Regulation of the Expression of DAPK1 by SUMO Pathway

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Western Blot
2.3. Statistical Analysis
2.4. Prediction of SUMOylation Sites
2.5. Antibodies and Chemicals
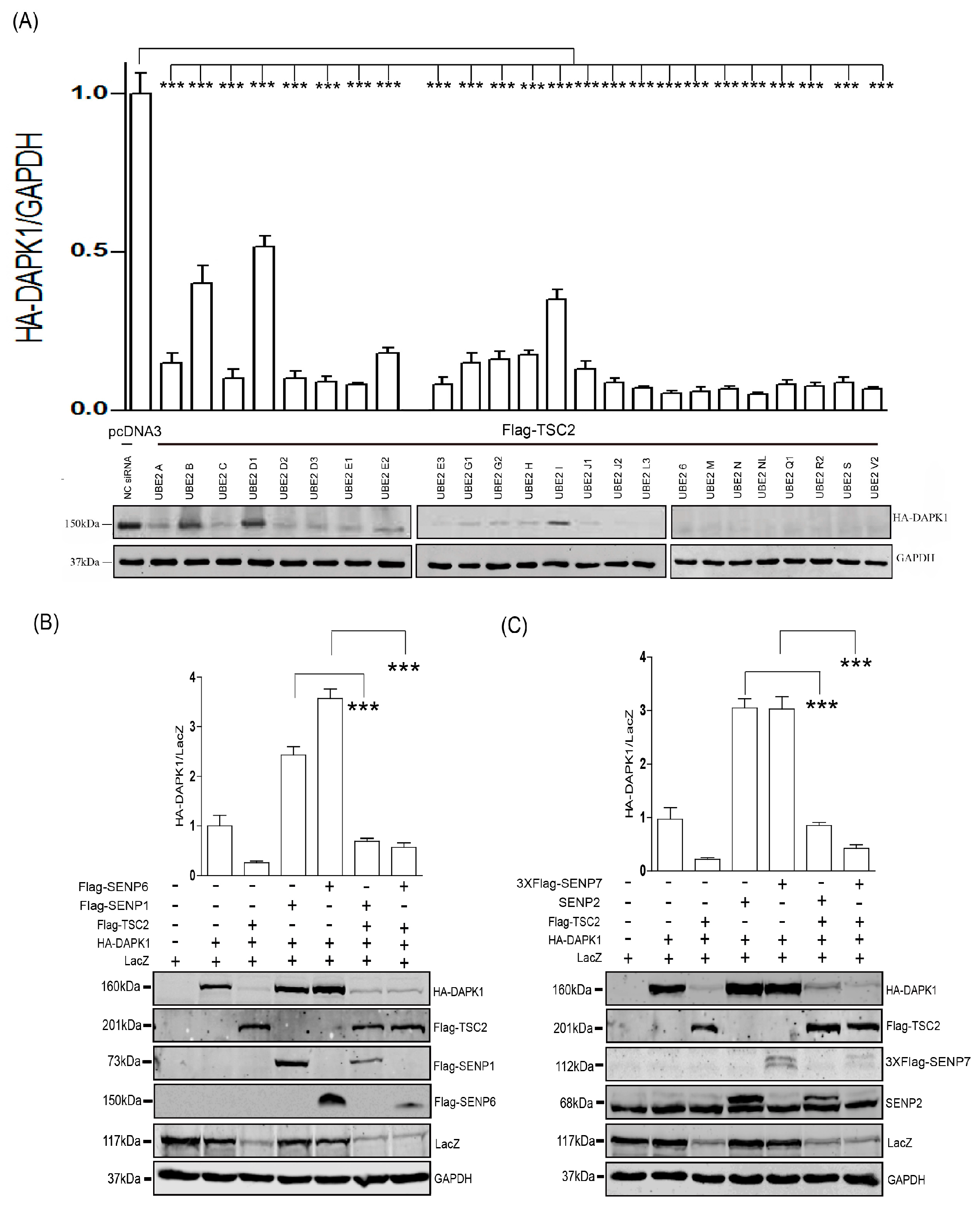
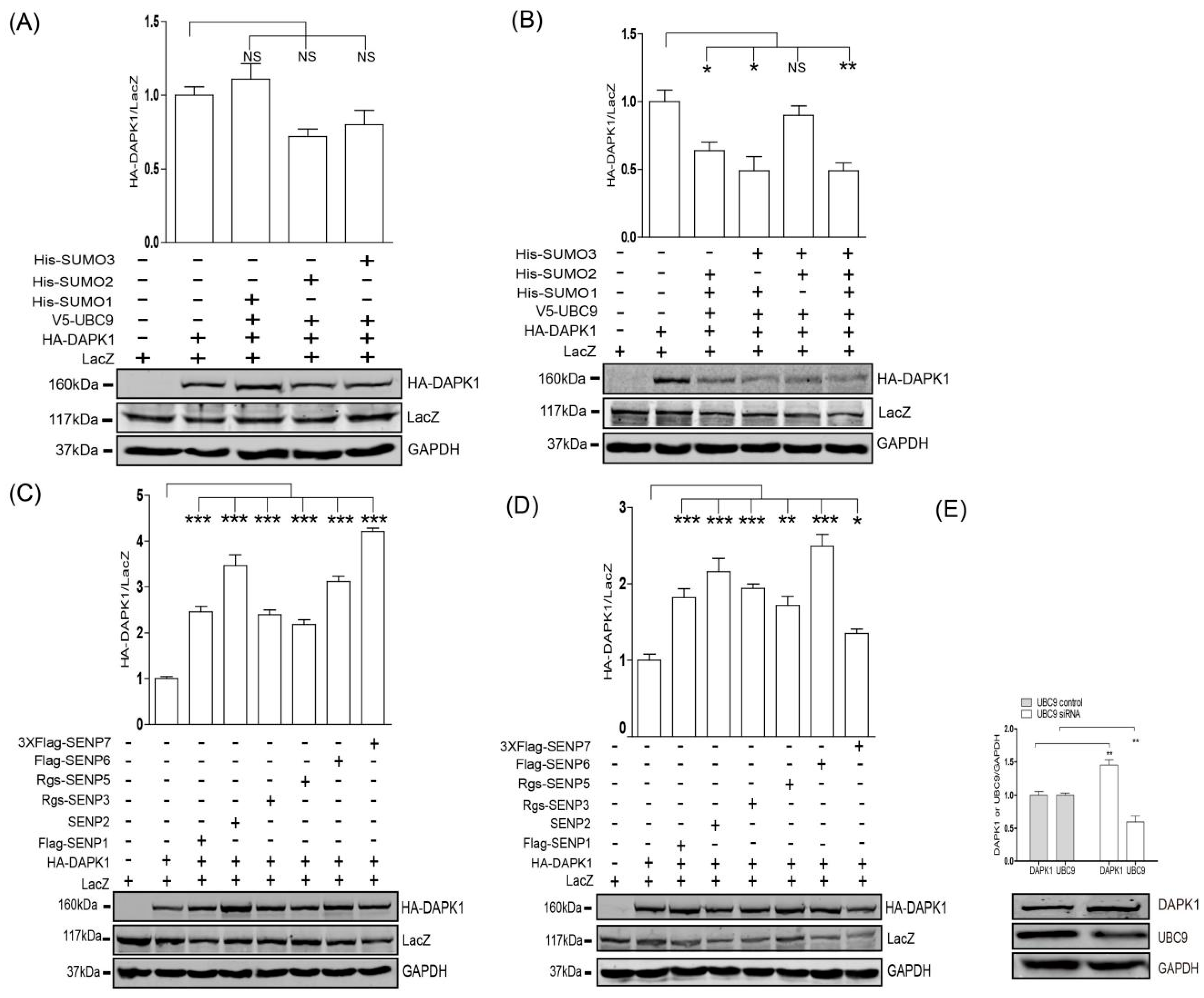
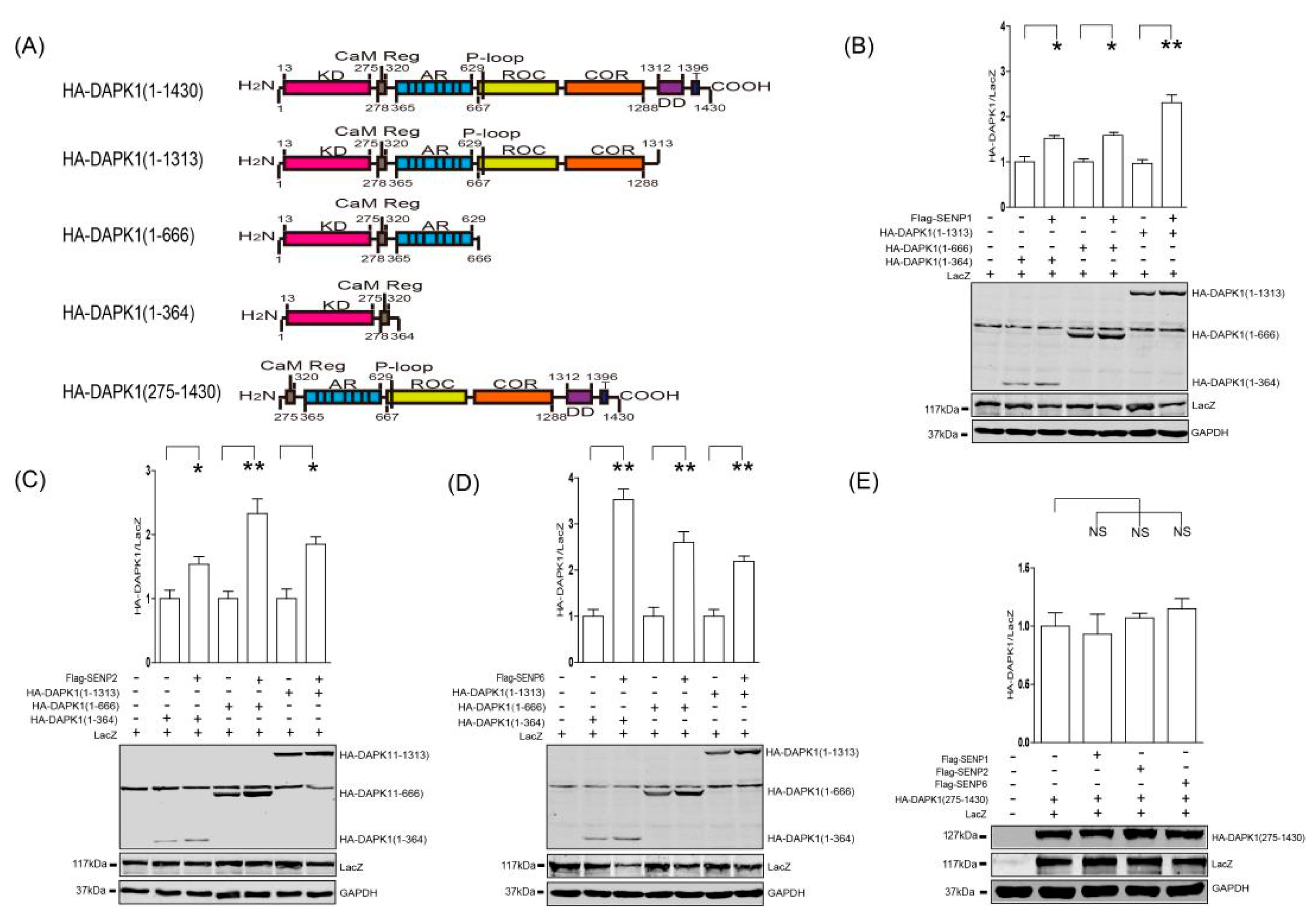
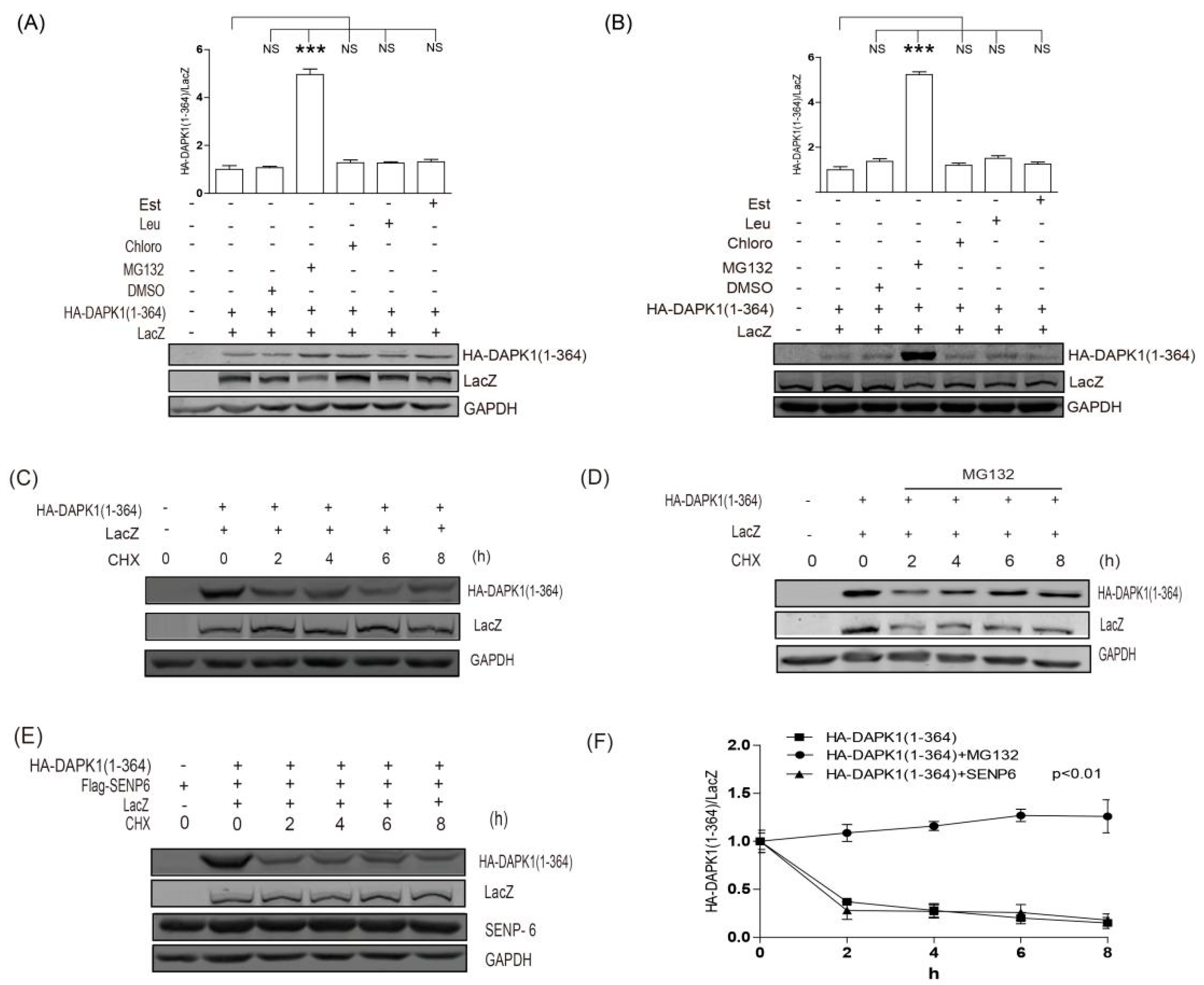
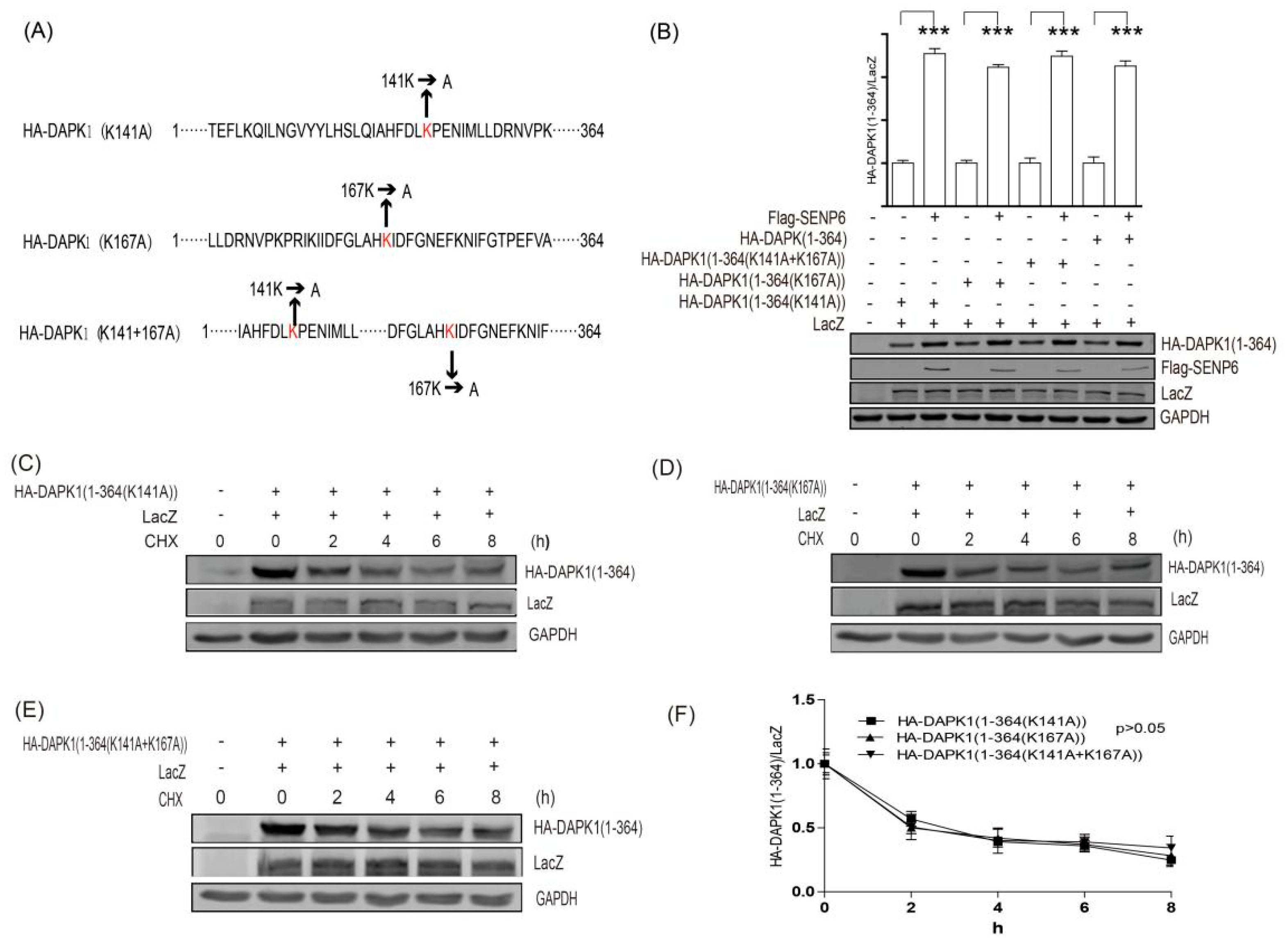
3. Results
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bialik, S.; Kimchi, A. The DAP-kinase interactome. Apoptosis 2014, 19, 316–328. [Google Scholar] [CrossRef]
- Wu, B.; Yao, H.; Wang, S.; Xu, R. DAPK1 modulates a curcumin-induced G2/M arrest and apoptosis by regulating STAT3, NF-κB, and caspase-3 activation. Biochem. Biophys. Res. Commun. 2013, 434, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Byun, H.J.; Kim, B.R.; Lee, K.H.; Park, S.Y.; Rho, S.B. DAPk1 inhibits NF-κB activation through TNF-α and INF-γ-induced apoptosis. Cell. Signal. 2012, 24, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.Y.; Chen, P.C.; Zhang, J.L.; Gao, Z.S.; Neves, H.; Zhang, S.D.; Wen, Q.; Chen, W.D.; Kwok, H.F.; Lin, Y. The prognostic significance of DAPK1 in bladder cancer. PLoS ONE 2017, 12, e0175290. [Google Scholar] [CrossRef]
- Niklinska, W.; Naumnik, W.; Sulewska, A.; Kozłowski, M.; Pankiewicz, W.; Milewski, R. Prognostic significance of DAPK and RASSF1A promoter hypermethylation in non-small cell lung cancer (NSCLC). Folia Histochem. Cytobiol. 2009, 47, 275–280. [Google Scholar] [CrossRef]
- Shiloh, R.; Bialik, S.; Kimchi, A. The DAPK family: A structure–function analysis. Apoptosis 2014, 19, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, L.; Guo, L.; Hupp, T.R.; Lin, Y. Evaluating DAPK as a therapeutic target. Apoptosis 2014, 19, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.F.; Chuang, Y.T.; Hsieh, W.C.; Chang, P.Y.; Liu, H.Y.; Mo, S.T.; Hsu, T.S.; Miaw, S.C.; Chen, R.H.; Kimchi, A. Tumour suppressor death-associated protein kinase targets cytoplasmic HIF-1α for Th17 suppression. Nat. Commun. 2016, 7, 11904. [Google Scholar] [CrossRef] [Green Version]
- Chuang, M.; Hsiao, T.I.; Tong, A.; Xu, S.; Chisholm, A.D. DAPK interacts with Patronin and the microtubule cytoskeleton in epidermal development and wound repair. Elife 2016, 5, e15833. [Google Scholar] [CrossRef]
- Liu, W.L.; Yang, H.C.; Hsu, C.S.; Wang, C.C.; Wang, T.S.; Kao, J.H.; Chen, D.S. Pegylated IFN-α suppresses hepatitis C virus by promoting the DAPK-mTOR pathway. Proc. Natl. Acad. Sci. USA 2016, 113, 14799–14804. [Google Scholar] [CrossRef] [Green Version]
- Benderska, N.; Schneider-Stock, R. Transcription control of DAPK. Apoptosis 2014, 19, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J. Downregulation of Death-Associated Protein Kinase 1 (DAPK1) in Chronic Lymphocytic Leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanovska, J.; Zlobec, I.; Forster, S.; Karamitopoulou, E.; Dawson, H.; Koelzer, V.H.; Agaimy, A.; Garreis, F.; Söder, S.; Laqua, W.; et al. DAPK loss in colon cancer tumor buds: Implications for migration capacity of disseminating tumor cells. Oncotarget 2015, 6, 36774–36788. [Google Scholar] [CrossRef]
- Chan, M.W.; Chan, L.W.; Tang, N.L.; Tong, J.H.; Lo, K.W.; Lee, T.L.; Cheung, H.Y.; Wong, W.S.; Chan, P.S.; Lai, F.M.; et al. Hypermethylation of multiple genes in tumor tissues and voided urine in urinary bladder cancer patients. Clin. Cancer Res. 2002, 8, 464–470. [Google Scholar]
- Gallagher, P.J.; Blue, E.K. Post-translational regulation of the cellular levels of DAPK. Apoptosis 2014, 19, 306–315. [Google Scholar] [CrossRef]
- Jin, Y.; Blue, E.K.; Gallagher, P.J. Control of death-associated protein kinase (DAPK) activity by phosphorylation and proteasomal degradation. J. Biol. Chem. 2006, 281, 39033–39040. [Google Scholar] [CrossRef]
- Zhang, L.; Nephew, K.P.; Gallagher, P.J. Regulation of death-associated protein kinase. Stabilization by HSP90 heterocomplexes. J. Biol. Chem. 2007, 282, 11795–11804. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Yuan, W.C.; Ho, H.C.; Chen, C.H.; Shih, H.M.; Chen, R.H. The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. EMBO J. 2014, 29, 1748–1761. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Yao, L.; Paul, H.; Susanne, P.; Jack, S.; Ted, H.; Craig, S. Tuberous sclerosis-2 (TSC2) regulates the stability of death-associated protein kinase-1 (DAPK) through a lysosome-dependent degradation pathway. FEBS J. 2011, 278, 354–370. [Google Scholar]
- Lin, Y.; Stevens, C.; Harrison, B.; Pathuri, S.; Amin, E.; Hupp, T.R. The alternative splice variant of DAPK-1, s-DAPK-1, induces proteasome-independent DAPK-1 destabilization. Mol. Cell. Biochem. 2009, 328, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Stevens, C.; Hupp, T. Identification of a Dominant Negative Functional Domain on DAPK-1 That Degrades DAPK-1 Protein and Stimulates TNFR-1-mediated Apoptosis. J. Biol. Chem. 2007, 282, 16792–16802. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef]
- Huang, C.J.; Wu, D.; Khan, F.A.; Huo, L.J. DeSUMOylation: An Important Therapeutic Target and Protein Regulatory Event. DNA Cell Biol. 2015, 34, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.I.; Baek, S.H. Chapter 7 Small Ubiquitin-Like Modifiers in Cellular Malignancy and Metastasis. Int. Rev. Cell Mol. Biol. 2009, 273, 265–311. [Google Scholar]
- Krumova, P.; Weishaupt, J.H. Sumoylation in neurodegenerative diseases. Cell. Mol. Life Sci. 2013, 70, 2123–2138. [Google Scholar] [CrossRef]
- Bohren, K.M.; Varsha, N.; Song, J.H.; Gabbay, K.H.; David, O. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 2004, 279, 27233–27238. [Google Scholar] [CrossRef]
- Liang, Y.C.; Lee, C.C.; Yao, Y.L.; Lai, C.C.; Schmitz, M.L.; Yang, W.M. SUMO5, a Novel Poly-SUMO Isoform, Regulates PML Nuclear Bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef]
- Nan, S.L.; Marie-Claude, G.; Jaffray, E.G.; Hay, R.T. Characterization of SENP7, a SUMO-2/3-specific isopeptidase. Biochem. J. 2009, 421, 223–230. [Google Scholar] [Green Version]
- Vertegaal, A.; Andersen, J.; Ogg, S.; Hay, R.; Mann, M.; Lamond, A. Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol. Cell. Proteom. 2006, 5, 2298–2310. [Google Scholar] [CrossRef]
- Bacco, A.D.; Ouyang, J.; Lee, H.; Catic, A.; Ploegh, H.; Gill, G. The SUMO-specific protease SENP5 is required for cell division. Mol. Cell. Biol. 2006, 26, 4489–4498. [Google Scholar] [CrossRef] [PubMed]
- Garone, L.; Fitton, J.E.; Steiner, R.F. Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. J. Biol. Chem. 2006, 281, 15869–15877. [Google Scholar]
- Chawon, Y.; Yonggang, W.; Debaditya, M.; Peter, B.; Nagamalleswari, K.; Alfred, Y.; Wilkinson, K.D.; Mary, D. Nucleolar protein B23/nucleophosmin regulates the vertebrate SUMO pathway through SENP3 and SENP5 proteases. J. Cell Biol. 2008, 183, 589–595. [Google Scholar] [Green Version]
- Lima, C.D.; David, R. Structure of the human SENP7 catalytic domain and poly-SUMO deconjugation activities for SENP6 and SENP7. J. Biol. Chem. 2008, 283, 32045–32055. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xie, Y.; Zheng, Y.; Jiang, S.; Liu, W.; Mu, W.; Liu, Z.; Zhao, Y.; Xue, Y.; Ren, J. GPS-SUMO: A tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 2014, 42, W325–W330. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Geoffroy, M.C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- Franch, H.A.; Sooparb, S.; Du, J.; Brown, N.S. A mechanism regulating proteolysis of specific proteins during renal tubular cell growth. J. Biol. Chem. 2001, 276, 19126–19131. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.; Nielsen, M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Hongjae, S.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef]
- Liu, X.H.; Sun, M.; Nie, F.Q.; Ge, Y.B.; Zhang, E.B.; Yin, D.D.; Kong, R.; Xia, R.; Lu, K.H.; Li, J.H.; et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol. Cancer 2014, 13, 92. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zhang, X.; Chen, L.; Weng, S.; Xia, Y.; Ye, Y.; Li, K.; Liao, Z.; Chen, P.; Alsamman, K.; et al. Regulation of the Expression of DAPK1 by SUMO Pathway. Biomolecules 2019, 9, 151. https://doi.org/10.3390/biom9040151
Wang Q, Zhang X, Chen L, Weng S, Xia Y, Ye Y, Li K, Liao Z, Chen P, Alsamman K, et al. Regulation of the Expression of DAPK1 by SUMO Pathway. Biomolecules. 2019; 9(4):151. https://doi.org/10.3390/biom9040151
Chicago/Turabian StyleWang, Qingshui, Xiuli Zhang, Ling Chen, Shuyun Weng, Yun Xia, Yan Ye, Ke Li, Ziqiang Liao, Pengchen Chen, Khaldoon Alsamman, and et al. 2019. "Regulation of the Expression of DAPK1 by SUMO Pathway" Biomolecules 9, no. 4: 151. https://doi.org/10.3390/biom9040151
APA StyleWang, Q., Zhang, X., Chen, L., Weng, S., Xia, Y., Ye, Y., Li, K., Liao, Z., Chen, P., Alsamman, K., Meng, C., Stevens, C., Hupp, T. R., & Lin, Y. (2019). Regulation of the Expression of DAPK1 by SUMO Pathway. Biomolecules, 9(4), 151. https://doi.org/10.3390/biom9040151