Structural and Dynamical Order of a Disordered Protein: Molecular Insights into Conformational Switching of PAGE4 at the Systems Level

 ,
,  , , ,
, , ,  ,
,  , add
Show full author list
, add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. AAWSEM: A Coarse-Grained Modeling Framework to Simulate Intrinsically Disordered Proteins
3. AAWSEM-Based Simulations of PAGE4 Reveal Two Different Kinds of Order Underneath Its Disordered Cloak
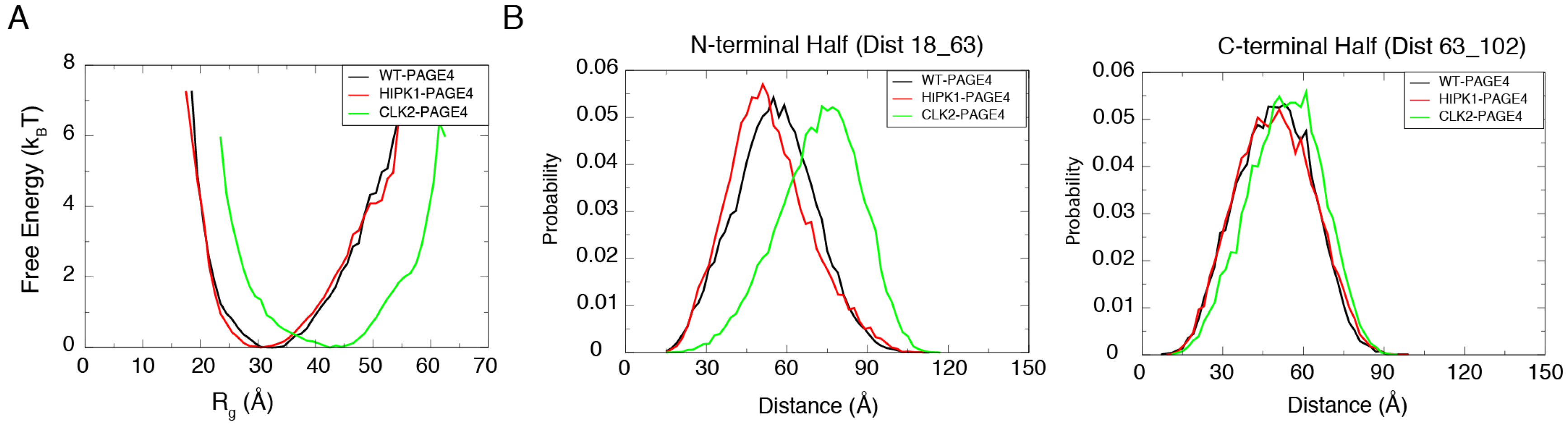
3.1. Simulation Reproduces the Expansion of PAGE4 upon Hyper-Phosphorylation
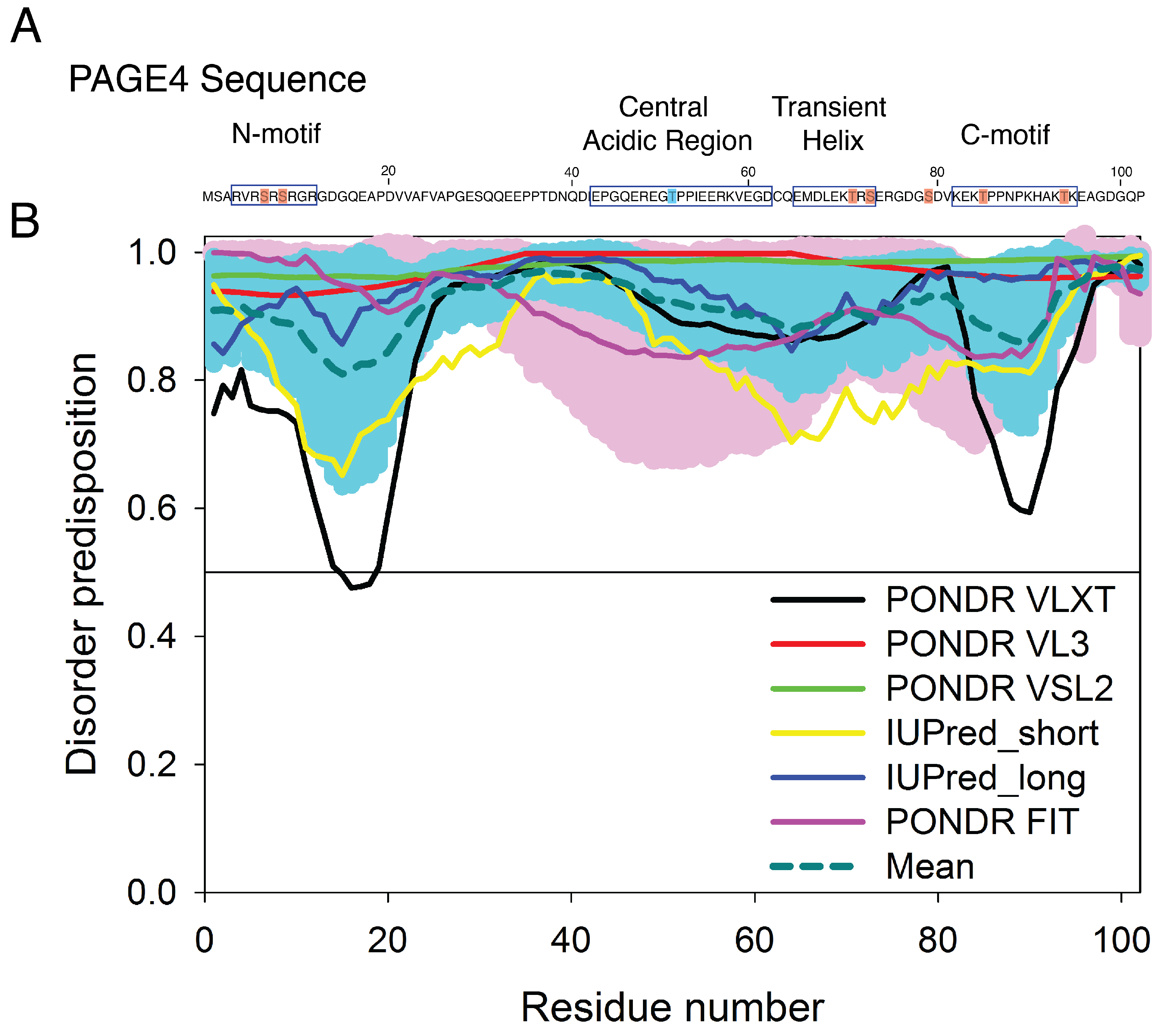
3.2. Simulations Reveal Structural Order in PAGE4
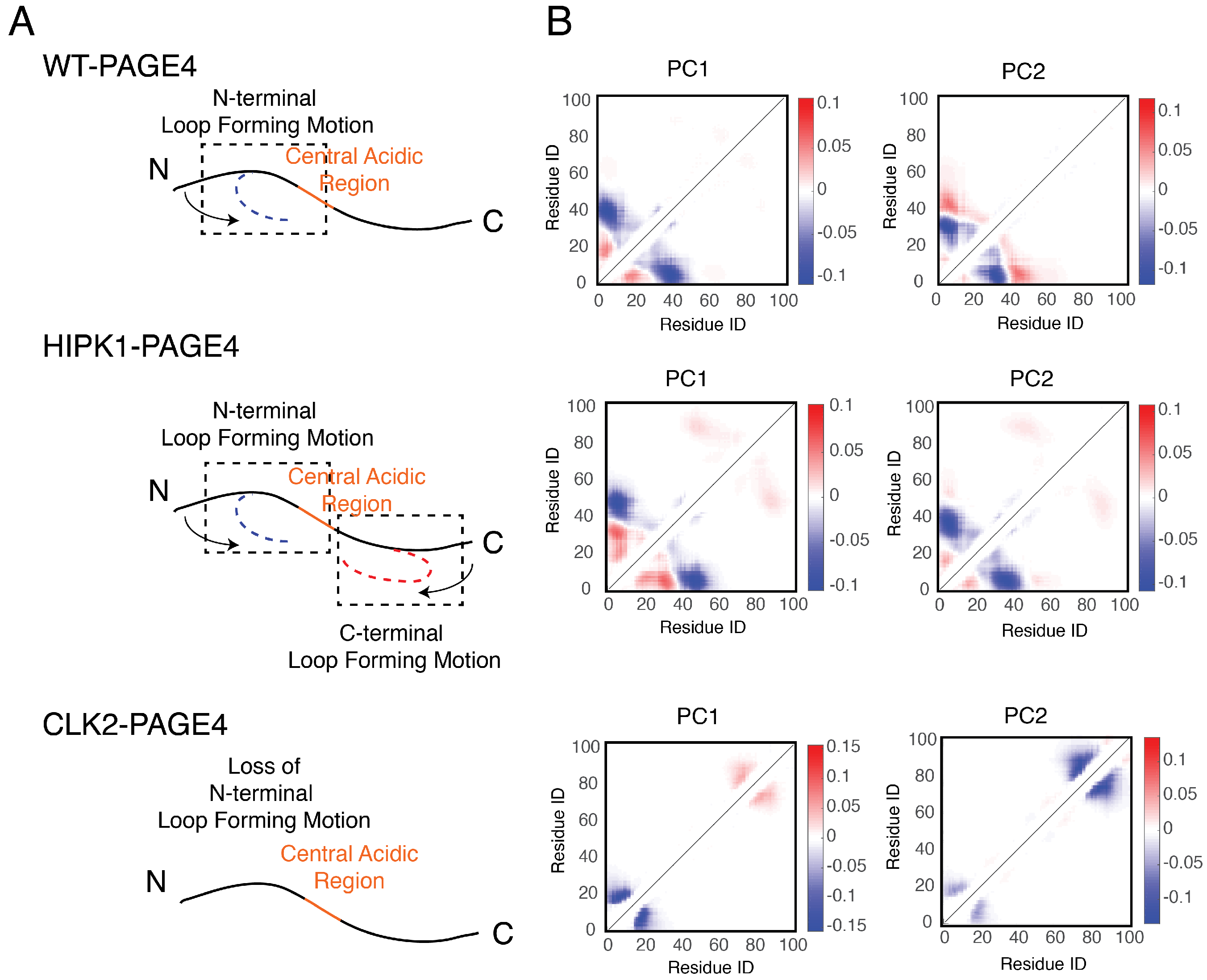
3.3. Collective Motions of PAGE4 Are Associated with Its Functions
4. From Structure to Function: PAGE4 Conformational Switching May Underlie Therapy Resistance in Prostate Cancer (PCa)
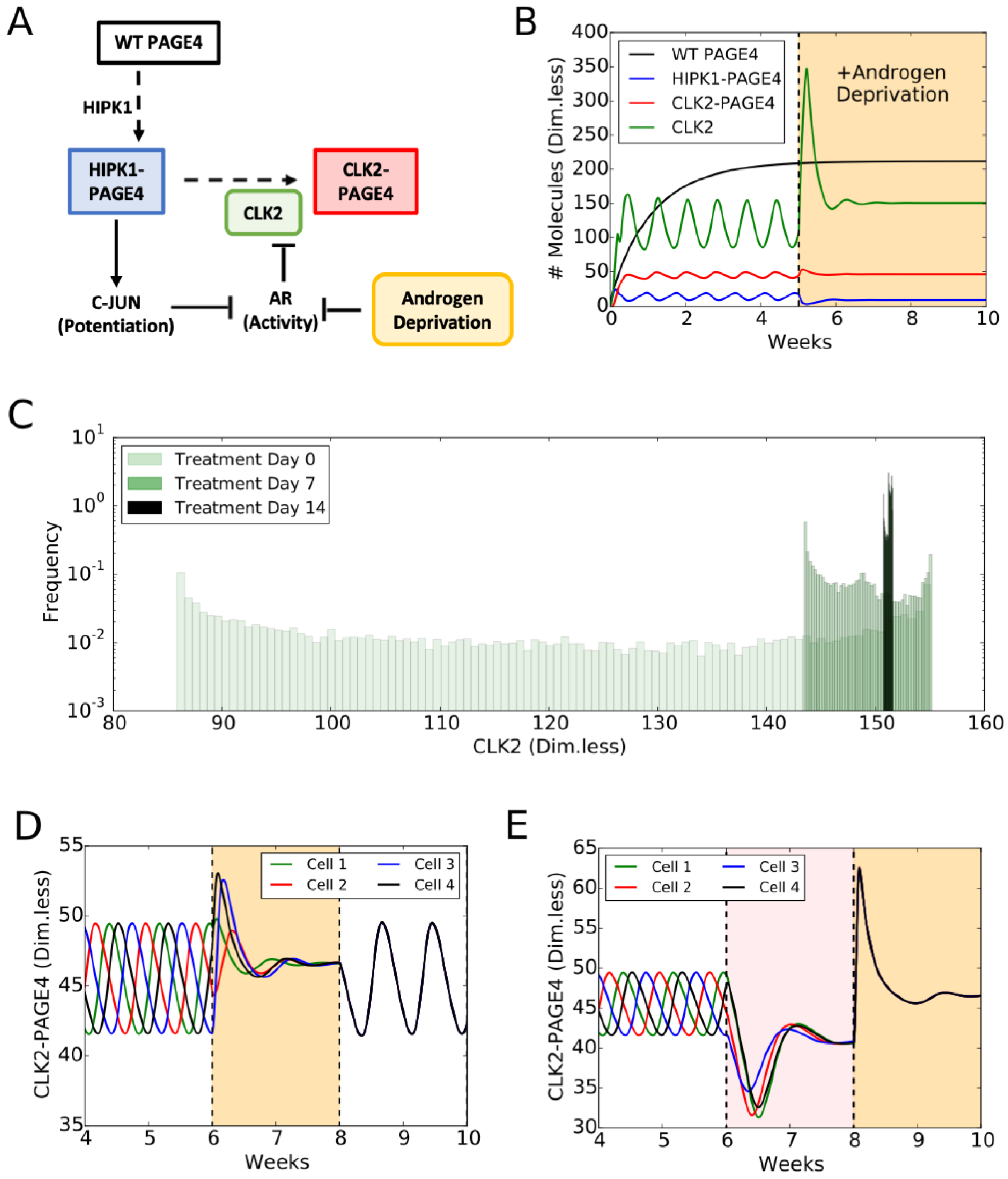
4.1. PAGE4 Conformational Switching Can Give Rise to Oscillations between an Androgen-Dependent Cell Phenotype and an Androgen-Independent Cell Phenotype
4.2. Androgen Deprivation Restricts the Phenotypic Heterogeneity by Damping PAGE4 Oscillations
5. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Ann. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Kinoshita, K.; Nakamura, H. Hub Promiscuity in Protein-Protein Interaction Networks. Int. J. Mol. Sci. 2010, 11, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006, 2, e100. [Google Scholar] [CrossRef] [PubMed]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R. Physical Biology of the Cell, 2nd ed.; Garland Science: London, UK; New York, NY, USA, 2013. [Google Scholar]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Biol. 2018, 430, 2309–2320. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 2018, 18, 1700193. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Protein intrinsic disorder-based liquid–liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv. Colloid Interface Sci. 2017, 239, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 2015, 589, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Whitford, P.C.; Sanbonmatsu, K.Y.; Onuchic, J.N. Biomolecular dynamics: order-disorder transitions and energy landscapes. Rep. Progr. Phys. Phys. Soc. 2012, 75, 076601. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Solomon, T.L.; He, Y.; Chen, Y.; Bryan, P.N.; Orban, J. Structural metamorphism and polymorphism in proteins on the brink of thermodynamic stability: Continuum of Order/Disorder Transitions. Protein Sci. 2018, 27, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Bryan, P.N.; Orban, J. Proteins that switch folds. Curr. Opin. Struct. Biol. 2010, 20, 482–488. [Google Scholar] [CrossRef]
- Bryan, P.N.; Orban, J. Implications of protein fold switching. Curr. Opin. Struct. Biol. 2013, 23, 314–316. [Google Scholar] [CrossRef]
- Goodchild, S.C.; Curmi, P.M.G.; Brown, L.J. Structural gymnastics of multifunctional metamorphic proteins. Biophys. Rev. 2011, 3, 143. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Oldfield, C.J.; Meng, J.; Romero, P.; Uversky, V.N.; Dunker, A.K. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468–13477. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Miskei, M.; Gregus, A.; Sharma, R.; Duro, N.; Zsolyomi, F.; Fuxreiter, M. Fuzziness enables context dependence of protein interactions. FEBS Lett. 2017, 591, 2682–2695. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, K.; Lebrun, P.; Tompa, P. To be disordered or not to be disordered: is that still a question for proteins in the cell? Cell. Mol. Life Sci. 2017, 74, 3185–3204. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, J.B.; Nunez-Castilla, J.; Siltberg-Liberles, J. Evolution of intrinsic disorder in eukaryotic proteins. Cell. Mol. Life Sci. 2017, 74, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Dunker, A.; Weninger, K.; Orban, J. Prostate-associated gene 4 (PAGE4), an intrinsically disordered cancer/testis antigen, is a novel therapeutic target for prostate cancer. Asian J. Androl. 2016, 18, 695. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Jolly, M.K.; Dorff, T.; Lau, C.; Weninger, K.; Orban, J.; Kulkarni, P. Prostate-Associated Gene 4 (PAGE4): Leveraging the Conformational Dynamics of a Dancing Protein Cloud as a Therapeutic Target. J. Clin. Med. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, K.; Qiu, R.; Mooney, S.M.; Rao, S.; Shiraishi, T.; Sacho, E.; Huang, H.; Shapiro, E.; Weninger, K.R.; Kulkarni, P. The Stress-response protein prostate-associated gene 4, interacts with c-Jun and potentiates its transactivation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Mooney, S.M.; Qiu, R.; Kim, J.J.; Sacho, E.J.; Rajagopalan, K.; Johng, D.; Shiraishi, T.; Kulkarni, P.; Weninger, K.R. Cancer/Testis Antigen PAGE4, a Regulator of c-Jun Transactivation, Is Phosphorylated by Homeodomain-Interacting Protein Kinase 1, a Component of the Stress-Response Pathway. Biochemistry 2014, 53, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Sadar, M.D.; Bruchovsky, N.; Saatcioglu, F.; Rennie, P.S.; Sato, S.; Lange, P.H.; Gleave, M.E. Androgenic induction of prostate-specific antigen gene is repressed by protein-protein interaction between the androgen receptor and AP-1/c-Jun in the human prostate cancer cell line LNCaP. J. Biol. Chem. 1997, 272, 17485–17494. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Jolly, M.K.; Jia, D.; Mooney, S.M.; Bhargava, A.; Kagohara, L.T.; Chen, Y.; Hao, P.; He, Y.; Veltri, R.W.; et al. Phosphorylation-induced conformational dynamics in an intrinsically disordered protein and potential role in phenotypic heterogeneity. Proc. Nat. Acad. Sci. USA 2017, 114, E2644–E2653. [Google Scholar] [CrossRef]
- Lin, X.; Roy, S.; Jolly, M.K.; Bocci, F.; Schafer, N.P.; Tsai, M.Y.; Chen, Y.; He, Y.; Grishaev, A.; Weninger, K.; et al. PAGE4 and Conformational Switching: Insights from Molecular Dynamics Simulations and Implications for Prostate Cancer. J. Mol. Biol. 2018, 430, 2422–2438. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Xu, D.; Yang, J.; Walker, S.; Zhang, Y. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.K.; Obradovic, Z. Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Informat. Workshop Genome Informat. 1999, 10, 30–40. [Google Scholar]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 2005, 61, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformat. 2006, 7, 208. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Walsh, I.; Giollo, M.; Di Domenico, T.; Ferrari, C.; Zimmermann, O.; Tosatto, S.C.E. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015, 31, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Kurgan, L. Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J. Biomol. Struct. Dyn. 2014, 32, 448–464. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, Y.; Mooney, S.M.; Rajagopalan, K.; Bhargava, A.; Sacho, E.; Weninger, K.; Bryan, P.N.; Kulkarni, P.; Orban, J. Phosphorylation-induced Conformational Ensemble Switching in an Intrinsically Disordered Cancer/Testis Antigen. J. Biol. Chem. 2015, 290, 25090–25102. [Google Scholar] [CrossRef] [PubMed]
- Bernadó, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. BioSyst. 2012, 8, 151–167. [Google Scholar] [CrossRef]
- Schneider, R.; Huang, J.R.; Yao, M.; Communie, G.; Ozenne, V.; Mollica, L.; Salmon, L.; Ringkjøbing Jensen, M.; Blackledge, M. Towards a robust description of intrinsic protein disorder using nuclear magnetic resonance spectroscopy. Mol. BioSyst. 2012, 8, 58–68. [Google Scholar] [CrossRef]
- Schuler, B.; Soranno, A.; Hofmann, H.; Nettels, D. Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys. 2016, 45, 207–231. [Google Scholar] [CrossRef]
- Jemth, P.; Karlsson, E.; Vögeli, B.; Guzovsky, B.; Andersson, E.; Hultqvist, G.; Dogan, J.; Güntert, P.; Riek, R.; Chi, C.N. Structure and dynamics conspire in the evolution of affinity between intrinsically disordered proteins. Sci. Adv. 2018, 4, eaau4130. [Google Scholar] [CrossRef]
- Zheng, W.; Borgia, A.; Buholzer, K.; Grishaev, A.; Schuler, B.; Best, R.B. Probing the Action of Chemical Denaturant on an Intrinsically Disordered Protein by Simulation and Experiment. J. Am. Chem. Soc. 2016, 138, 11702–11713. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.; Kim, Y.C.; Best, R.B.; Mittal, J. Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol. 2018, 14, e1005941. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.; Best, R.B.; Kim, Y.C.; Mittal, J. Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 9929–9934. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wolynes, P.G.; Papoian, G.A. AWSEM-IDP: A Coarse-Grained Force Field for Intrinsically Disordered Proteins. J. Phys. Chem. B 2018, 122, 11115–11125. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Zong, C.; Hamelberg, D.; McCammon, J.A.; Wolynes, P.G. The folding energy landscape and phosphorylation: modeling the conformational switch of the NFAT regulatory domain. FASEB J. 2005, 19, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Lätzer, J.; Shen, T.; Wolynes, P.G. Conformational Switching upon Phosphorylation: A Predictive Framework Based on Energy Landscape Principles. Biochemistry 2008, 47, 2110–2122. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Latham, A.P.; Zhang, B. Improving Coarse-Grained Protein Force Fields with Small-Angle X-ray Scattering Data. J. Phys. Chem. B 2019, 123, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Bryngelson, J.D.; Wolynes, P.G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 7524–7528. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.A.; Luthey-Schulten, Z.A.; Wolynes, P.G. Optimal protein-folding codes from spin-glass theory. Proc. Natl. Acad. Sci. USA 1992, 89, 4918–4922. [Google Scholar] [CrossRef]
- Davtyan, A.; Schafer, N.P.; Zheng, W.; Clementi, C.; Wolynes, P.G.; Papoian, G.A. AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinformatically Based Local Structure Biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [Google Scholar] [CrossRef]
- Zheng, W.; Schafer, N.P.; Davtyan, A.; Papoian, G.A.; Wolynes, P.G. Predictive energy landscapes for protein-protein association. Proc. Natl. Acad. Sci. USA 2012, 109, 19244–19249. [Google Scholar] [CrossRef]
- Kim, B.L.; Schafer, N.P.; Wolynes, P.G. Predictive energy landscapes for folding -helical transmembrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111, 11031–11036. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lin, X.; Zheng, W.; Onuchic, J.N.; Wolynes, P.G. Protein Folding and Structure Prediction from the Ground Up: The Atomistic Associative Memory, Water Mediated, Structure and Energy Model. J. Phys. Chem. B 2016, 120, 8557–8565. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lin, X.; Lu, W.; Onuchic, J.N.; Wolynes, P.G. Protein Folding and Structure Prediction from the Ground Up II: AAWSEM for α/β Proteins. J. Phys. Chem. B 2017, 121, 3473–3482. [Google Scholar] [CrossRef] [PubMed]
- Sirovetz, B.J.; Schafer, N.P.; Wolynes, P.G. Protein structure prediction: making AWSEM AWSEM-ER by adding evolutionary restraints. Proteins Struct. Funct. Bioinformat. 2017, 85, 2127–2142. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lin, X.; Lu, W.; Schafer, N.P.; Onuchic, J.N.; Wolynes, P.G. Template-Guided Protein Structure Prediction and Refinement Using Optimized Folding Landscape Force Fields. J. Chem. Theory Comput. 2018, 14, 6102–6116. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tsai, M.Y.; Chen, M.; Wolynes, P.G. Exploring the aggregation free energy landscape of the amyloid-β protein (1–40). Proc. Natl. Acad. Sci. USA 2016, 113, 11835–11840. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tsai, M.; Zheng, W.; Wolynes, P.G. The Aggregation Free Energy Landscapes of Polyglutamine Repeats. J. Am. Chem. Soc. 2016, 138, 15197–15203. [Google Scholar] [CrossRef]
- Potoyan, D.A.; Zheng, W.; Komives, E.A.; Wolynes, P.G. Molecular stripping in the NF-κB/IκB/DNA genetic regulatory network. Proc. Natl. Acad. Sci. USA 2016, 113, 110–115. [Google Scholar] [CrossRef]
- Potoyan, D.A.; Bueno, C.; Zheng, W.; Komives, E.A.; Wolynes, P.G. Resolving the NFκB Heterodimer Binding Paradox: Strain and Frustration Guide the Binding of Dimeric Transcription Factors. J. Am. Chem. Soc. 2017, 139, 18558–18566. [Google Scholar] [CrossRef]
- Tsai, M.Y.; Zhang, B.; Zheng, W.; Wolynes, P.G. Molecular Mechanism of Facilitated Dissociation of Fis Protein from DNA. J. Am. Chem. Soc. 2016, 138, 13497–13500. [Google Scholar] [CrossRef]
- Koretke, K.K.; Luthey-Schulten, Z.; Wolynes, P.G. Self-consistently optimized energy functions for protein structure prediction by molecular dynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 2932–2937. [Google Scholar] [CrossRef] [PubMed]
- Schafer, N.P.; Kim, B.L.; Zheng, W.; Wolynes, P.G. Learning To Fold Proteins Using Energy Landscape Theory. Israel J. Chem. 2014, 54, 1311–1337. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, M.S.; Wolynes, P.G. Toward protein tertiary structure recognition by means of associative memory hamiltonians. Science 1989, 246, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.Y.; Zheng, W.; Balamurugan, D.; Schafer, N.P.; Kim, B.L.; Cheung, M.S.; Wolynes, P.G. Electrostatics, structure prediction, and the energy landscapes for protein folding and binding: Electrostatic Energy Landscapes for Folding and Binding. Protein Sci. 2016, 25, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Genet. 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N.; Wolynes, P.G. Fly-Casting in Protein—DNA Binding: Frustration between Protein Folding and Electrostatics Facilitates Target Recognition. J. Am. Chem. Soc. 2007, 129, 738–739. [Google Scholar] [CrossRef]
- Trizac, E.; Levy, Y.; Wolynes, P.G. Capillarity theory for the fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 2746–2750. [Google Scholar] [CrossRef]
- Kratiras, Z.; Konstantinidis, C.; Skriapas, K. A review of continuous vs intermittent androgen deprivation therapy: Redefining the gold standard in the treatment of advanced prostate cancer. Myths, facts and new data on a perpetual dispute. Int. Braz J. Urol. 2014, 40, 3–15. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Antonarakis, E.S.; Wang, H.; Ajiboye, A.S.; Spitz, A.; Cao, H.; Luo, J.; Haffner, M.C.; Yegnasubramanian, S.; Carducci, M.A.; et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: Results from a pilot clinical study. Sci. Transl. Med. 2015, 7, 269ra2. [Google Scholar] [CrossRef] [PubMed]
- Goldbeter, A. Dissipative structures in biological systems: bistability, oscillations, spatial patterns and waves. Philos. Trans. R. Soc. A 2018, 376, 20170376. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jolly, M.K.; Kulkarni, P.; Levine, H. Phenotypic Plasticity and Cell Fate Decisions in Cancer: Insights from Dynamical Systems Theory. Cancers 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Buchan, N.C.; Goldenberg, S.L. Intermittent androgen suppression for prostate cancer. Nat. Rev. Urol. 2010, 7, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Whitford, P.C.; Miyashita, O.; Levy, Y.; Onuchic, J.N. Conformational transitions of adenylate kinase: Switching by cracking. J. Mol. Biol. 2007, 366, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Whitford, P.C.; Gosavi, S.; Onuchic, J.N. Conformational Transitions in Adenylate Kinase: ALLOSTERIC COMMUNICATION REDUCES MISLIGATION. J. Biol. Chem. 2008, 283, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Noel, J.K.; Schug, A.; Verma, A.; Wenzel, W.; Garcia, A.E.; Onuchic, J.N. Mirror Images as Naturally Competing Conformations in Protein Folding. J. Phys. Chem. B 2012, 116, 6880–6888. [Google Scholar] [CrossRef]
- Schug, A.; Whitford, P.C.; Levy, Y.; Onuchic, J.N. Mutations as trapdoors to two competing native conformations of the Rop-dimer. Proc. Natl. Acad. Sci. USA 2007, 104, 17674–17679. [Google Scholar] [CrossRef]
- Lin, X.; Eddy, N.R.; Noel, J.K.; Whitford, P.C.; Wang, Q.; Ma, J.; Onuchic, J.N. Order and disorder control the functional rearrangement of influenza hemagglutinin. Proc. Natl. Acad. Sci. USA 2014, 111, 12049–12054. [Google Scholar] [CrossRef]
- Miyashita, O.; Onuchic, J.N.; Wolynes, P.G. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 12570–12575. [Google Scholar] [CrossRef] [PubMed]
- Sasai, M.; Wolynes, P.G. Stochastic gene expression as a many-body problem. Proc. Natl. Acad. Sci. USA 2003, 100, 2374–2379. [Google Scholar] [CrossRef]
- Raj, A.; van Oudenaarden, A. Nature, Nurture, or Chance: Stochastic Gene Expression and Its Consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef]
- Jia, D.; Jolly, M.K.; Tripathi, S.C.; Den Hollander, P.; Huang, B.; Lu, M.; Celiktas, M.; Ramirez-Peña, E.; Ben-Jacob, E.; Onuchic, J.N.; et al. Distinguishing mechanisms underlying EMT tristability. Cancer Converg. 2017, 1. [Google Scholar] [CrossRef]
- Mahmoudabadi, G.; Rajagopalan, K.; Getzenberg, R.H.; Hannenhalli, S.; Rangarajan, G.; Kulkarni, P. Intrinsically disordered proteins and conformational noise: Implications in cancer. Cell Cycle 2013, 12, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic Plasticity, Bet-Hedging, and Androgen Independence in Prostate Cancer: Role of Non-Genetic Heterogeneity. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sharma, N.; Giri, R. Therapeutic Interventions of Cancers Using Intrinsically Disordered Proteins as Drug Targets: c-Myc as Model System. Cancer Informat. 2017, 16, 1176935117699408. [Google Scholar] [CrossRef]
- Mooney, S.M.; Jolly, M.K.; Levine, H.; Kulkarni, P. Phenotypic plasticity in prostate cancer: role of intrinsically disordered proteins. Asian J. Androl. 2016, 18, 704–710. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Kulkarni, P.; Bocci, F.; Schafer, N.P.; Roy, S.; Tsai, M.-Y.; He, Y.; Chen, Y.; Rajagopalan, K.; Mooney, S.M.; et al. Structural and Dynamical Order of a Disordered Protein: Molecular Insights into Conformational Switching of PAGE4 at the Systems Level. Biomolecules 2019, 9, 77. https://doi.org/10.3390/biom9020077
Lin X, Kulkarni P, Bocci F, Schafer NP, Roy S, Tsai M-Y, He Y, Chen Y, Rajagopalan K, Mooney SM, et al. Structural and Dynamical Order of a Disordered Protein: Molecular Insights into Conformational Switching of PAGE4 at the Systems Level. Biomolecules. 2019; 9(2):77. https://doi.org/10.3390/biom9020077
Chicago/Turabian StyleLin, Xingcheng, Prakash Kulkarni, Federico Bocci, Nicholas P. Schafer, Susmita Roy, Min-Yeh Tsai, Yanan He, Yihong Chen, Krithika Rajagopalan, Steven M. Mooney, and et al. 2019. "Structural and Dynamical Order of a Disordered Protein: Molecular Insights into Conformational Switching of PAGE4 at the Systems Level" Biomolecules 9, no. 2: 77. https://doi.org/10.3390/biom9020077
APA StyleLin, X., Kulkarni, P., Bocci, F., Schafer, N. P., Roy, S., Tsai, M.-Y., He, Y., Chen, Y., Rajagopalan, K., Mooney, S. M., Zeng, Y., Weninger, K., Grishaev, A., Onuchic, J. N., Levine, H., Wolynes, P. G., Salgia, R., Rangarajan, G., Uversky, V., ... Jolly, M. K. (2019). Structural and Dynamical Order of a Disordered Protein: Molecular Insights into Conformational Switching of PAGE4 at the Systems Level. Biomolecules, 9(2), 77. https://doi.org/10.3390/biom9020077