Extreme Fuzziness: Direct Interactions between Two IDPs


Abstract
:1. Introduction
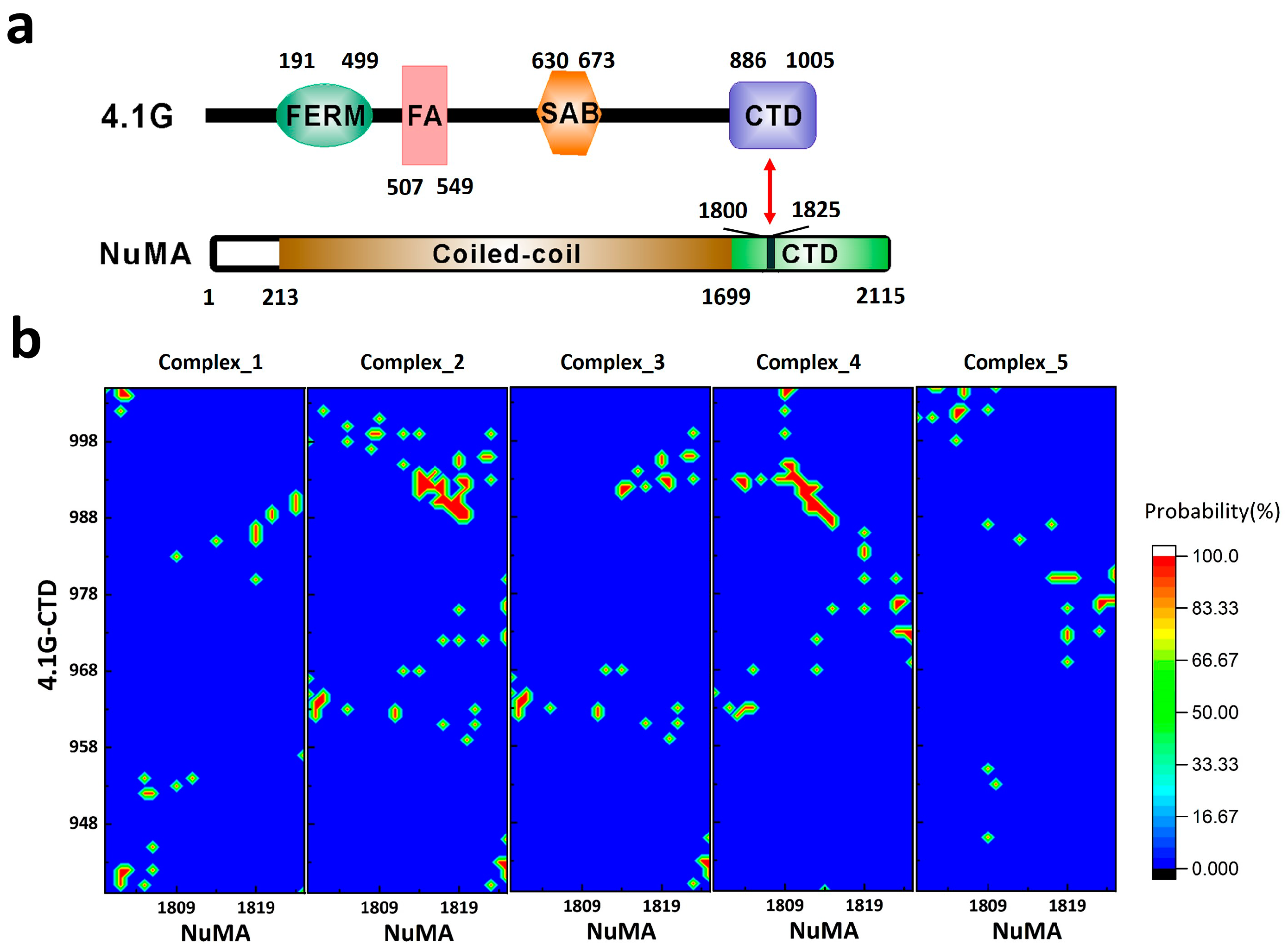
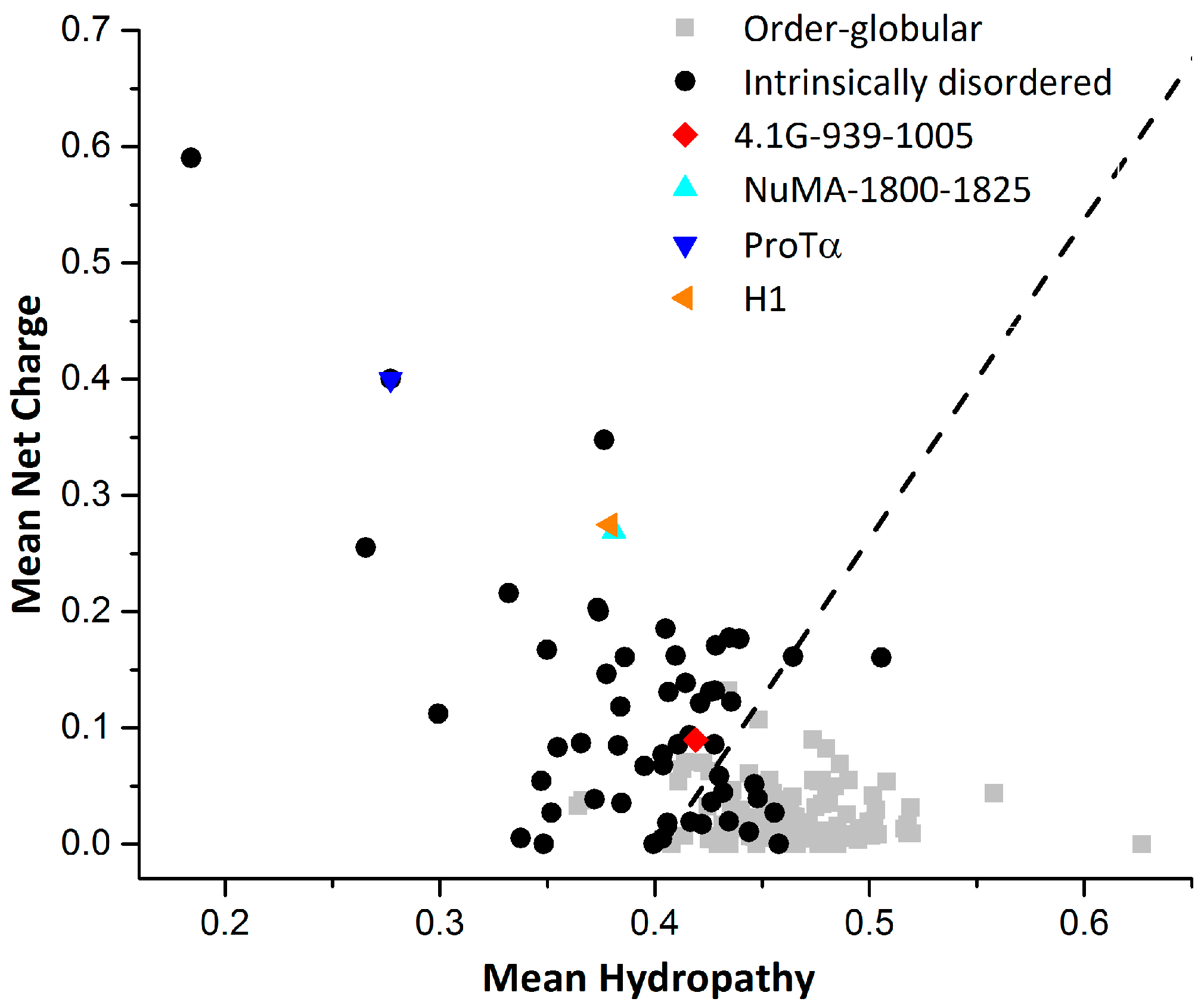
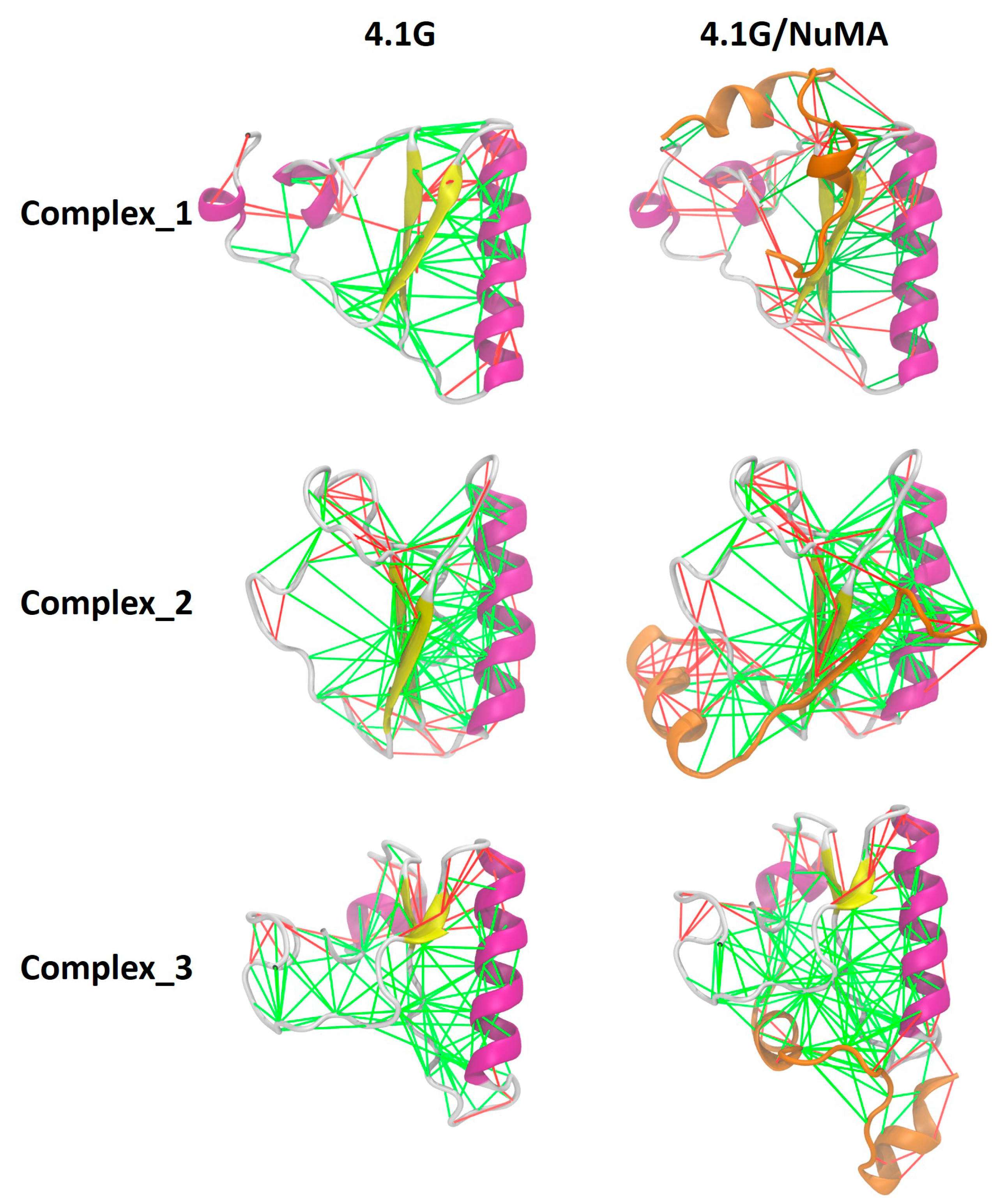
2. Interaction between 4.1G-CTD and NuMA
3. Interaction between ProTα and H1
4. How Unique are Extremely Fuzzy Complexes?
5. Conclusions
Funding
Conflicts of Interest
References
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Linderstrøm-Lang, K.U.; Schellman, J.A. Protein Structure and Enzyme Activity. Enzyme 1959, 1, 443–510. [Google Scholar]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, M.; Dobson, C.M. Structural biology. Dynamic visions of enzymatic reactions. Science 2006, 313, 1586–1587. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Minezaki, Y.; Homma, K.; Nishikawa, K. Genome-wide survey of transcription factors in prokaryotes reveals many bacteria-specific families not found in archaea. DNA Res. 2005, 12, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg, C.; Hilvert, D. Protein conformational disorder and enzyme catalysis. In Dynamics in Enzyme Catalysis; Springer: Berlin/Heidelberg, Germany, 2013; Volume 337, pp. 41–67. [Google Scholar]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M. Fuzziness in Protein Interactions-A Historical Perspective. J. Mol. Biol. 2018, 430, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.A.; Ferkey, D.M.; Mao, F.; Kimelman, D.; Xu, W. Tcf4 can specifically recognize beta-catenin using alternative conformations. Nat. Struct. Biol. 2001, 8, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Delaforge, E.; Kragelj, J.; Tengo, L.; Palencia, A.; Milles, S.; Bouvignies, G.; Salvi, N.; Blackledge, M.; Jensen, M.R. Deciphering the Dynamic Interaction Profile of an Intrinsically Disordered Protein by NMR Exchange Spectroscopy. J. Am. Chem. Soc. 2018, 140, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.P.; Remenyi, A.; Good, M.C.; Bashor, C.J.; Falick, A.M.; Lim, W.A. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 2006, 311, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, I.; Perez-Alvarado, G.C.; Parker, D.; Dyson, H.J.; Montminy, M.R.; Wright, P.E. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell 1997, 91, 741–752. [Google Scholar] [CrossRef]
- Mittag, T.; Orlicky, S.; Choy, W.Y.; Tang, X.; Lin, H.; Sicheri, F.; Kay, L.E.; Tyers, M.; Forman-Kay, J.D. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 17772–17777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Arbesu, M.; Iruela, G.; Fuentes, H.; Teixeira, J.M.C.; Pons, M. Intramolecular Fuzzy Interactions Involving Intrinsically Disordered Domains. Front. Mol. Biosci. 2018, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.G.; Teilum, K.; Kragelund, B.B. Behaviour of intrinsically disordered proteins in protein-protein complexes with an emphasis on fuzziness. Cell. Mol. Life Sci. 2017, 74, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.Y.J.; Birol, M.; Rhoades, E. IDPs in macromolecular complexes: The roles of multivalent interactions in diverse assemblies. Curr. Opin. Struct. Biol. 2018, 49, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Hough, L.E.; Dutta, K.; Sparks, S.; Temel, D.B.; Kamal, A.; Tetenbaum-Novatt, J.; Rout, M.P.; Cowburn, D. The molecular mechanism of nuclear transport revealed by atomic-scale measurements. eLife 2015, 4, e10027. [Google Scholar] [CrossRef] [PubMed]
- Milles, S.; Mercadante, D.; Aramburu, I.V.; Jensen, M.R.; Banterle, N.; Koehler, C.; Tyagi, S.; Clarke, J.; Shammas, S.L.; Blackledge, M.; et al. Plasticity of an ultrafast interaction between nucleoporins and nuclear transport receptors. Cell 2015, 163, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Muenzner, J.; Traub, L.M.; Kelly, B.T.; Graham, S.C. Cellular and viral peptides bind multiple sites on the N-terminal domain of clathrin. Traffic 2017, 18, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Cano, K.E.; Wang, L.; Ilangovan, U.; Hinck, A.P.; Sousa, R.; Lafer, E.M. Nuclear Magnetic Resonance Structural Mapping Reveals Promiscuous Interactions between Clathrin-Box Motif Sequences and the N-Terminal Domain of the Clathrin Heavy Chain. Biochemistry 2015, 54, 2571–2580. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Ilangovan, U.; Schirf, V.; Demeler, B.; Sousa, R.; Hinck, A.P.; Lafer, E.M. Dynamic interactions between clathrin and locally structured elements in a disordered protein mediate clathrin lattice assembly. J. Mol. Biol. 2010, 404, 274–290. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.; Aivazian, D.; Stern, L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry 2004, 43, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B.; Zhuravleva, A.V.; Orekhov, V.Y. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie 2007, 89, 419–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nourse, A.; Mittag, T. The cytoplasmic domain of the T-cell receptor zeta subunit does not form disordered dimers. J. Mol. Biol. 2014, 426, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Song, J.; Chan, H.S. Theoretical perspectives on nonnative interactions and intrinsic disorder in protein folding and binding. Curr. Opin. Struct. Biol. 2015, 30, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, D.; Liu, J.; Feng, Y.; Weng, J.; Li, Y.; Gao, X.; Liu, J.; Wang, W. The Dynamic Multisite Interactions between Two Intrinsically Disordered Proteins. Angew. Chem. Int. Ed. Engl. 2017, 56, 7515–7519. [Google Scholar] [CrossRef] [PubMed]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.J.; Lu, H.C.; Bennett, P.M. The Protein 4.1 family: hub proteins in animals for organizing membrane proteins. Biochim. Biophys. Acta 2014, 1838, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Kiyomitsu, T.; Cheeseman, I.M. Cortical dynein and asymmetric membrane elongation coordinately position the spindle in anaphase. Cell 2013, 154, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Seldin, L.; Poulson, N.D.; Foote, H.P.; Lechler, T. NuMA localization, stability, and function in spindle orientation involve 4.1 and Cdk1 interactions. Mol. Biol. Cell. 2013, 24, 3651–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Zhou, B.R.; Bai, Y. Binding Affinity and Function of the Extremely Disordered Protein Complex Containing Human Linker Histone H1.0 and Its Chaperone ProTalpha. Biochemistry 2018. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.K.; Singh, P.; Roy, S.; Bhat, R. Comparative Analysis of the Conformation, Aggregation, Interaction, and Fibril Morphologies of Human α-, β-, and γ-Synuclein Proteins. Biochemistry 2018, 57, 3830–3848. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Yang, X.; Baum, J. Interactions between the Intrinsically Disordered Proteins beta-Synuclein and α-Synuclein. Proteomics 2018, 18, e1800109. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Hill, T.L. Effect of rotation on the diffusion-controlled rate of ligand-protein association. Proc. Natl. Acad. Sci. USA 1975, 72, 4918–4922. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Weber, G. Dynamics and time-averaged chemical potential of proteins: Importance in oligomer association. Proc. Natl. Acad. Sci. USA 1982, 79, 5268–5271. [Google Scholar] [CrossRef] [PubMed]
- Berg, O.G. Time-averaged chemical potential of proteins and the detailed-balance principle (an alternative viewpoint). Proc. Natl. Acad. Sci. USA 1983, 80, 5302–5303. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, N.; Sharp, K. Protein-solvent interactions. Chem. Rev. 2006, 106, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-X.; Bates, P.A. Modeling protein association mechanisms and kinetics. Curr. Opin. Struct. Biol. 2013, 23, 887–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.X.; Pang, X.; Lu, C. Rate constants and mechanisms of intrinsically disordered proteins binding to structured targets. Phys. Chem. Chem. Phys. 2012, 14, 10466–10476. [Google Scholar] [CrossRef] [PubMed]
- Dogan, J.; Gianni, S.; Jemth, P. The binding mechanisms of intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2014, 16, 6323–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Parra, R.G.; Schafer, N.P.; Radusky, L.G.; Tsai, M.Y.; Guzovsky, A.B.; Wolynes, P.G.; Ferreiro, D.U. Protein Frustratometer 2: A tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 2016, 44, W356–W360. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M. Fold or not to fold upon binding-does it really matter? Curr. Opin. Struct. Biol. 2018, 54, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Mollica, L.; Bessa, L.M.; Hanoulle, X.; Jensen, M.R.; Blackledge, M.; Schneider, R. Binding Mechanisms of Intrinsically Disordered Proteins: Theory, Simulation, and Experiment. Front. Mol. Biosci. 2016, 3, 52. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B. Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.A.; Shea, J.E. Simulations of disordered proteins and systems with conformational heterogeneity. Curr. Opin. Struct. Biol. 2017, 43, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. Force field development and simulations of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2018, 48, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Stanley, N.; Esteban-Martin, S.; De Fabritiis, G. Progress in studying intrinsically disordered proteins with atomistic simulations. Prog. Biophys. Mol. Biol. 2015, 119, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Towards the physical basis of how intrinsic disorder mediates protein function. Arch. Biochem. Biophys. 2012, 524, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Miskei, M.; Antal, C.; Fuxreiter, M. FuzDB: Database of fuzzy complexes, a tool to develop stochastic structure-function relationships for protein complexes and higher-order assemblies. Nucleic Acids Res. 2017, 45, D228–D235. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Sun, Q.; Zhang, W.; Liu, Y.; Lai, L. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2018, 24, 217–227. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Binding Energy Components (kJ/mol) | |
|---|---|
| ΔEvdW | −206.2 ± 2.2 |
| ΔEele | −1496.4 ± 8.9 |
| ΔGpolar | 1653.3 ± 11.9 |
| ΔGnonpolar | −36.9 ± 0.2 |
| ΔGbind | −86.0 ± 4.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wang, D. Extreme Fuzziness: Direct Interactions between Two IDPs. Biomolecules 2019, 9, 81. https://doi.org/10.3390/biom9030081
Wang W, Wang D. Extreme Fuzziness: Direct Interactions between Two IDPs. Biomolecules. 2019; 9(3):81. https://doi.org/10.3390/biom9030081
Chicago/Turabian StyleWang, Wenning, and Dongdong Wang. 2019. "Extreme Fuzziness: Direct Interactions between Two IDPs" Biomolecules 9, no. 3: 81. https://doi.org/10.3390/biom9030081
APA StyleWang, W., & Wang, D. (2019). Extreme Fuzziness: Direct Interactions between Two IDPs. Biomolecules, 9(3), 81. https://doi.org/10.3390/biom9030081