Evidence of Destabilization of the Human Thymidylate Synthase (hTS) Dimeric Structure Induced by the Interface Mutation Q62R

 ,
, 
 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning and Site-Directed Mutagenesis
2.2. Protein Expression and Purification
2.3. Enzymatic Activity Assays
2.4. Circular Dichroism (CD) Thermal Denaturation Analysis
2.5. Protein Crystallization
2.6. Data Collection, Structure Solution and Refinement
2.7. Protein Data Bank (PDB) Deposition
3. Results
3.1. Variant Production
3.2. Kinetic Characterization
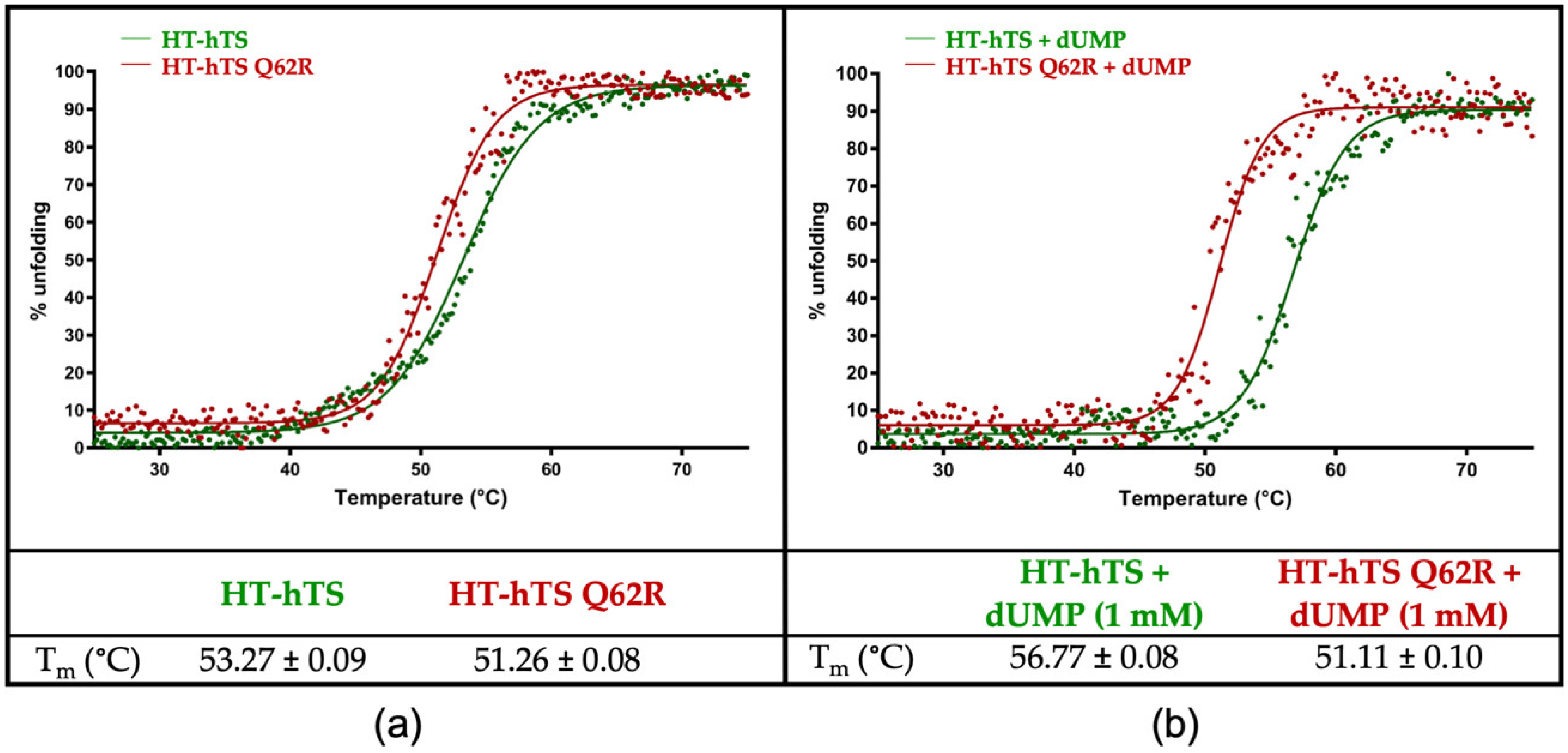
3.3. Circular Dichroism (CD) Thermal Denaturation Analysis
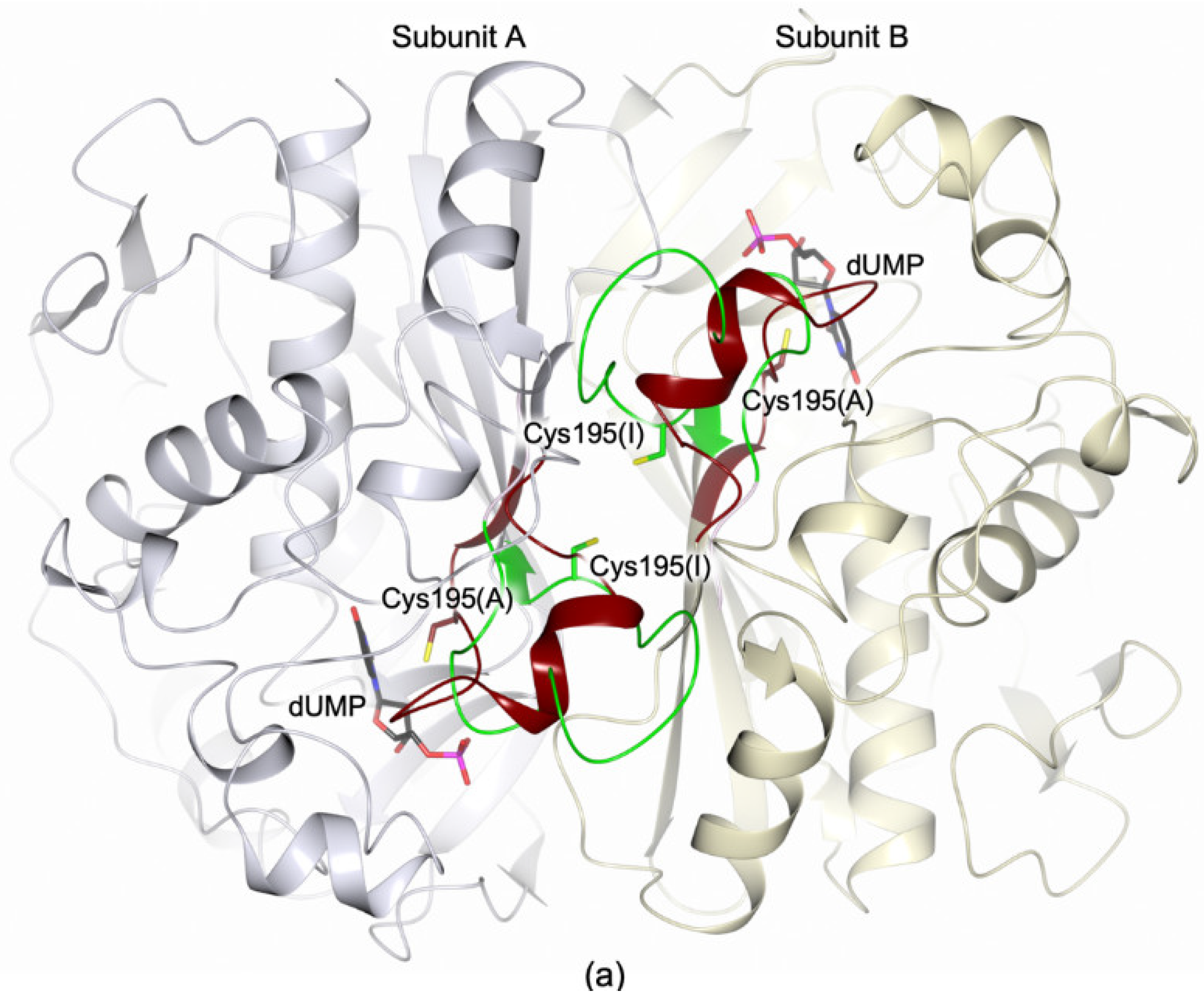
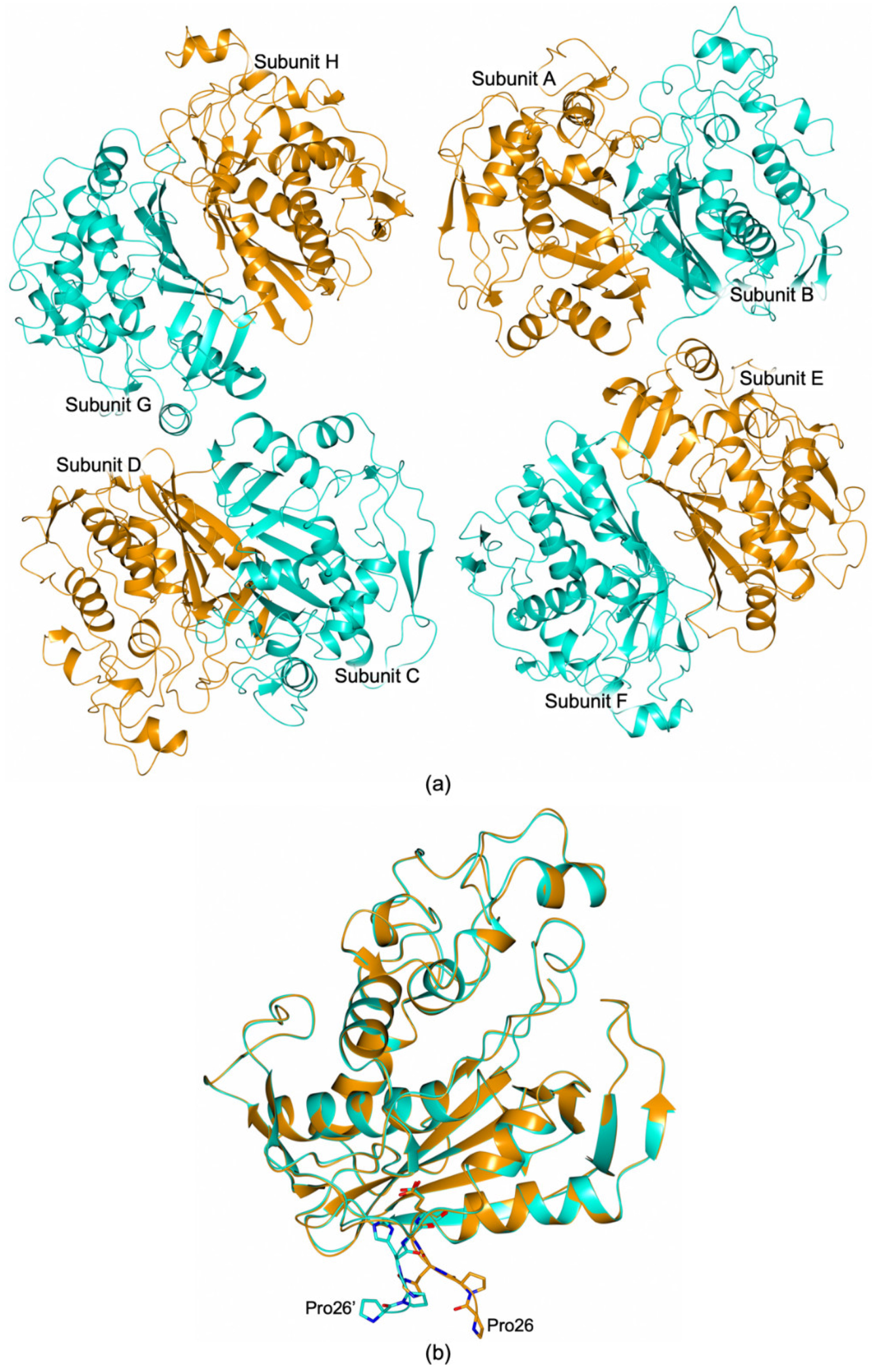
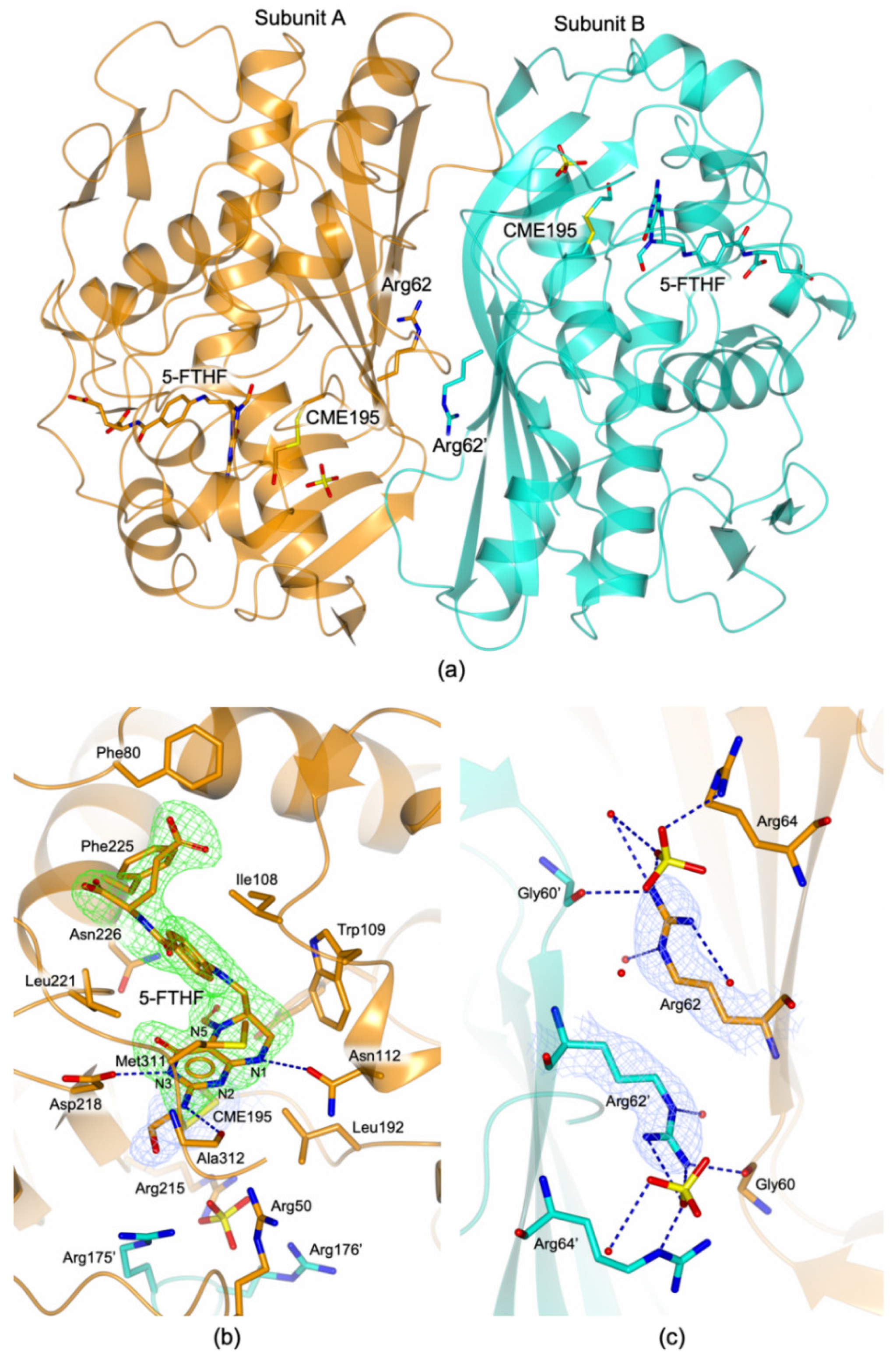
3.4. Structural Characterization of the HT-hTS Variant Q62R
3.4.1. The HT-hTS Q62R Active Site
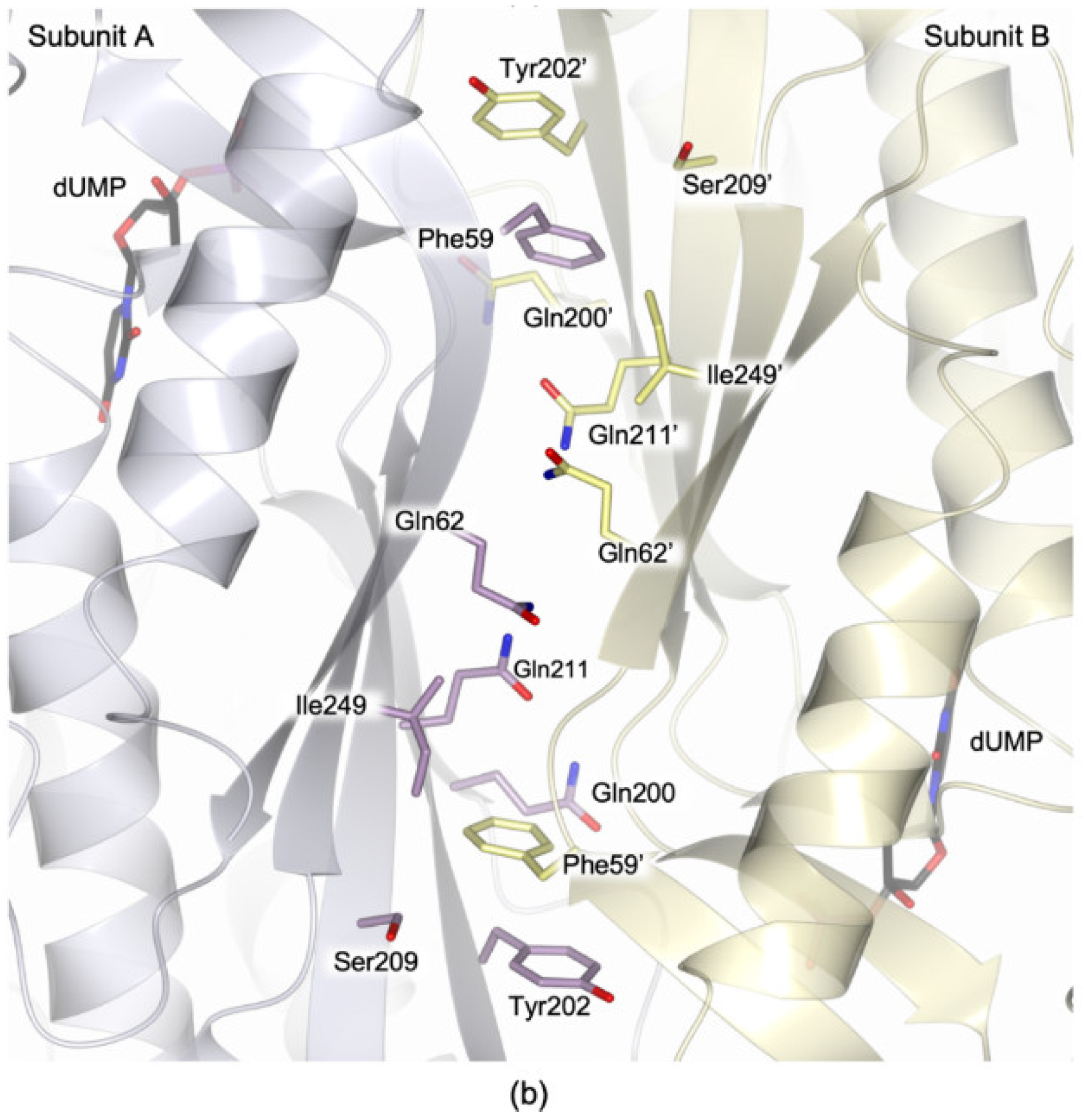
3.4.2. The Arg62 Pocket
4. Discussion
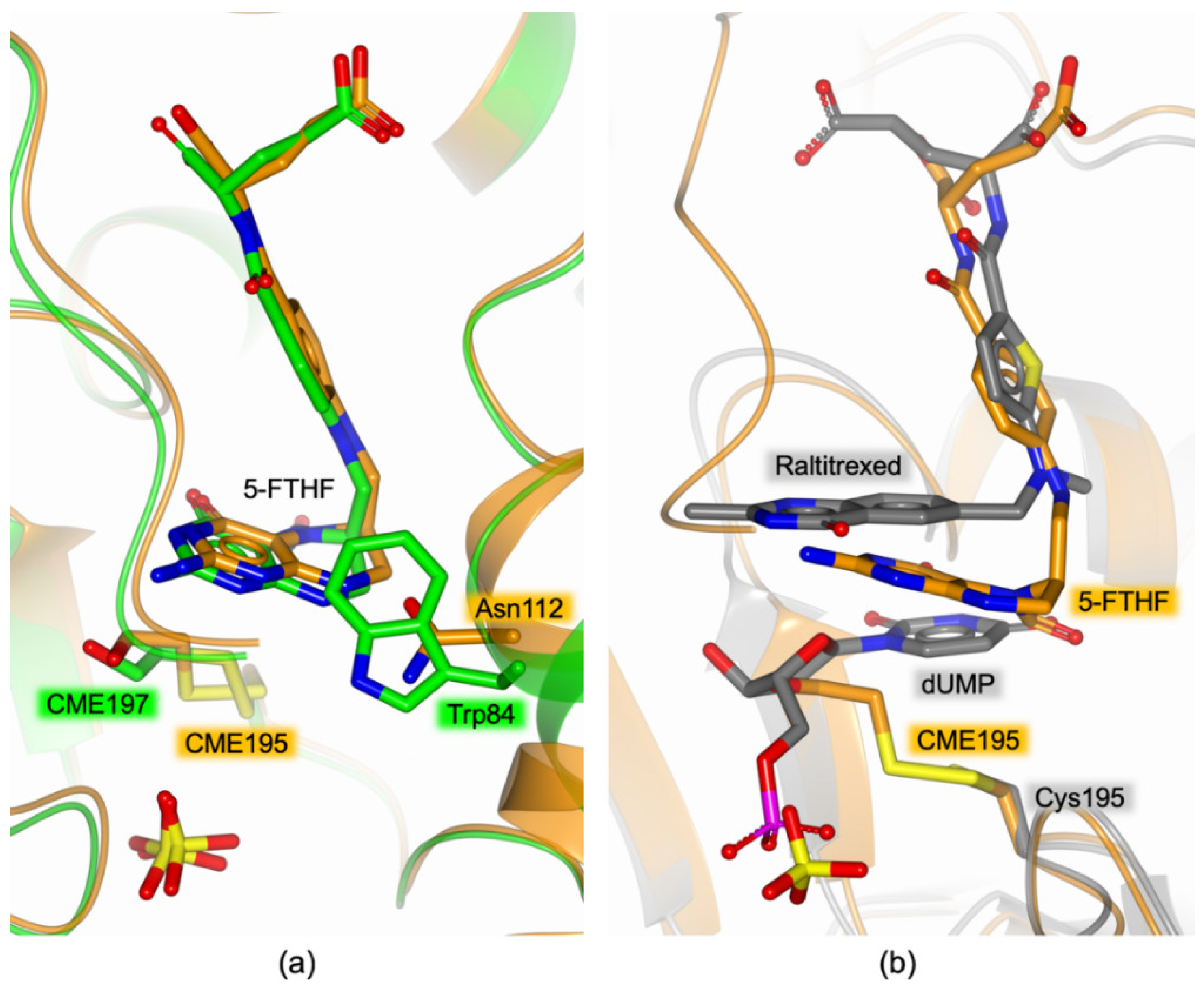
4.1. 5-FTHF Binding in the HT-hTS Q62R Active Site
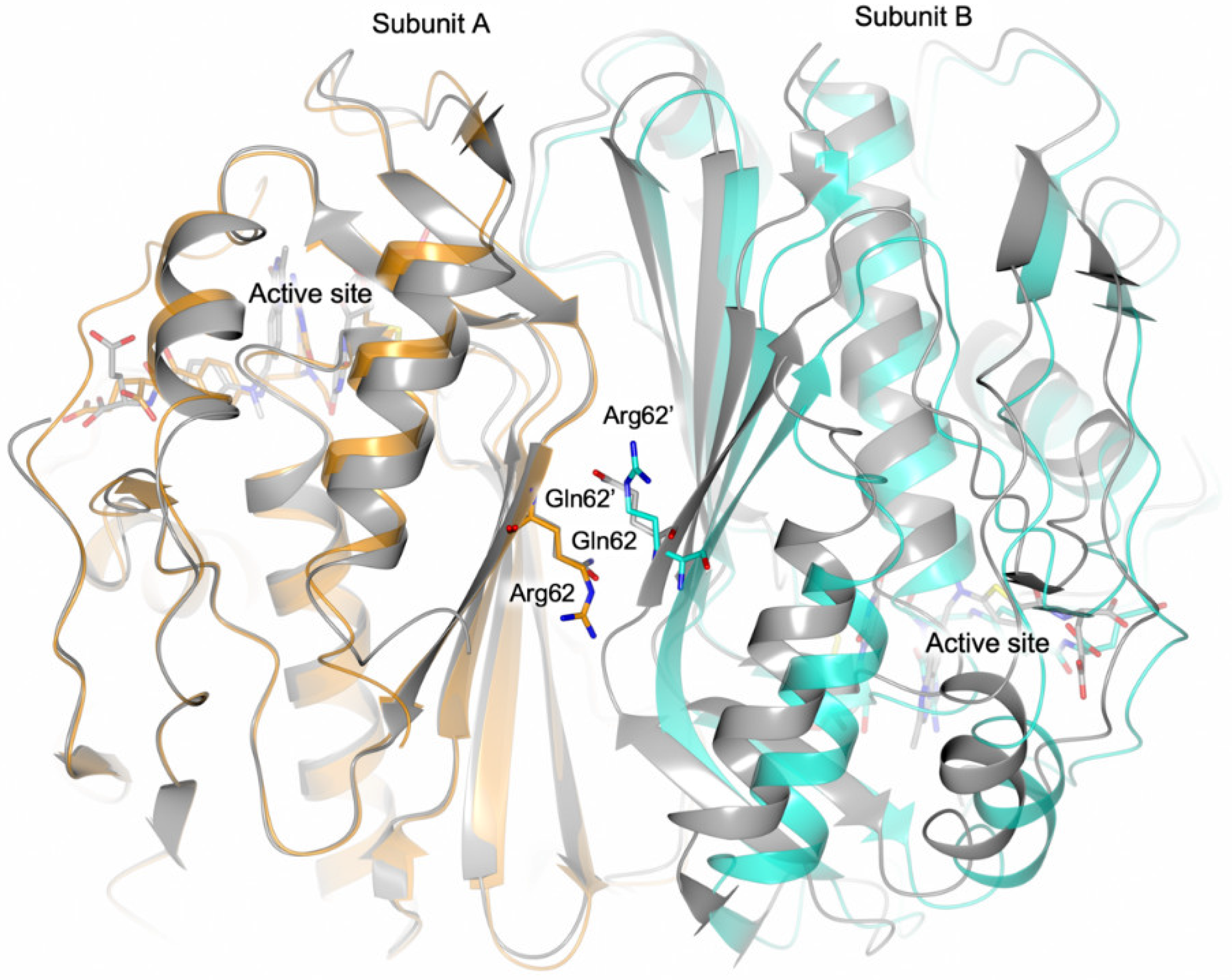
4.2. Effect of the Interface Point Mutation Q62R on the hTS Dimer Stability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kholodar, S.A.; Kohen, A. Noncovalent Intermediate of Thymidylate Synthase: Fact or Fiction? J. Am. Chem. Soc. 2016, 138, 8056–8059. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.A.; Houghton, P.J. Cellular responses to antimetabolite anticancer agents: Cytostasis versus cytotoxicity. In Progress in Cell Cycle Research; Springer: Boston, MA, USA, 1996; pp. 175–185. ISBN 978-1-4613-7693-4. [Google Scholar]
- Taddia, L.; D’Arca, D.; Ferrari, S.; Marraccini, C.; Severi, L.; Ponterini, G.; Assaraf, Y.G.; Marverti, G.; Costi, M.P. Inside the biochemical pathways of thymidylate synthase perturbed by anticancer drugs: Novel strategies to overcome cancer chemoresistance. Drug Resist. Updates 2015, 23, 20–54. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Severi, L.; Pozzi, C.; Quotadamo, A.; Ponterini, G.; Losi, L.; Marverti, G.; Costi, M.P. Chapter Seventeen—Human Thymidylate Synthase Inhibitors Halting Ovarian Cancer Growth. In Vitamins and Hormones; Litwack, G., Ed.; Ovarian Cycle; Academic Press: Cambridge, MA, USA, 2018; Volume 107, pp. 473–513. [Google Scholar]
- Liu, J.; Schmitz, J.C.; Lin, X.; Tai, N.; Yan, W.; Farrell, M.; Bailly, M.; Chen, T.; Chu, E. Thymidylate synthase as a translational regulator of cellular gene expression. Biochim. Biophys. Acta 2002, 1587, 174–182. [Google Scholar] [CrossRef]
- Schiffer, C.A.; Clifton, I.J.; Davisson, V.J.; Santi, D.V.; Stroud, R.M. Crystal structure of human thymidylate synthase: A structural mechanism for guiding substrates into the active site. Biochemistry 1995, 34, 16279–16287. [Google Scholar] [CrossRef]
- Phan, J.; Koli, S.; Minor, W.; Dunlap, R.B.; Berger, S.H.; Lebioda, L. Human Thymidylate Synthase Is in the Closed Conformation When Complexed with dUMP and Raltitrexed, an Antifolate Drug. Biochemistry 2001, 40, 1897–1902. [Google Scholar] [CrossRef]
- Chu, E.; Allegra, C.J. The role of thymidylate synthase as an RNA binding protein. Bioessays 1996, 18, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.H.; Berger, F.G.; Lebioda, L. Effects of ligand binding and conformational switching on intracellular stability of human thymidylate synthase. Biochim. Biophys. Acta 2004, 1696, 15–22. [Google Scholar] [CrossRef]
- Lin, X.; Liu, J.; Maley, F.; Chu, E. Role of cysteine amino acid residues on the RNA binding activity of human thymidylate synthase. Nucleic Acids Res. 2003, 31, 4882–4887. [Google Scholar] [CrossRef]
- Voeller, D.M.; Zajac-Kaye, M.; Fisher, R.J.; Allegra, C.J. The identification of thymidylate synthase peptide domains located in the interface region that bind thymidylate synthase mRNA. Biochem. Biophys. Res. Commun. 2002, 297, 24–31. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Tochowicz, A.; Pozzi, C.; Cardinale, D.; Ferrari, S.; Boum, Y.; Mangani, S.; Stroud, R.M.; Saxena, P.; Myllykallio, H.; et al. Hotspots in an Obligate Homodimeric Anticancer Target. Structural and Functional Effects of Interfacial Mutations in Human Thymidylate Synthase. J. Med. Chem. 2015, 58, 3572–3581. [Google Scholar] [CrossRef]
- Chen, D.; Jansson, A.; Sim, D.; Larsson, A.; Nordlund, P. Structural analyses of human thymidylate synthase reveal a site that may control conformational switching between active and inactive states. J. Biol. Chem. 2017, 292, 13449–13458. [Google Scholar] [CrossRef]
- Cardinale, D.; Guaitoli, G.; Tondi, D.; Luciani, R.; Henrich, S.; Salo-Ahen, O.M.H.; Ferrari, S.; Marverti, G.; Guerrieri, D.; Ligabue, A.; et al. Protein–protein interface-binding peptides inhibit the cancer therapy target human thymidylate synthase. Proc. Natl. Acad. Sci. USA 2011, 108, E542–E549. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Benvenuti, M.; Mangani, S. Crystallization of soluble proteins in vapor diffusion for X-ray crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Winn, M.D.; Isupov, M.N.; Murshudov, G.N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 122–133. [Google Scholar] [CrossRef]
- Painter, J.; Merritt, E.A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Cryst. 2006, 39, 109–111. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. Validation of protein models derived from experiment. Curr. Opin. Struct. Biol. 1998, 8, 631–639. [Google Scholar] [CrossRef]
- Potterton, L.; McNicholas, S.; Krissinel, E.; Gruber, J.; Cowtan, K.; Emsley, P.; Murshudov, G.N.; Cohen, S.; Perrakis, A.; Noble, M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2288–2294. [Google Scholar] [CrossRef]
- Pedersen-Lane, J.; Maley, G.F.; Chu, E.; Maley, F. High-Level Expression of Human Thymidylate Synthase. Protein Expr. Purif. 1997, 10, 256–262. [Google Scholar] [CrossRef]
- Stover, P.; Schirch, V. The metabolic role of leucovorin. Trends Biochem. Sci. 1993, 18, 102–106. [Google Scholar] [CrossRef]
- Pozzi, C.; Ferrari, S.; Cortesi, D.; Luciani, R.; Stroud, R.M.; Catalano, A.; Costi, M.P.; Mangani, S. The structure of Enterococcus faecalis thymidylate synthase provides clues about folate bacterial metabolism. Acta Cryst. D 2012, 68, 1232–1241. [Google Scholar] [CrossRef]
- Brunn, N.D.; Dibrov, S.M.; Kao, M.B.; Ghassemian, M.; Hermann, T. Analysis of mRNA recognition by human thymidylate synthase. Biosci. Rep. 2014, 34, e00168. [Google Scholar] [CrossRef]
- Lin, Q.; Parker, M.J.; Taguchi, A.T.; Ravichandran, K.; Kim, A.; Kang, G.; Shao, J.; Drennan, C.L.; Stubbe, J. Glutamate 52-β at the α/β subunit interface of Escherichia coli class Ia ribonucleotide reductase is essential for conformational gating of radical transfer. J. Biol. Chem. 2017, 292, 9229–9239. [Google Scholar] [CrossRef]
- Sayre, P.H.; Finer-Moore, J.S.; Fritz, T.A.; Biermann, D.; Gates, S.B.; MacKellar, W.C.; Patel, V.F.; Stroud, R.M. Multi-targeted antifolates aimed at avoiding drug resistance form covalent closed inhibitory complexes with human and Escherichia coli thymidylate synthases. J. Mol. Biol. 2001, 313, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Almqvist, H.; Axelsson, H.; Jafari, R.; Dan, C.; Mateus, A.; Haraldsson, M.; Larsson, A.; Molina, D.M.; Artursson, P.; Lundbäck, T.; et al. CETSA screening identifies known and novel thymidylate synthase inhibitors and slow intracellular activation of 5-fluorouracil. Nat. Commun. 2016, 7, 11040. [Google Scholar] [CrossRef]
- Almog, R.; Waddling, C.A.; Maley, F.; Maley, G.F.; Van Roey, P. Crystal structure of a deletion mutant of human thymidylate synthase Δ(7–29) and its ternary complex with Tomudex and dUMP. Protein Sci. 2001, 10, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KM (dUMP) (μM) | KM (mTHF) (μM) | kcat (s−1) | kcat/KM (dUMP) (μM−1 s−1) | kcat/KM (mTHF) (μM−1 s−1) | |
|---|---|---|---|---|---|
| HT-hTS | 10 ± 1 | 6 ± 1 | 1.00 ± 0.01 | 1.00 × 10−7 | 1.67 × 10−7 |
| HT-hTS Q62R | 8 ± 1 | 16 ± 1 | 0.16 ± 0.06 | 0.20 × 10−7 | 0.10 × 10−7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozzi, C.; Lopresti, L.; Santucci, M.; Costi, M.P.; Mangani, S. Evidence of Destabilization of the Human Thymidylate Synthase (hTS) Dimeric Structure Induced by the Interface Mutation Q62R. Biomolecules 2019, 9, 134. https://doi.org/10.3390/biom9040134
Pozzi C, Lopresti L, Santucci M, Costi MP, Mangani S. Evidence of Destabilization of the Human Thymidylate Synthase (hTS) Dimeric Structure Induced by the Interface Mutation Q62R. Biomolecules. 2019; 9(4):134. https://doi.org/10.3390/biom9040134
Chicago/Turabian StylePozzi, Cecilia, Ludovica Lopresti, Matteo Santucci, Maria Paola Costi, and Stefano Mangani. 2019. "Evidence of Destabilization of the Human Thymidylate Synthase (hTS) Dimeric Structure Induced by the Interface Mutation Q62R" Biomolecules 9, no. 4: 134. https://doi.org/10.3390/biom9040134
APA StylePozzi, C., Lopresti, L., Santucci, M., Costi, M. P., & Mangani, S. (2019). Evidence of Destabilization of the Human Thymidylate Synthase (hTS) Dimeric Structure Induced by the Interface Mutation Q62R. Biomolecules, 9(4), 134. https://doi.org/10.3390/biom9040134