, J.; Silhavy, J.L.; Kara, M.; Khalil, R.O.; Ben-Omran, T.; et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 2012, 338, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Topol, A.; English, J.A.; Flaherty, E.; Rajarajan, P.; Hartley, B.J.; Gupta, S.; Desland, F.; Zhu, S.; Goff, T.; Friedman, L.; et al. Increased abundance of translation machinery in stem cell-derived neural progenitor cells from four schizophrenia patients. Transl. Psychiatr. 2015, 5, e662. [Google Scholar] [CrossRef] [PubMed]
- Stefánsson, H.; Rujescu, D.; Cichon, S.; Pietiläinen, O.P.H.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236. [Google Scholar] [CrossRef]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008, 455, 237–241. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; Pasciuto, E.; Li, K.W.; Fernández, E.; Di Marino, D.; Buzzi, A.; Ostroff, L.E.; Klann, E.; Zwartkruis, F.J.T.; Komiyama, N.H.; et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 2013, 79, 1169–1182. [Google Scholar] [CrossRef]
- Napoli, I.; Mercaldo, V.; Boyl, P.P.; Eleuteri, B.; Zalfa, F.; De Rubeis, S.; Di Marino, D.; Mohr, E.; Massimi, M.; Falconi, M.; et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 2008, 134, 1042–1054. [Google Scholar] [CrossRef]
- Kirkpatrick, S.L.; Goldberg, L.R.; Yazdani, N.; Babbs, R.K.; Wu, J.; Reed, E.R.; Jenkins, D.F.; Bolgioni, A.F.; Landaverde, K.I.; Luttik, K.P.; et al. Cytoplasmic FMR1-Interacting Protein 2 Is a Major Genetic Factor Underlying Binge Eating. Biol. Psychiatr. 2017, 81, 757–769. [Google Scholar] [CrossRef]
- Endo, R.; Takashima, N.; Nekooki-Machida, Y.; Komi, Y.; Hui, K.K.-W.; Takao, M.; Akatsu, H.; Murayama, S.; Sawa, A.; Tanaka, M. TAR DNA-Binding Protein 43 and Disrupted in Schizophrenia 1 Coaggregation Disrupts Dendritic Local Translation and Mental Function in Frontotemporal Lobar Degeneration. Biol. Psychiatr. 2018, 84, 509–521. [Google Scholar] [CrossRef]
- Trinh, M.A.; Kaphzan, H.; Wek, R.C.; Pierre, P.; Cavener, D.R.; Klann, E. Brain-specific disruption of the eIF2α kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep. 2012, 1, 676–688. [Google Scholar] [CrossRef]
- Aguilar-Valles, A.; Haji, N.; De Gregorio, D.; Matta-Camacho, E.; Eslamizade, M.J.; Popic, J.; Sharma, V.; Cao, R.; Rummel, C.; Tanti, A.; et al. Translational control of depression-like behavior via phosphorylation of eukaryotic translation initiation factor 4E. Nat. Commun. 2018, 9, 2459. [Google Scholar] [CrossRef]
- Allen, N.J. Astrocyte regulation of synaptic behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 439–463. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dissing-Olesen, L.; MacVicar, B.A.; Stevens, B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015, 36, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.-C.; Suen, K.-C.; Ma, C.-H.; Elyaman, W.; Ng, H.-K.; Hugon, J. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J. Neurochem. 2002, 83, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.C.; Wong, A.K.Y.; Ng, H.-K.; Hugon, J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 2002, 13, 2429–2432. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.; Onuki, R.; Katayama, T.; Manabe, T.; Kudo, T.; Taira, K.; Tohyama, M. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson’s disease and Huntington’s disease. Neurochem. Int. 2005, 46, 11–18. [Google Scholar] [CrossRef]
- Peel, A.L.; Bredesen, D.E. Activation of the cell stress kinase PKR in Alzheimer’s disease and human amyloid precursor protein transgenic mice. Neurobiol. Dis. 2003, 14, 52–62. [Google Scholar] [CrossRef]
- Peel, A.L.; Rao, R.V.; Cottrell, B.A.; Hayden, M.R.; Ellerby, L.M.; Bredesen, D.E. Double-stranded RNA-dependent protein kinase, PKR, binds preferentially to Huntington’s disease (HD) transcripts and is activated in HD tissue. Hum. Mol. Genet. 2001, 10, 1531–1538. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Lapalus, P.; Gray, F.; Laplanche, J.-L.; Hugon, J.; Groupe d’Investigation du Liquide Céphalorachidien Study Network. Increased cerebrospinal fluid levels of double-stranded RNA-dependant protein kinase in Alzheimer’s disease. Biol. Psychiatr. 2012, 71, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim. Biophys. Acta 2012, 1822, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Tible, M.; Mouton-Liger, F.; Schmitt, J.; Giralt, A.; Farid, K.; Thomasseau, S.; Gourmaud, S.; Paquet, C.; Rondi Reig, L.; Meurs, E.; et al. PKR knockout in the 5xFAD model of Alzheimer’s disease reveals beneficial effects on spatial memory and brain lesions. Aging Cell 2019, 18, e12887. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Paccalin, M.; Lafay-Chebassier, C.; Chalon, S.; Ingrand, I.; Pinguet, J.; Pontcharraud, R.; Guillard, O.; Fauconneau, B.; Page, G. Pharmacological inhibition of PKR in APPswePS1dE9 mice transiently prevents inflammation at 12 months of age but increases Aβ42 levels in the late stages of the Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Page, G.; Rioux Bilan, A.; Ingrand, S.; Lafay-Chebassier, C.; Pain, S.; Perault Pochat, M.C.; Bouras, C.; Bayer, T.; Hugon, J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience 2006, 139, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhou, X.; Zimmermann, H.R.; Cavener, D.R.; Klann, E.; Ma, T. Repression of the eIF2α kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2016, 41, 19–24. [Google Scholar] [CrossRef]
- Segev, Y.; Barrera, I.; Ounallah-Saad, H.; Wibrand, K.; Sporild, I.; Livne, A.; Rosenberg, T.; David, O.; Mints, M.; Bramham, C.R.; et al. PKR Inhibition Rescues Memory Deficit and ATF4 Overexpression in ApoE ε4 Human Replacement Mice. J. Neurosci. 2015, 35, 12986–12993. [Google Scholar] [CrossRef]
- Johnson, G.; Gotlib, J.; Haroutunian, V.; Bierer, L.; Nairn, A.C.; Merril, C.; Wallace, W. Increased phosphorylation of elongation factor 2 in Alzheimer’s disease. Brain Res. Mol. Brain Res. 1992, 15, 319–326. [Google Scholar] [CrossRef]
- Ryazanov, A.G.; Shestakova, E.A.; Natapov, P.G. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 1988, 334, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Beckelman, B.C.; Yang, W.; Kasica, N.P.; Zimmermann, H.R.; Zhou, X.; Keene, C.D.; Ryazanov, A.G.; Ma, T. Genetic reduction of eEF2 kinase alleviates pathophysiology in Alzheimer’s disease model mice. J. Clin. Invest. 2019, 129, 820–833. [Google Scholar] [CrossRef]
- Xu, J.; Patassini, S.; Rustogi, N.; Riba-Garcia, I.; Hale, B.D.; Phillips, A.M.; Waldvogel, H.; Haines, R.; Bradbury, P.; Stevens, A.; et al. Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun. Biol. 2019, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Markesbery, W.R.; Chen, Q.; Li, F.; Keller, J.N. Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 2005, 25, 9171–9175. [Google Scholar] [CrossRef] [PubMed]
- Langstrom, N.S.; Anderson, J.P.; Lindroos, H.G.; Winblad, B.; Wallace, W.C. Alzheimer’s disease-associated reduction of polysomal mRNA translation. Brain Res. Mol. Brain Res. 1989, 5, 259–269. [Google Scholar] [CrossRef]
- Beckelman, B.C.; Day, S.; Zhou, X.; Donohue, M.; Gouras, G.K.; Klann, E.; Keene, C.D.; Ma, T. Dysregulation of Elongation Factor 1A Expression is Correlated with Synaptic Plasticity Impairments in Alzheimer’s Disease. J. Alzheimers Dis. 2016, 54, 669–678. [Google Scholar] [CrossRef]
- Beckelman, B.C.; Zhou, X.; Keene, C.D.; Ma, T. Impaired Eukaryotic Elongation Factor 1A Expression in Alzheimer’s Disease. Neurodegener. Dis. 2016, 16, 39–43. [Google Scholar] [CrossRef]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.-J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef]
- Kapur, M.; Monaghan, C.E.; Ackerman, S.L. Regulation of mRNA Translation in Neurons-A Matter of Life and Death. Neuron 2017, 96, 616–637. [Google Scholar] [CrossRef]
- Ishimura, R.; Nagy, G.; Dotu, I.; Zhou, H.; Yang, X.-L.; Schimmel, P.; Senju, S.; Nishimura, Y.; Chuang, J.H.; Ackerman, S.L. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 2014, 345, 455–459. [Google Scholar] [CrossRef]
- Pisareva, V.P.; Skabkin, M.A.; Hellen, C.U.T.; Pestova, T.V.; Pisarev, A.V. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J. 2011, 30, 1804–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimura, R.; Nagy, G.; Dotu, I.; Chuang, J.H.; Ackerman, S.L. Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife 2016, 5, 568. [Google Scholar] [CrossRef] [PubMed]
- Jaberi, E.; Rohani, M.; Shahidi, G.A.; Nafissi, S.; Arefian, E.; Soleimani, M.; Rasooli, P.; Ahmadieh, H.; Daftarian, N.; KaramiNejadRanjbar, M.; et al. Identification of mutation in GTPBP2 in patients of a family with neurodegeneration accompanied by iron deposition in the brain. Neurobiol. Aging 2016, 38, 216.e11–216.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli-Avella, A.M.; Garcia-Aznar, J.M.; Brandau, O.; Al-Hakami, F.; Yüksel, Z.; Marais, A.; Grüning, N.-M.; Abbasi Moheb, L.; Paknia, O.; Alshaikh, N.; et al. Biallelic inactivating variants in the GTPBP2 gene cause a neurodevelopmental disorder with severe intellectual disability. Eur. J. Hum. Genet. 2018, 26, 592–598. [Google Scholar] [CrossRef]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-W.; Tanaka, M. Genome-wide Translation Profiling by Ribosome-Bound tRNA Capture. Cell Rep. 2018, 23, 608–621. [Google Scholar] [CrossRef] [Green Version]
- Joazeiro, C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019, 33, 439. [Google Scholar] [CrossRef]
- Shoemaker, C.J.; Eyler, D.E.; Green, R. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop off to initiate no-go decay. Science 2010, 330, 369–372. [Google Scholar] [CrossRef]
- Hu, W.; Sweet, T.J.; Chamnongpol, S.; Baker, K.E.; Coller, J. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature 2009, 461, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Tesina, P.; Heckel, E.; Cheng, J.; Fromont-Racine, M.; Buschauer, R.; Kater, L.; Beatrix, B.; Berninghausen, O.; Jacquier, A.; Becker, T.; et al. Structure of the 80S ribosome-Xrn1 nuclease complex. Nat. Struct. Mol. Biol. 2019, 26, 275–280. [Google Scholar] [CrossRef]
- Heuer, A.; Gerovac, M.; Schmidt, C.; Trowitzsch, S.; Preis, A.; Kötter, P.; Berninghausen, O.; Becker, T.; Beckmann, R.; Tampé, R. Structure of the 40S-ABCE1 post-splitting complex in ribosome recycling and translation initiation. Nat. Struct. Mol. Biol. 2017, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Li, S.; Kato, M.; Ikeuchi, K.; Ichimura, A.; Matsuo, Y.; Inada, T. Sequential Ubiquitination of Ribosomal Protein uS3 Triggers the Degradation of Non-functional 18S rRNA. Cell Rep. 2019, 26, 3400–3415.e7. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ikeuchi, K.; Saeki, Y.; Iwasaki, S.; Schmidt, C.; Udagawa, T.; Sato, F.; Tsuchiya, H.; Becker, T.; Tanaka, K.; et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat. Commun. 2017, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Juszkiewicz, S.; Hegde, R.S. Initiation of Quality Control during Poly(A) Translation Requires Site-Specific Ribosome Ubiquitination. Mol. Cell 2017, 65, 743–750.e4. [Google Scholar] [CrossRef] [Green Version]
- Bengtson, M.H.; Joazeiro, C.A.P. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 2010, 467, 470–473. [Google Scholar] [CrossRef]
- Brandman, O.; Stewart-Ornstein, J.; Wong, D.; Larson, A.; Williams, C.C.; Li, G.-W.; Zhou, S.; King, D.; Shen, P.S.; Weibezahn, J.; et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 2012, 151, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Lyumkis, D.; Oliveira dos Passos, D.; Tahara, E.B.; Webb, K.; Bennett, E.J.; Vinterbo, S.; Potter, C.S.; Carragher, B.; Joazeiro, C.A.P. Structural basis for translational surveillance by the large ribosomal subunit-associated protein quality control complex. Proc. Natl. Acad. Sci. USA 2014, 111, 15981–15986. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.S.; Park, J.; Qin, Y.; Li, X.; Parsawar, K.; Larson, M.H.; Cox, J.; Cheng, Y.; Lambowitz, A.M.; Weissman, J.S.; et al. Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 2015, 347, 75–78. [Google Scholar] [CrossRef]
- Shao, S.; Brown, A.; Santhanam, B.; Hegde, R.S. Structure and Assembly Pathway of the Ribosome Quality Control Complex. Mol. Cell 2015, 57, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Kostova, K.K.; Hickey, K.L.; Osuna, B.A.; Hussmann, J.A.; Frost, A.; Weinberg, D.E.; Weissman, J.S. CAT-tailing as a fail-safe mechanism for efficient degradation of stalled nascent polypeptides. Science 2017, 357, 414–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitron, C.S.; Brandman, O. CAT tails drive degradation of stalled polypeptides on and off the ribosome. Nat. Struct. Molec. Biol. 2019, 26, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Choe, Y.-J.; Park, S.-H.; Hassemer, T.; Körner, R.; Vincenz-Donnelly, L.; Hayer-Hartl, M.; Hartl, F.U. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature 2016, 531, 191–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defenouillère, Q.; Zhang, E.; Namane, A.; Mouaikel, J.; Jacquier, A.; Fromont-Racine, M. Rqc1 and Ltn1 Prevent C-terminal Alanine-Threonine Tail (CAT-tail)-induced Protein Aggregation by Efficient Recruitment of Cdc48 on Stalled 60S Subunits. J. Biol. Chem. 2016, 291, 12245–12253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonashiro, R.; Tahara, E.B.; Bengtson, M.H.; Khokhrina, M.; Lorenz, H.; Chen, K.-C.; Kigoshi-Tansho, Y.; Savas, J.N.; Yates, J.R.; Kay, S.A.; et al. The Rqc2/Tae2 subunit of the ribosome-associated quality control (RQC) complex marks ribosome-stalled nascent polypeptide chains for aggregation. Elife 2016, 5, e11794. [Google Scholar] [CrossRef]
- Verma, R.; Reichermeier, K.M.; Burroughs, A.M.; Oania, R.S.; Reitsma, J.M.; Aravind, L.; Deshaies, R.J. Vms1 and ANKZF1 peptidyl-tRNA hydrolases release nascent chains from stalled ribosomes. Nature 2018, 557, 446. [Google Scholar] [CrossRef]
- Defenouillère, Q.; Yao, Y.; Mouaikel, J.; Namane, A.; Galopier, A.; Decourty, L.; Doyen, A.; Malabat, C.; Saveanu, C.; Jacquier, A.; et al. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc. Natl. Acad. Sci. USA 2013, 110, 5046–5051. [Google Scholar] [CrossRef] [Green Version]
- Kuroha, K.; Zinoviev, A.; Hellen, C.U.T.; Pestova, T.V. Release of Ubiquitinated and Non-ubiquitinated Nascent Chains from Stalled Mammalian Ribosomal Complexes by ANKZF1 and Ptrh1. Mol. Cell 2018, 72, 286–302.e8. [Google Scholar] [CrossRef] [Green Version]
- Defenouillère, Q.; Namane, A.; Mouaikel, J.; Jacquier, A.; Fromont-Racine, M. The ribosome-bound quality control complex remains associated to aberrant peptides during their proteasomal targeting and interacts with Tom1 to limit protein aggregation. Mol. Biol. Cell 2017, 28, 1165–1176. [Google Scholar] [CrossRef] [Green Version]
- Sung, M.-K.; Porras-Yakushi, T.R.; Reitsma, J.M.; Huber, F.M.; Sweredoski, M.J.; Hoelz, A.; Hess, S.; Deshaies, R.J. A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. Elife 2016, 5, 3429. [Google Scholar] [CrossRef]
- Sung, M.-K.; Reitsma, J.M.; Sweredoski, M.J.; Hess, S.; Deshaies, R.J. Ribosomal proteins produced in excess are degraded by the ubiquitin-proteasome system. Mol. Biol. Cell 2016, 27, 2642–2652. [Google Scholar] [CrossRef] [PubMed]
- Froyen, G.; Belet, S.; Martinez, F.; Santos-Rebouças, C.B.; Declercq, M.; Verbeeck, J.; Donckers, L.; Berland, S.; Mayo, S.; Rosello, M.; et al. Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements. Am. J. Hum. Genet. 2012, 91, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Froyen, G.; Corbett, M.; Vandewalle, J.; Järvelä, I.; Lawrence, O.; Meldrum, C.; Bauters, M.; Govaerts, K.; Vandeleur, L.; Van Esch, H.; et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 2008, 82, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Moortgat, S.; Berland, S.; Aukrust, I.; Maystadt, I.; Baker, L.; Benoit, V.; Caro-Llopis, A.; Cooper, N.S.; Debray, F.-G.; Faivre, L.; et al. HUWE1 variants cause dominant X-linked intellectual disability: A clinical study of 21 patients. Eur. J. Hum. Genet. 2018, 26, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Hong, N.A.; Masuda, C.A.; Jenkins, B.V.; Nelms, K.A.; Goodnow, C.C.; Glynne, R.J.; Wu, H.; Masliah, E.; Joazeiro, C.A.P.; et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 2097–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anazi, S.; Maddirevula, S.; Faqeih, E.; Alsedairy, H.; Alzahrani, F.; Shamseldin, H.E.; Patel, N.; Hashem, M.; Ibrahim, N.; Abdulwahab, F.; et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol. Psychiatr. 2017, 22, 615–624. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Nguyen, L.; Cleary, J.D.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation: Molecular Mechanisms and Contribution to Neurological Disease. Annu. Rev. Neurosci. 2019, 42, 227–247. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.-M.; Schludi, M.H.; van der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates that Initiate In Vitro and In Vivo Neuronal Death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.-F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 2016, 19, 668–677. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ito, D.; Honda, T.; Kubo, K.-I.; Noda, M.; Nakajima, K.; Suzuki, N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet. 2015, 24, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. Eneuro 2017, 4, ENEURO.0249-16.2017. [Google Scholar] [CrossRef]
- Hoem, G.; Bowitz Larsen, K.; Øvervatn, A.; Brech, A.; Lamark, T.; Sjøttem, E.; Johansen, T. The FMRpolyGlycine Protein Mediates Aggregate Formation and Toxicity Independent of the CGG mRNA Hairpin in a Cellular Model for FXTAS. Front Genet. 2019, 10, 249. [Google Scholar] [CrossRef]
- Guo, Q.; Lehmer, C.; Martínez-Sánchez, A.; Rudack, T.; Beck, F.; Hartmann, H.; Pérez-Berlanga, M.; Frottin, F.; Hipp, M.S.; Hartl, F.U.; et al. In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 2018, 172, 696–705.e12. [Google Scholar] [CrossRef] [Green Version]
- Kramer, N.J.; Haney, M.S.; Morgens, D.W.; Jovičić, A.; Couthouis, J.; Li, A.; Ousey, J.; Ma, R.; Bieri, G.; Tsui, C.K.; et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 2018, 50, 603–612. [Google Scholar] [CrossRef]
- Oh, S.Y.; He, F.; Krans, A.; Frazer, M.; Taylor, J.P.; Paulson, H.L.; Todd, P.K. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum. Mol. Genet. 2015, 24, 4317–4326. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Jansen-West, K.; Xu, Y.-F.; Gendron, T.F.; Bieniek, K.F.; Lin, W.-L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014, 128, 505–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.-H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Jovičić, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W.; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Wang, H.; Xia, Q.; Li, K.; Li, K.; Jiang, X.; Xu, G.; Wang, G.; Ying, Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 2015, 24, 2426–2441. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.D.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H.; et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell 2017, 65, 1044–1055.e5. [Google Scholar] [CrossRef]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17. [Google Scholar] [CrossRef] [Green Version]
- Chew, J.; Cook, C.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Daughrity, L.M.; Castanedes-Casey, M.; Kurti, A.; Stankowski, J.N.; Disney, M.D.; et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol. Neurodegener. 2019, 14, 9–15. [Google Scholar] [CrossRef]
- Lin, Y.; Mori, E.; Kato, M.; Xiang, S.; Wu, L.; Kwon, I.; McKnight, S.L. Toxic PR Poly-Dipeptides Encoded by the C9orf72 Repeat Expansion Target LC Domain Polymers. Cell 2016, 167, 789–802.e12. [Google Scholar] [CrossRef] [Green Version]
- White, M.R.; Mitrea, D.M.; Zhang, P.; Stanley, C.B.; Cassidy, D.E.; Nourse, A.; Phillips, A.H.; Tolbert, M.; Taylor, J.P.; Kriwacki, R.W. C9orf72 Poly(PR) Dipeptide Repeats Disturb Biomolecular Phase Separation and Disrupt Nucleolar Function. Mol. Cell 2019, 74, 713–728.e6. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.M.; Anderson, P.J.; Ivanov, P. ALS/FTD-Associated C9ORF72 Repeat RNA Promotes Phase Transitions In Vitro and in Cells. Cell Rep. 2017, 21, 3573–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.G.; Green, K.M.; Krans, A.; Rodriguez, C.M.; Linsalata, A.E.; Goldstrohm, A.C.; Todd, P.K. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol. Cell 2016, 62, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.; Wang, S.; Mestre, A.A.; Fu, C.; Makarem, A.; Xian, F.; Hayes, L.R.; Lopez-Gonzalez, R.; Drenner, K.; Jiang, J.; et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2α phosphorylation. Nat. Commun. 2018, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun. 2017, 8, 2005. [Google Scholar] [CrossRef]
- Sonobe, Y.; Ghadge, G.; Masaki, K.; Sendoel, A.; Fuchs, E.; Roos, R.P. Translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol. Dis. 2018, 116, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Starck, S.R.; Jiang, V.; Pavon-Eternod, M.; Prasad, S.; McCarthy, B.; Pan, T.; Shastri, N. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science 2012, 336, 1719–1723. [Google Scholar] [CrossRef]
- Yamada, S.B.; Gendron, T.F.; Niccoli, T.; Genuth, N.R.; Grosely, R.; Shi, Y.; Glaria, I.; Kramer, N.J.; Nakayama, L.; Fang, S.; et al. RPS25 is required for efficient RAN translation of C9orf72 and other neurodegenerative disease-associated nucleotide repeats. Nat. Neurosci. 2019, 22, 1383–1388. [Google Scholar] [CrossRef]
- Hertz, M.I.; Landry, D.M.; Willis, A.E.; Luo, G.; Thompson, S.R. Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol. Cell. Biol. 2013, 33, 1016–1026. [Google Scholar] [CrossRef]
- Muhs, M.; Yamamoto, H.; Ismer, J.; Takaku, H.; Nashimoto, M.; Uchiumi, T.; Nakashima, N.; Mielke, T.; Hildebrand, P.W.; Nierhaus, K.H.; et al. Structural basis for the binding of IRES RNAs to the head of the ribosomal 40S subunit. Nucleic Acids Res. 2011, 39, 5264–5275. [Google Scholar] [CrossRef]
- Landry, D.M.; Hertz, M.I.; Thompson, S.R. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 2009, 23, 2753–2764. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, T.; Yamamoto, H.; Uchiumi, T.; Nakashima, N. Eukaryotic ribosomal protein RPS25 interacts with the conserved loop region in a dicistroviral intergenic internal ribosome entry site. Nucleic Acids Res. 2007, 35, 1514–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabet, R.; Schaeffer, L.; Freyermuth, F.; Jambeau, M.; Workman, M.; Lee, C.-Z.; Lin, C.-C.; Jiang, J.; Jansen-West, K.; Abou-Hamdan, H.; et al. CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat. Commun. 2018, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.G.; Goldman, D.H.; Choi, J.; Nwaezeapu, C.; Liang, D.; Green, K.M.; Goldstrohm, A.C.; Todd, P.K.; Green, R.; Wilusz, J.E. Ribosome queuing enables non-AUG translation to be resistant to multiple protein synthesis inhibitors. Genes Dev. 2019, 33, 871–885. [Google Scholar] [CrossRef] [Green Version]
- Murthy, A.C.; Dignon, G.L.; Kan, Y.; Zerze, G.H.; Parekh, S.H.; Mittal, J.; Fawzi, N.L. Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019, 26, 637–648. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nüske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 2018, 359, eaao5654. [Google Scholar] [CrossRef] [Green Version]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Kim, T.H.; Tsang, B.; Vernon, R.M.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 2019, 365, 825–829. [Google Scholar] [CrossRef]
- Ford, L.; Ling, E.; Kandel, E.R.; Fioriti, L. CPEB3 inhibits translation of mRNA targets by localizing them to P bodies. Proc. Natl. Acad. Sci. USA 2019, 51, 201815275. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Elbaum-Garfinkle, S.; Langdon, E.M.; Taylor, N.; Occhipinti, P.; Bridges, A.A.; Brangwynne, C.P.; Gladfelter, A.S. RNA Controls PolyQ Protein Phase Transitions. Mol. Cell 2015, 60, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jove Navarro, M.; Kashida, S.; Chouaib, R.; Souquere, S.; Pierron, G.; Weil, D.; Gueroui, Z. RNA is a critical element for the sizing and the composition of phase-separated RNA-protein condensates. Nat. Commun. 2019, 10, 3230. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Protter, D.S.W.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, R.J.; Zaccara, S.; Klein, P.; Olarerin-George, A.; Namkoong, S.; Pickering, B.F.; Patil, D.P.; Kwak, H.; Lee, J.H.; Jaffrey, S.R. m6A enhances the phase separation potential of mRNA. Nature 2019, 571, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Vale, R.D. RNA phase transitions in repeat expansion disorders. Nature 2017, 546, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Bolognesi, B.; Lorenzo Gotor, N.; Dhar, R.; Cirillo, D.; Baldrighi, M.; Tartaglia, G.G.; Lehner, B. A Concentration-Dependent Liquid Phase Separation Can Cause Toxicity upon Increased Protein Expression. Cell Rep. 2016, 16, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, L.M.; Safren, N.; Pithadia, A.S.; Gerson, J.E.; Dulchavsky, M.; Fischer, S.; Patel, R.; Lantis, G.; Ashraf, N.; Kim, J.H.; et al. Mutant UBQLN2 promotes toxicity by modulating intrinsic self-assembly. Proc. Natl. Acad. Sci. USA 2018, 115, E10495–E10504. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e9. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Clatterbuck-Soper, S.F.; Jackrel, M.E.; Shorter, J.; Mili, S. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J. Cell Biol. 2017, 216, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.S.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.W.; Kato, M.; Xie, S.; Wu, L.C.; Mirzaei, H.; Pei, J.; Chen, M.; Xie, Y.; Allen, J.; Xiao, G.; et al. Cell-free formation of RNA granules: Bound RNAs identify features and components of cellular assemblies. Cell 2012, 149, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171, 615–627.e16. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, 35. [Google Scholar] [CrossRef]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 2019, 10, 2006. [Google Scholar] [CrossRef] [PubMed]
- López-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 2018, 100, 816–830.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiihashi, G.; Ito, D.; Yagi, T.; Nihei, Y.; Ebine, T.; Suzuki, N. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain 2016, 139, 2380–2394. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 2018, 173, 677–692.e20. [Google Scholar] [CrossRef] [Green Version]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Jin, H.; Kasarskis, E.J.; Zhu, H. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. USA 2018, 115, E11904–E11913. [Google Scholar] [CrossRef] [Green Version]
- Tsang, B.; Arsenault, J.; Vernon, R.M.; Lin, H.; Sonenberg, N.; Wang, L.-Y.; Bah, A.; Forman-Kay, J.D. Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc. Natl. Acad. Sci. USA 2019, 116, 4218–4227. [Google Scholar] [CrossRef] [Green Version]
- Gopal, P.P.; Nirschl, J.J.; Klinman, E.; Holzbaur, E.L.F. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E2466–E2475. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.C.Y.; Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol. Cell 2018, 69, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Hung, V.; Udeshi, N.D.; Lam, S.S.; Loh, K.H.; Cox, K.J.; Pedram, K.; Carr, S.A.; Ting, A.Y. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 2016, 11, 456–475. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Fazal, F.M.; Han, S.; Kaewsapsak, P.; Parker, K.R.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-seq. Cell 2018, 178, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Padrón, A.; Iwasaki, S.; Ingolia, N.T. Proximity RNA Labeling by APEX-Seq Reveals the Organization of Translation Initiation Complexes and Repressive RNA Granules. Mol. Cell 2019, 75, 875–887.e5. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.-C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.-Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef]
- Zhang, P.; Fan, B.; Yang, P.; Temirov, J.; Messing, J.; Kim, H.J.; Taylor, J.P. OptoGranules reveal the evolution of stress granules to ALS-FTD pathology. bioRxiv 2018, 348870. [Google Scholar] [Green Version]
- Udagawa, T.; Farny, N.G.; Jakovcevski, M.; Kaphzan, H.; Alarcon, J.M.; Anilkumar, S.; Ivshina, M.; Hurt, J.A.; Nagaoka, K.; Nalavadi, V.C.; et al. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat. Med. 2013, 19, 1473–1477. [Google Scholar] [CrossRef] [Green Version]
- Cinesi, C.; Aeschbach, L.; Yang, B.; Dion, V. Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat. Commun. 2016, 7, 13272. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.S.; Saxena, T.; Oliveira, R.; Méndez-Gómez, H.R.; Cleary, J.D.; Denes, L.T.; McConnell, O.; Arboleda, J.; Xia, G.; Swanson, M.S.; et al. Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9. Mol. Cell 2017, 68, 479–490.e5. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.; Nelles, D.A.; Pirie, E.; Blue, S.M.; Marina, R.J.; Wang, H.; Chaim, I.A.; Thomas, J.D.; Zhang, N.; Nguyen, V.; et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 2017, 170, 899–912.e10. [Google Scholar] [CrossRef] [PubMed]
- Hautbergue, G.M.; Castelli, L.M.; Ferraiuolo, L.; Sanchez-Martinez, A.; Cooper-Knock, J.; Higginbottom, A.; Lin, Y.-H.; Bauer, C.S.; Dodd, J.E.; Myszczynska, M.A.; et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat. Commun. 2017, 8, 16063. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Sato, N.; Ueyama, M.; Fujikake, N.; Sellier, C.; Kanegami, A.; Tokuda, E.; Zamiri, B.; Gall-Duncan, T.; Mirceta, M.; et al. Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron 2017, 94, 108–124.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef] [Green Version]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol. Cell 2018, 71, 703–717. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hui, K.K.; Chen, Y.-K.; Endo, R.; Tanaka, M. Translation from the Ribosome to the Clinic: Implication in Neurological Disorders and New Perspectives from Recent Advances. Biomolecules 2019, 9, 680. https://doi.org/10.3390/biom9110680
Hui KK, Chen Y-K, Endo R, Tanaka M. Translation from the Ribosome to the Clinic: Implication in Neurological Disorders and New Perspectives from Recent Advances. Biomolecules. 2019; 9(11):680. https://doi.org/10.3390/biom9110680
Chicago/Turabian StyleHui, Kelvin K., Yi-Kai Chen, Ryo Endo, and Motomasa Tanaka. 2019. "Translation from the Ribosome to the Clinic: Implication in Neurological Disorders and New Perspectives from Recent Advances" Biomolecules 9, no. 11: 680. https://doi.org/10.3390/biom9110680
APA StyleHui, K. K., Chen, Y. -K., Endo, R., & Tanaka, M. (2019). Translation from the Ribosome to the Clinic: Implication in Neurological Disorders and New Perspectives from Recent Advances. Biomolecules, 9(11), 680. https://doi.org/10.3390/biom9110680