UV-Vis Spectroelectrochemistry of Oleuropein, Tyrosol, and p-Coumaric Acid Individually and in an Equimolar Combination. Differences in LC-ESI-MS2 Profiles of Oxidation Products and Their Neuroprotective Properties


 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Electrochemical Procedures
2.2.1. Square Wave Voltammetry (SWV) of Phenolic Compounds
2.2.2. Spectroelectrochemical Measurements
2.2.3. Linear Sweep Voltammetry (LSV)
2.2.4. Electrolysis
2.2.5. Electrolysis of Ole, Tyr, and p-Cou Individually
2.2.6. Simultaneous and Sequential Electrolysis of Ole, Tyr and p-Cou in Combination
2.3. HPLC-DAD-ESI-MS2 Process
2.4. HPLC-DAD-ESI-MS2 Analysis
2.5. Cell Culture and Cell Viability Assays
2.6. Cell Viability Assays
3. Results and Discussion
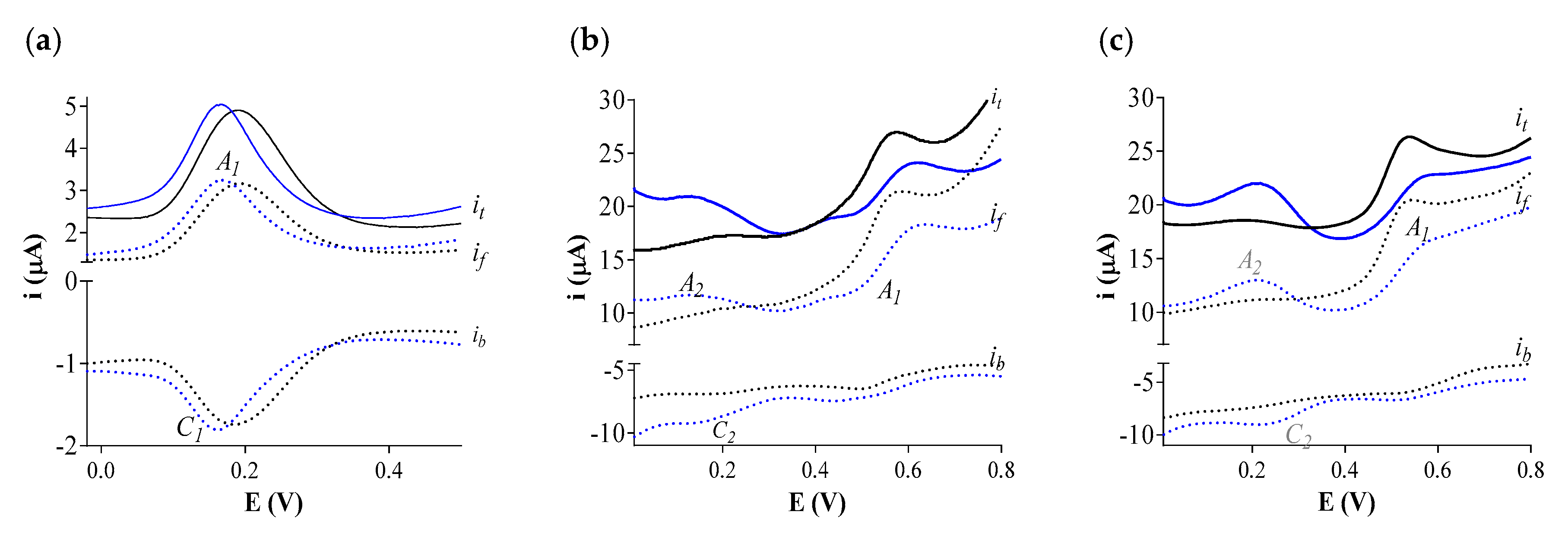
3.1. Square Wave Voltammetry (SWV) of the Three ArOH-EVOO Standards
3.2. Spectroelectrochemical Analysis of Three ArOH-EVOO Standards Individually and in the Mix Samples
3.3. The Remaining Content of the Molecules after Electrolysis Individually and in the Mix by LC-UV-ESI-MS2
3.4. Characterization of Products by LC-MS2 after the Electrochemical Oxidation of Ole
3.5. Characterization of Products by LC-MS2 after Electrochemical Oxidation of Tyr
3.6. Characterization of Products by LC-MS2 after Electrochemical Oxidation of p-Cou
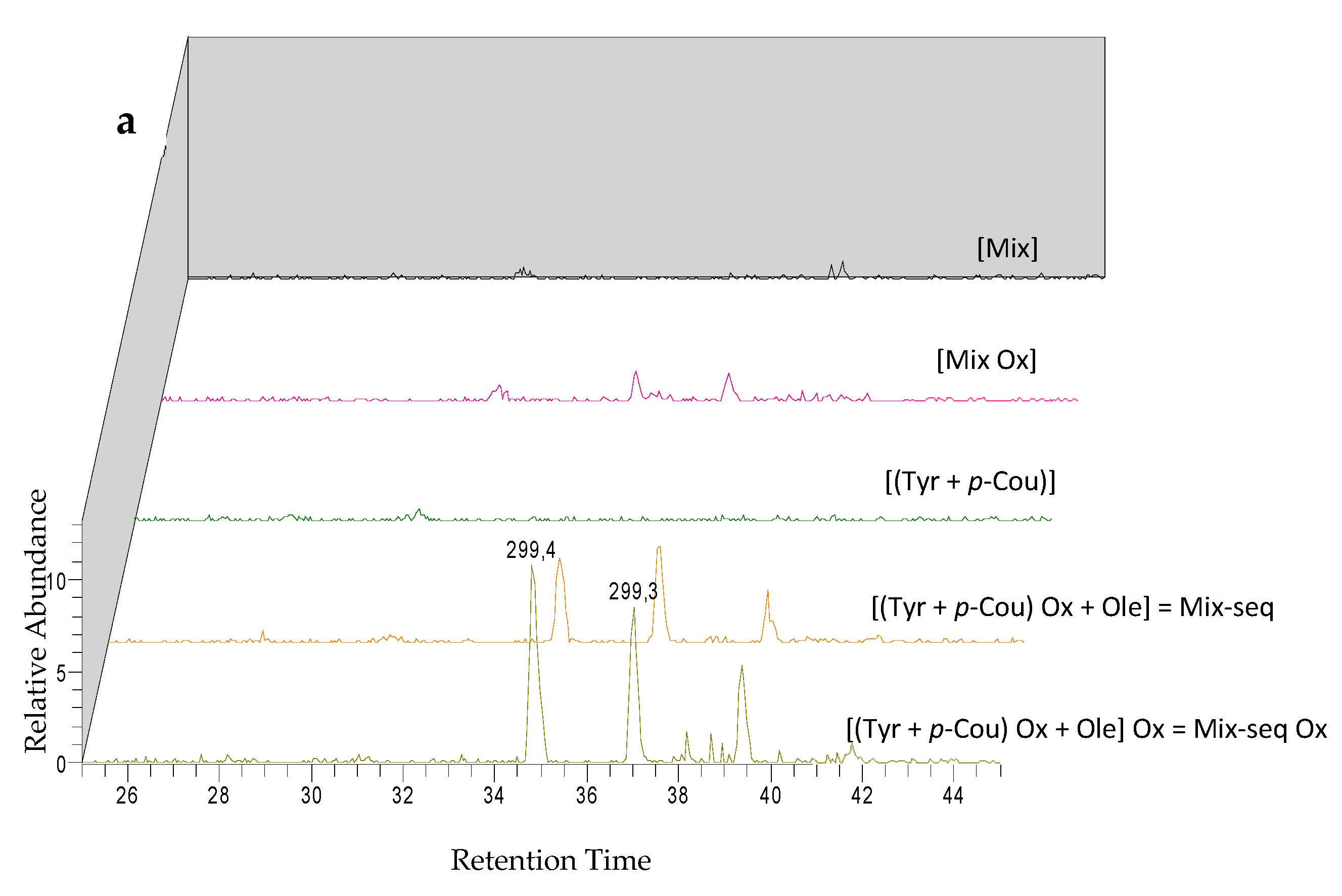
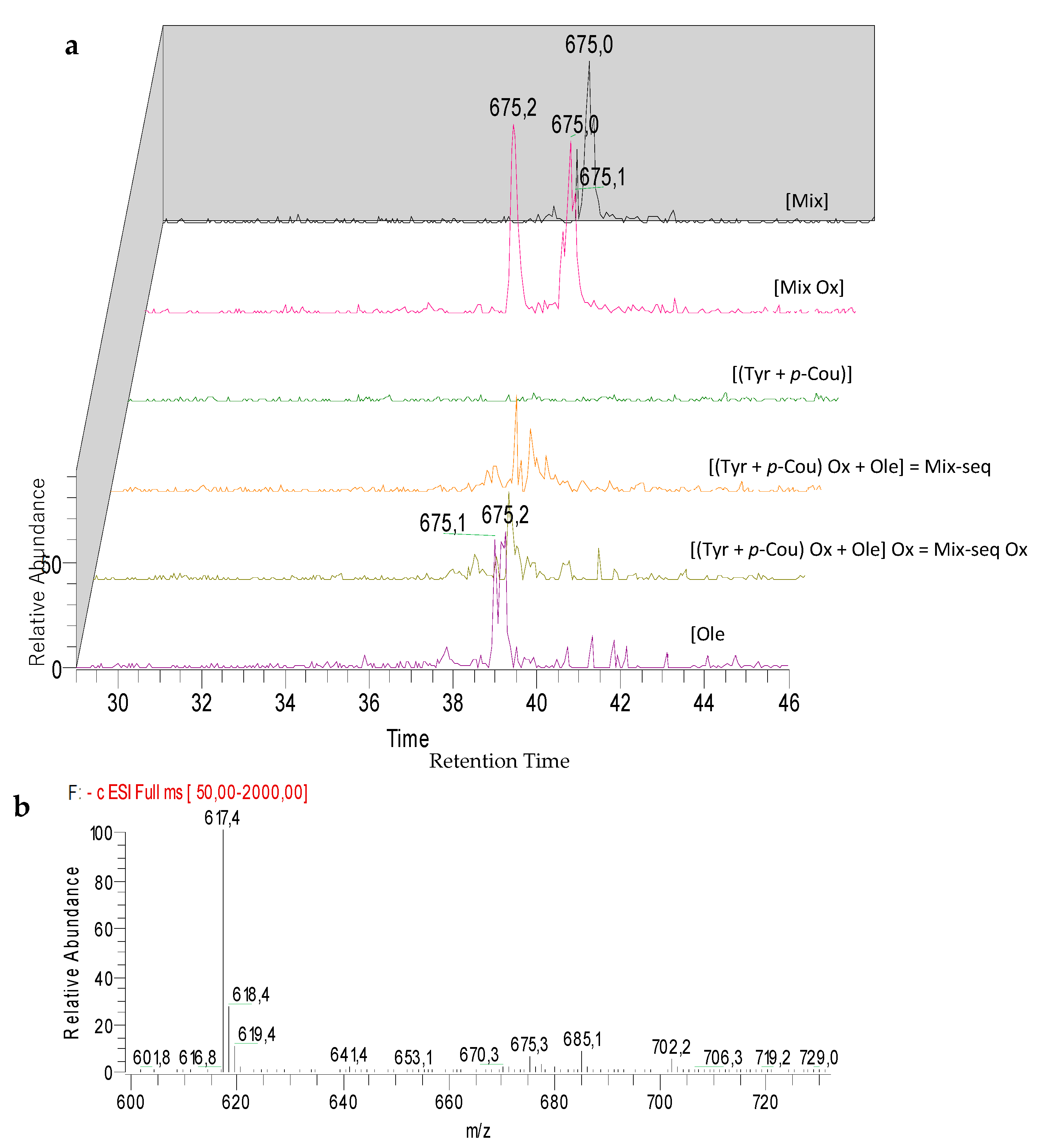
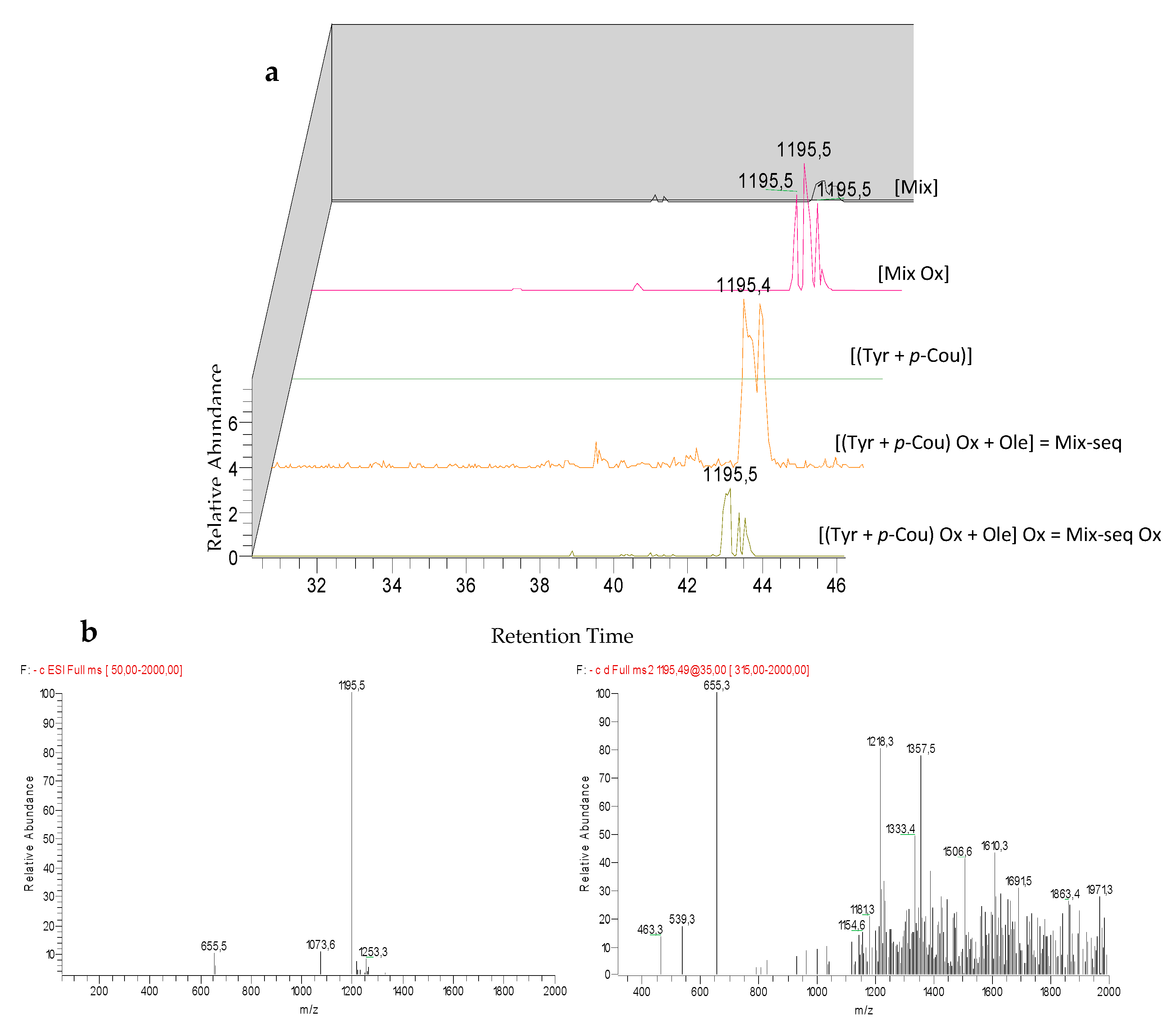
3.7. Comparison of the Chemical Profiles between Mix and the Sequentially Electrolyzed Mix (Mix-seq)
3.8. Neuroprotective Effect of Oxidized Ole and Mix
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benavente-Garcia, O.; Castillo, J.; Lorente, J.; Ortuno, A.; Del Rio, J.A. Antioxidant activity of phenolics extracted from Olea europaea L. leaves. Food Chem. 2000, 68, 457–462. [Google Scholar] [CrossRef]
- Roche, M.; Dufour, C.; Mora, N.; Dangles, O. Antioxidant activity of olive phenols: Mechanistic investigation and characterization of oxidation products by mass spectrometry. Org. Biomol. Chem. 2005, 3, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Umeno, A.; Takashima, M.; Murotomi, K.; Nakajima, Y.; Koike, T.; Matsuo, T.; Yoshida, Y. Radical-scavenging activity and antioxidative effects of olive leaf components oleuropein and hydroxytyrosol in comparison with homovanillic alcohol. J. Oleo Sci. 2015, 64, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Apak, R.; Gorinstein, S.; Böhm, V.; Schaich, K.M.; Özyürek, M.; Güçlü, K. Methods of measurement and evaluation of natural antioxidant capacity/activity (IUPAC Technical Report). Pure Appl. Chem. 2013, 85, 957–998. [Google Scholar] [CrossRef]
- Mateos, R.; Trujillo, M.; Pereira-Caro, G.; Madrona, A.; Cert, A.; Espartero, J.L. New lipophilic tyrosyl esters. Comparative antioxidant evaluation with hydroxytyrosyl esters. J. Agric. Food Chem. 2008, 56, 10960–10966. [Google Scholar] [CrossRef]
- Mathew, S.; Abraham, T.E.; Zakaria, Z.A. Reactivity of phenolic compounds towards free radicals under in vitro conditions. J. Food Sci. Technol. 2015, 52, 5790–5798. [Google Scholar] [CrossRef]
- Madrona, A.; Pereira-Caro, G.; Bravo, L.; Mateos, R.; Espartero, J.L. Preparation and antioxidant activity of tyrosyl and homovanillyl ethers. Food Chem. 2011, 129, 1169–1178. [Google Scholar] [CrossRef]
- Espinosa, R.R.; Inchingolo, R.; Alencar, S.M.; Rodriguez-Estrada, M.T.; Castro, I.A. Antioxidant activity of phenolic compounds added to a functional emulsion containing omega-3 fatty acids and plant sterol esters. Food Chem. 2015, 182, 95–104. [Google Scholar] [CrossRef]
- Lambert de Malezieu, M.; Courtel, P.; Sleno, L.; Abasq, M.-L.; Ramassamy, C. Synergistic properties of bioavailable phenolic compounds from olive oil: Electron transfer and neuroprotective properties. Nutr. Neurosci. 2019, 1–14. [Google Scholar] [CrossRef]
- Antolovich, M.; Bedgood, D.R.; Bishop, A.G.; Jardine, D.; Prenzler, P.D.; Robards, K. LC-MS investigation of oxidation products of phenolic antioxidants. J. Agric. Food Chem. 2004, 52, 962–971. [Google Scholar] [CrossRef]
- Cardoso Susana, M.; Guyot, S.; Marnet, N.; Lopes-da-Silva José, A.; Renard Catherine, M.G.C.; Coimbra Manuel, A. Characterisation of phenolic extracts from olive pulp and olive pomace by electrospray mass spectrometry. J. Sci. Food Agric. 2005, 85, 21–32. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Guyot, S.; Marnet, N.; Lopes-da-Silva, J.A.; Silva, A.M.S.; Renard, C.M.G.C.; Coimbra, M.A. Identification of oleuropein oligomers in olive pulp and pomace. J. Sci. Food Agric. 2006, 86, 1495–1502. [Google Scholar] [CrossRef]
- Obied, H.K.; Bedgood, D.R.; Prenzler, P.D.; Robards, K. Chemical screening of olive biophenol extracts by hyphenated liquid chromatography. Anal. Chim. Acta 2007, 603, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, I.; Esposto, S.; Taticchi, A.; Selvaggini, R.; Veneziani, G.; Urbani, S.; Servili, M. HPLC–ESI-MS investigation of tyrosol and hydroxytyrosol oxidation products in virgin olive oil. Food Chem. 2011, 125, 21–28. [Google Scholar] [CrossRef]
- Jerman Klen, T.; Golc Wondra, A.; Vrhovšek, U.; Mozetič Vodopivec, B. Phenolic profiling of olives and olive oil process-derived matrices using UPLC-DAD-ESI-QTOF-HRMS analysis. J. Agric. Food Chem. 2015, 63, 3859–3872. [Google Scholar] [CrossRef]
- Gentile, L.; Uccella, N.A.; Sivakumar, G. Soft-MS and computational mapping of oleuropein. Int. J. Mol. Sci. 2017, 18, 992. [Google Scholar] [CrossRef]
- St-Laurent-Thibault, C.; Arseneault, M.; Longpre, F.; Ramassamy, C. Tyrosol and hydroxytyrosol, two main components of olive oil, protect N2a cells against amyloid-beta-induced toxicity. Involvement of the NF-kappaB signaling. Curr. Alzheimer Res. 2011, 8, 543–551. [Google Scholar] [CrossRef]
- Pasban-Aliabadi, H.; Esmaeili-Mahani, S.; Sheibani, V.; Abbasnejad, M.; Mehdizadeh, A.; Yaghoobi, M.M. Inhibition of 6-hydroxydopamine-induced PC12 cell apoptosis by olive (Olea europaea L.) leaf extract is performed by its main component oleuropein. Rejuvenation Res. 2013, 16, 134–142. [Google Scholar] [CrossRef]
- Peng, S.; Zhang, B.; Yao, J.; Duan, D.; Fang, J. Dual protection of hydroxytyrosol, an olive oil polyphenol, against oxidative damage in PC12 cells. Food Funct. 2015, 6, 2091–2100. [Google Scholar] [CrossRef]
- Sun, W.; Wang, X.; Hou, C.; Yang, L.; Li, H.; Guo, J.; Huo, C.; Wang, M.; Miao, Y.; Liu, J.; et al. Oleuropein improves mitochondrial function to attenuate oxidative stress by activating the Nrf2 pathway in the hypothalamic paraventricular nucleus of spontaneously hypertensive rats. Neuropharmacology 2017, 113, 556–566. [Google Scholar] [CrossRef]
- Peyrat-Maillard, M.N.; Cuvelier, M.E.; Berset, C. Antioxidant activity of phenolic compounds in 2,2′-azobis (2-amidinopropane) dihydrochloride (AAPH)-induced oxidation: Synergistic and antagonistic effects. JAOCS 2003, 80, 1007–1012. [Google Scholar] [CrossRef]
- Belkacemi, A.; Ramassamy, C. Innovative anthocyanin/anthocyanidin formulation protects SK-N-SH cells against the amyloid-beta peptide-induced toxicity: Relevance to Alzheimer’s disease. Cent. Nerv. Syst. Agents Med. Chem. 2015, 16, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Belkacemi, A.; Ramassamy, C. Anthocyanins protect SK-N-SH cells against acrolein-induced toxicity by preserving the cellular redox state. J. Alzheimers Dis. 2016, 50, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Caruso, D.; Galli, C.; Viappiani, S.; Galli, G.; Sala, A. Olive oils rich in natural catecholic phenols decrease isoprostane excretion in humans. Biochem. Biophys. Res. Commun. 2000, 278, 797–799. [Google Scholar] [CrossRef]
- Corona, G.; Tzounis, X.; Assunta DessÌ, M.; Deiana, M.; Debnam, E.S.; Visioli, F.; Spencer, J.P.E. The fate of olive oil polyphenols in the gastrointestinal tract: Implications of gastric and colonic microflora-dependent biotransformation. Free Radic. Res. 2006, 40, 647–658. [Google Scholar] [CrossRef]
- de Bock, M.; Thorstensen, E.B.; Derraik, J.G.B.; Henderson, H.V.; Hofman, P.L.; Cutfield, W.S. Human absorption and metabolism of oleuropein and hydroxytyrosol ingested as olive ( Olea europaea L.) leaf extract. Mol. Nutr. Food Res. 2013, 57, 2079–2085. [Google Scholar] [CrossRef]
- Fási, L.; Di Meo, F.; Kuo, C.-Y.; Stojkovic Buric, S.; Martins, A.; Kúsz, N.; Béni, Z.; Dékány, M.; Balogh, G.T.; Pesic, M.; et al. Antioxidant-inspired drug discovery: Antitumor metabolite is formed in situ from a hydroxycinnamic acid derivative upon free-radical scavenging. J. Med. Chem. 2019, 62, 1657–1668. [Google Scholar] [CrossRef]
- Hunyadi, A. The mechanism(s) of action of antioxidants: From scavenging reactive oxygen/nitrogen species to redox signaling and the generation of bioactive secondary metabolites. Med. Res. Rev. 2019, 39, 2505–2533. [Google Scholar] [CrossRef]
- Guyot, S.; Marnet, N.; Laraba, D.; Sanoner, P.; Drilleau, J.-F. Reversed-phase HPLC following thiolysis for quantitative estimation and characterization of the four main classes of phenolic compounds in different tissue zones of a french cider apple variety (Malus domestica var. Kermerrien). J. Agric. Food Chem. 1998, 46, 1698–1705. [Google Scholar] [CrossRef]
- Enache, T.A.; Amine, A.; Brett, C.M.A.; Oliveira-Brett, A.M. Virgin olive oil ortho-phenols—electroanalytical quantification. Talanta 2013, 105, 179–186. [Google Scholar] [CrossRef]
- Paradiso, V.M.; Di Mattia, C.; Giarnetti, M.; Chiarini, M.; Andrich, L.; Caponio, F. Antioxidant behavior of olive phenolics in oil-in-water emulsions. J. Agric. Food Chem. 2016, 64, 5877–5886. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C. Electrochemical approach to the mechanistic study of proton-coupled electron transfer. Chem. Rev. 2008, 108, 2145–2179. [Google Scholar] [CrossRef] [PubMed]
- Enache, T.A.; Oliveira-Brett, A.M. Phenol and para-substituted phenols electrochemical oxidation pathways. J. Electroanal. Chem. 2011, 655, 9–16. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M. Concerted proton−electron transfers: Electrochemical and related approaches. Acc. Chem. Res. 2010, 43, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Cheynier, V.; Rigaud, J.; Moutounet, M. Oxidation kinetics of trans-caffeoyltartrate and its glutathione derivatives in grape musts. Phytochemistry 1990, 29, 1751–1753. [Google Scholar] [CrossRef]
- Guyot, S.; Vercauteren, J.; Cheynier, V. Structural determination of colourless and yellow dimers resulting from (+)-catechin coupling catalysed by grape polyphenoloxidase. Phytochemistry 1996, 42, 1279–1288. [Google Scholar] [CrossRef]
- Quirantes-Piné, R.; Lozano-Sánchez, J.; Herrero, M.; Ibáñez, E.; Segura-Carretero, A.; Fernández-Gutiérrez, A. HPLC-ESI-QTOF-MS as a powerful analytical tool for characterising phenolic compounds in olive-leaf extracts: Characterisation of phenolic compounds from olive leaves. Phytochem. Anal. 2013, 24, 213–223. [Google Scholar] [CrossRef]
- Rodríguez, G.; Lama, A.; Trujillo, M.; Espartero, J.L.; Fernández-Bolaños, J. Isolation of a powerful antioxidant from Olea europaea fruit-mill waste: 3,4-Dihydroxyphenylglycol. Lwt Food Sci. Technol. 2009, 42, 483–490. [Google Scholar] [CrossRef]
- Longo, E.; Morozova, K.; Scampicchio, M. Effect of light irradiation on the antioxidant stability of oleuropein. Food Chem. 2017, 237, 91–97. [Google Scholar] [CrossRef]
- Antenucci, S.; Panzella, L.; Farina, H.; Ortenzi, M.A.; Caneva, E.; Martinotti, S.; Ranzato, E.; Burlando, B.; d’Ischia, M.; Napolitano, A.; et al. Powering tyrosol antioxidant capacity and osteogenic activity by biocatalytic polymerization. RSC Adv. 2016, 6, 2993–3002. [Google Scholar] [CrossRef] [Green Version]
- Chakroun, H.; Bouaziz, M.; Yangui, T.; Blibech, I.; Dhouib, A.; Sayadi, S. Enzymatic transformation of tyrosol by Trametes trogii laccases: Identification of the product and study of its biological activities. J. Mol. Catal. B-Enzym. 2013, 87, 11–17. [Google Scholar] [CrossRef]
- Šmejkalová, D.; Piccolo, A.; Spiteller, M. Oligomerization of humic phenolic monomers by oxidative coupling under biomimetic catalysis. Environ. Sci. Technol. 2006, 40, 6955–6962. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, H.; Bouaziz, M.; Dhouib, A.; Sayadi, S. Enzymatic oxidative transformation of phenols by Trametes trogii laccases. Environ. Technol. 2012, 33, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Hapiot, P.; Neudeck, A.; Pinson, J.; Fulcrand, H.; Neta, P.; Rolando, C. Oxidation of caffeic acid and related hydroxycinnamic acids. J. Electroanal. Chem. 1996, 405, 169–176. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, C.; Shen, L.; Shin, H.-C.; Lee, K.B.; Ji, B. Comparative analysis of oxidative mechanisms of phloroglucinol and dieckol by electrochemical, spectroscopic, cellular and computational methods. RSC Adv. 2018, 8, 1963–1972. [Google Scholar] [CrossRef] [Green Version]
- Di Gennaro, P.; Sabatini, V.; Fallarini, S.; Pagliarin, R.; Sello, G. Polyphenol polymerization by an alternative oxidative microbial enzyme and characterization of the biological activity of oligomers. Biomed. Res. Int. 2018, 2018, 3828627. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | RT (min) | λmax (nm) | m/z | Ole 2 mM | Mix 2 mM | Mix-seq 2 mM | MS2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref | Ox | Ref | Ox | (Tyr + p-Cou) Ox + Ole | Ox | |||||||||
| O1 | 3,4-Dihydroxyphenylglycol | 4.1 | 280 | 169 | traces | + | − | + | − | + | 151 (−18) | 123 (−46) | ||
| O2 | 11-Methyl-oleoside | 22.0 | <240 | 403 | traces | + | traces | + | + | + | 223 (−180) | 179 (−224) | 359 (−CO2) | 161 (−242) |
| O3 | unknown | 30.8 | 587 | + | + | + | + | + | + | 543 (−44) | 403 (−184) | |||
| O4 | unknown | 32.4 | 288 | 573 | + | traces | traces | + | traces | - | 529 (−44) | 403 (−170) | 222 (−350) | |
| O5 | 10- Hydroxy-oleuropein | 35.1 | 555 | + | + | + | + | + | + | 537 (−H2O) | 393 (162) | 403 (−152) | ||
| O6 | Oleuropein + [EtOH] | 35.8 | 244/307 | 585 | + | + | + | + | + | + | 523 (−62) | 541 (−44) | 361 (−224) | 199 (−386) |
| 403 (−182) | ||||||||||||||
| O7 | unknown | 37.0 | 255 | 615 | + | + | + | + | + | + | 423 (−191) | 455 (−160) | 273 (−344) | 551 (−64) |
| O8 | unknown | 37.8 | 266/336 | 617 | traces | traces | - | + | + | + | 423 (−194) | 455 (−162) | 273 (−344) | 585 (−32) |
| O9 | unknown | 38.0 | 266/337 | 431 | + | traces | + | + | + | + | 269 (−162) | |||
| O10 | unknown | 38.3 | 267/344 | 567 | traces | + | traces | + | - | + | 373 (−194) | 403 (−164) | 223 (−344) | |
| O11 | unknown | 38.3 | 461 | + | + | + | + | + | + | 299 (−162) | 446 (−15) | 307 (−154) | ||
| O12 | Oleuropein | 38.4 | 278 | 539 | + | + | + | + | + | + | 377 (−162) | 307 (−232) | 275 (−264) | |
| O13 | unknown | 38.6 | - | 553 | + | + | traces | + | + | + | 403 (−150) | 223 (−330) | 179 (−374) | |
| O14 | Oleuropein diglucoside | 38.6 | 309 | 701 | + | + | + | + | + | + | 377 (−324) | 307 (394) | 275 (−426) | |
| O15 | Ole dimer | 39.5 | 1077 | traces | + | traces | + | traces | - | 673 (−404) | 813 (−264) | 539 (−538) | ||
| O16 | Ole quinone dimer derivative | 39.8 | 263/420 | 1091 | − | + | − | + | traces | + | 687 (−404) | 403 (−688) | 525 (−56) | |
| O17 | Oleuropein quinone | 39.9 | 295 | 537 | traces | + | traces | traces | traces | + | − | |||
| O18a | Oxidized lucidumoside C | 40.3 | 259/434 | 581 | − | + | − | + | + | + | 535 (-46) | 403 (−178) | ||
| O19 | Oxidized Ole trimer | 40.3 | 1647 | − | + | − | traces | − | + | 685 (−962) | 1109 (−538) | |||
| O20 | Lucidumoside C | 40.6 | 255/350 | 583 | − | + | − | − | + | + | 537 (−46) | |||
| O18b | Oxidized lucidumoside C | 41.0 | <240/412 | 581 | − | + | + | traces | + | + | ||||
| O21 | Ole trimer derivative | 41.2 | 280 | 1619 | − | + | − | + | + | + | 1575 (−CO2) | 1557 (−CO2, −H2O) | 1019 | |
| O22 | Iso Ole trimer derivative | 41.6 | 280 | 1619 | − | + | − | + | + | + | 1575 | 1557 | 1019 | |
| O23 | Iso Ole dimer | 41.7 | 1077 | + | - | + | - | traces | − | 813 (−264) | 673 (−404) | 539 (−538) | ||
| O24 | Ole trimer derivative | 42.6 | 281 | 1629 | − | + | − | + | + | + | 1019 | 1045 | 1585 | 1091 |
| O25 | Ole trimer derivative | 43.6 | 285 | 1601 | traces | + | − | + | traces | + | 1197 | 793 | ||
| Compound Name | RT (min) | λmax (nm) | m/z | Tyr 2mM | Mix 2mM | Mix-seq 2mM | MS2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref | Ox | Ref | Ox | (Tyr + p-Cou) Ox + Ole | Ox | ||||||||
| T1 | unknown | 10.4 | − | 1794 | − | + | − | − | − | − | |||
| T2 | unknown | 12.9 | 280 | 1820 | − | + | − | − | − | − | 1761 (−59) | ||
| T3 | Tyrosol | 14.6 | 275 | 502 | + | + | + | + | + | + | 365 (−Tyr) | 228 (−Tyr dim) | |
| 402 | + | + | + | + | + | + | 265 (−Tyr) | ||||||
| 273 | + | + | + | + | + | + | |||||||
| T4 | unknown | 17.8 | 285 | 1499 | − | + | − | − | − | − | 1449 (−50) | 1414 (−85) | 1240 (−259) |
| T5 | Tyrosol dimer | 34.0 | 280 | 273 | − | + | − | traces | + | + | 243 (−CH2O) | 255 (−H2O) | |
| T6 | Tyrosol trimer | 38.1 | nd | 409 | − | + | − | traces | + | + | 391 (−H2O) | 341 (−68) | 273 (−Tyr ox) |
| T7 | Tyrosol tetramer | 39.4 | 275–280 | 545 | − | + | − | − | traces | − | 409 (−Tyr ox) | ||
| T8 | Tyrosol trimer | 40.0 | 409 | + | − | − | − | − | 271 (−neutral Tyr) | 341 (−68) | 136 (−Tyr dim) | ||
| Compound Name | RT (min) | λmax (nm) | m/z | p-Cou 2mM | Mix 2mM | Mix-seq 2mM | MS2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref | Ox | Ref | Ox | (Tyr + p-Cou) Ox + Ole | Ox | |||||||
| C1 | p-coumaric acid | 28.9 | 308 | 163 | + | + | + | + | + | + | 119 (−CO2) | |
| C2 | iso-p-coumaric acid (m- or o-) | 31.8 | 295 | 163 | + | + | + | traces | + | + | 126 (−37) | 119 (−CO2) |
| C3 | unknown | 35.1 | − | 497 | + | + | − | − | − | − | ||
| C4 | p-coumaric acid dimer | 38.8 | 305 | 325 | − | + | − | − | traces | traces | 281 (−CO2) | 237 (−2CO2) |
| C5 | p-coumaric acid dimer | 40.0 | 305 | 325 | − | + | − | − | traces | traces | 281 (−CO2) | |
| C6 | unknown | 48.9 | − | 271 | + | + | − | − | + | − | 213 (−58) | |
| C7 | decarboxylated p-coumaric acid dimer | 41.8 | 300/316 | 281 | − | + | − | + | + | + | 237 (−CO2) | |
| C8 | decarboxylated p-coumaric acid dimer | 42.3 | 292/330 | 281 | − | + | + | + | + | + | ||
| Compound Name | RT (min) | λmax (nm) | m/z | Mix 2mM | Mix-seq 2mM | MS2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref | Ox | (Tyr + p-Cou) Ox + Ole | Ox | |||||||||
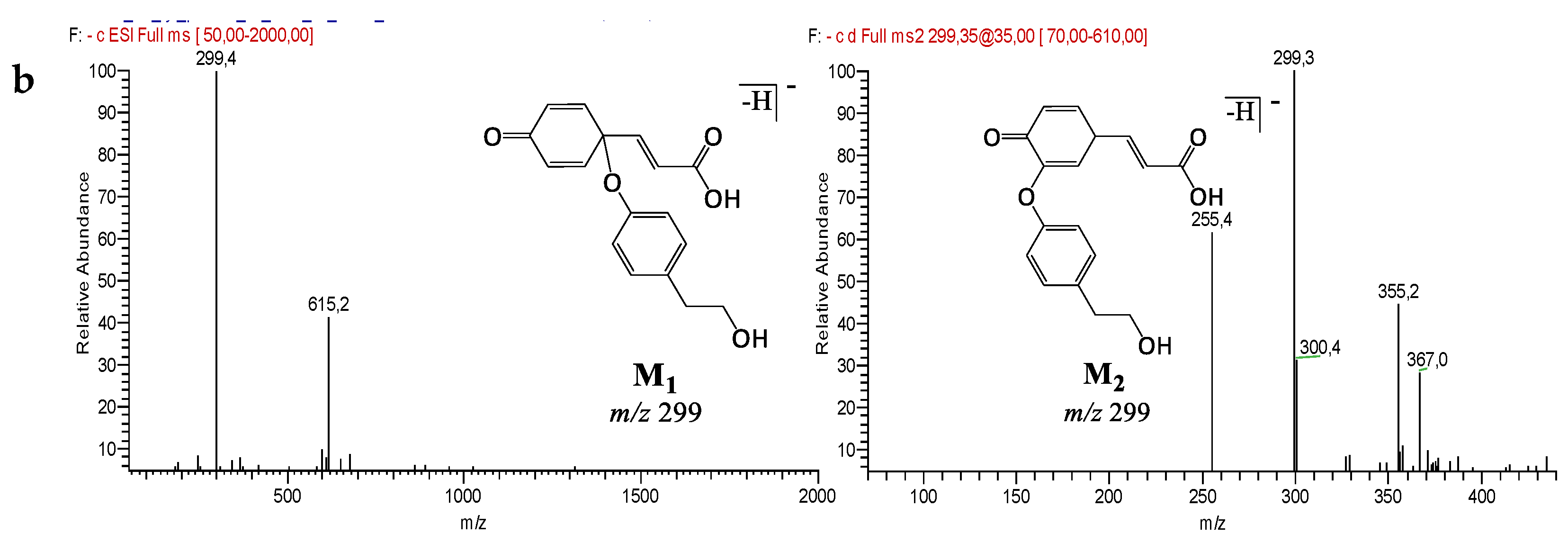
| M1 | Oxidized (Tyr - p-Cou) dimer | 34.9 | 299 | − | + | + | + | 255 (−CO2) | 135 (−164) | |||
| M2 | Oxidized (Tyr - p-Cou) dimer | 37.4 | 299 | − | + | + | + | 255 (−CO2) | 135 (−164) | |||
| M3 | Oxidized (Tyr - Ole) dimer | 37.9 | 675 | − | + | − | − | |||||
| M4 | unknown | 43.3 | 245/284 | 1195 | + | + | + | + | 655 (−Ole) | 791 (−11-methyl--oleoside) | 1033 (−glu) | 963 |
| 1163 (−O2) | 657 (−Ole Ox) | 403 | 539 | |||||||||
| M5 | unknown | 43.4 | 245/284 | 1195 | + | + | + | + | ||||
| M6 | unknown | 43.8 | 245/284 | 1195 | + | + | + | + | ||||
| M7 | unknown | 48.3 | 305 | 325 | − | + | + | + | 281 (−CO2) | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambert de Malezieu, M.; Ferron, S.; Sauvager, A.; Courtel, P.; Ramassamy, C.; Tomasi, S.; Abasq, M.-L. UV-Vis Spectroelectrochemistry of Oleuropein, Tyrosol, and p-Coumaric Acid Individually and in an Equimolar Combination. Differences in LC-ESI-MS2 Profiles of Oxidation Products and Their Neuroprotective Properties. Biomolecules 2019, 9, 802. https://doi.org/10.3390/biom9120802
Lambert de Malezieu M, Ferron S, Sauvager A, Courtel P, Ramassamy C, Tomasi S, Abasq M-L. UV-Vis Spectroelectrochemistry of Oleuropein, Tyrosol, and p-Coumaric Acid Individually and in an Equimolar Combination. Differences in LC-ESI-MS2 Profiles of Oxidation Products and Their Neuroprotective Properties. Biomolecules. 2019; 9(12):802. https://doi.org/10.3390/biom9120802
Chicago/Turabian StyleLambert de Malezieu, Morgane, Solenn Ferron, Aurélie Sauvager, Patricia Courtel, Charles Ramassamy, Sophie Tomasi, and Marie-Laurence Abasq. 2019. "UV-Vis Spectroelectrochemistry of Oleuropein, Tyrosol, and p-Coumaric Acid Individually and in an Equimolar Combination. Differences in LC-ESI-MS2 Profiles of Oxidation Products and Their Neuroprotective Properties" Biomolecules 9, no. 12: 802. https://doi.org/10.3390/biom9120802
APA StyleLambert de Malezieu, M., Ferron, S., Sauvager, A., Courtel, P., Ramassamy, C., Tomasi, S., & Abasq, M.-L. (2019). UV-Vis Spectroelectrochemistry of Oleuropein, Tyrosol, and p-Coumaric Acid Individually and in an Equimolar Combination. Differences in LC-ESI-MS2 Profiles of Oxidation Products and Their Neuroprotective Properties. Biomolecules, 9(12), 802. https://doi.org/10.3390/biom9120802