
The Structural Versatility of the BTB Domains of KCTD Proteins and Their Recognition of the GABAB Receptor




Abstract
:1. Introduction
2. Results
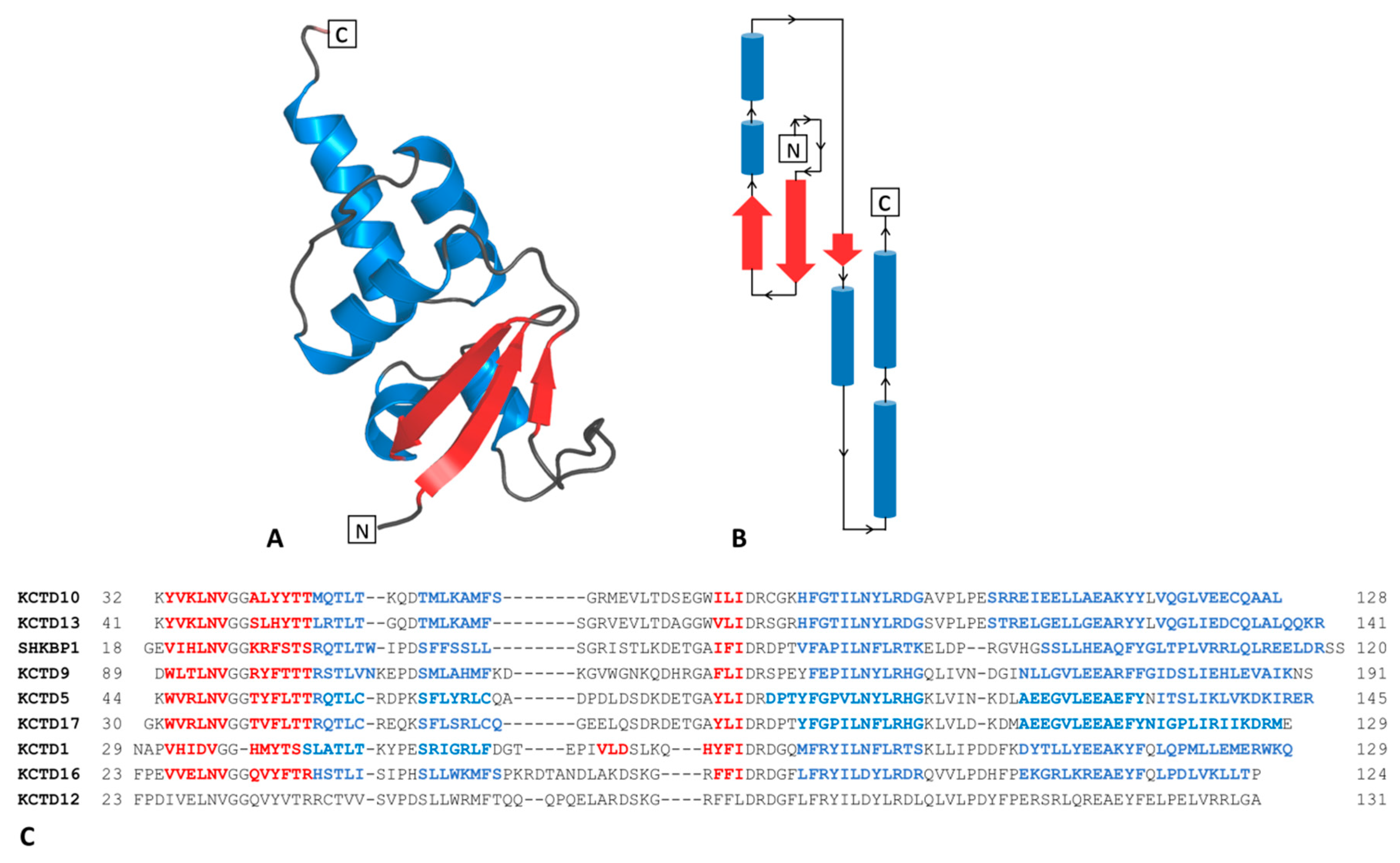
2.1. Analysis of the Structure and Stability of the KCTD-BTB Domains in Their Monomeric Forms
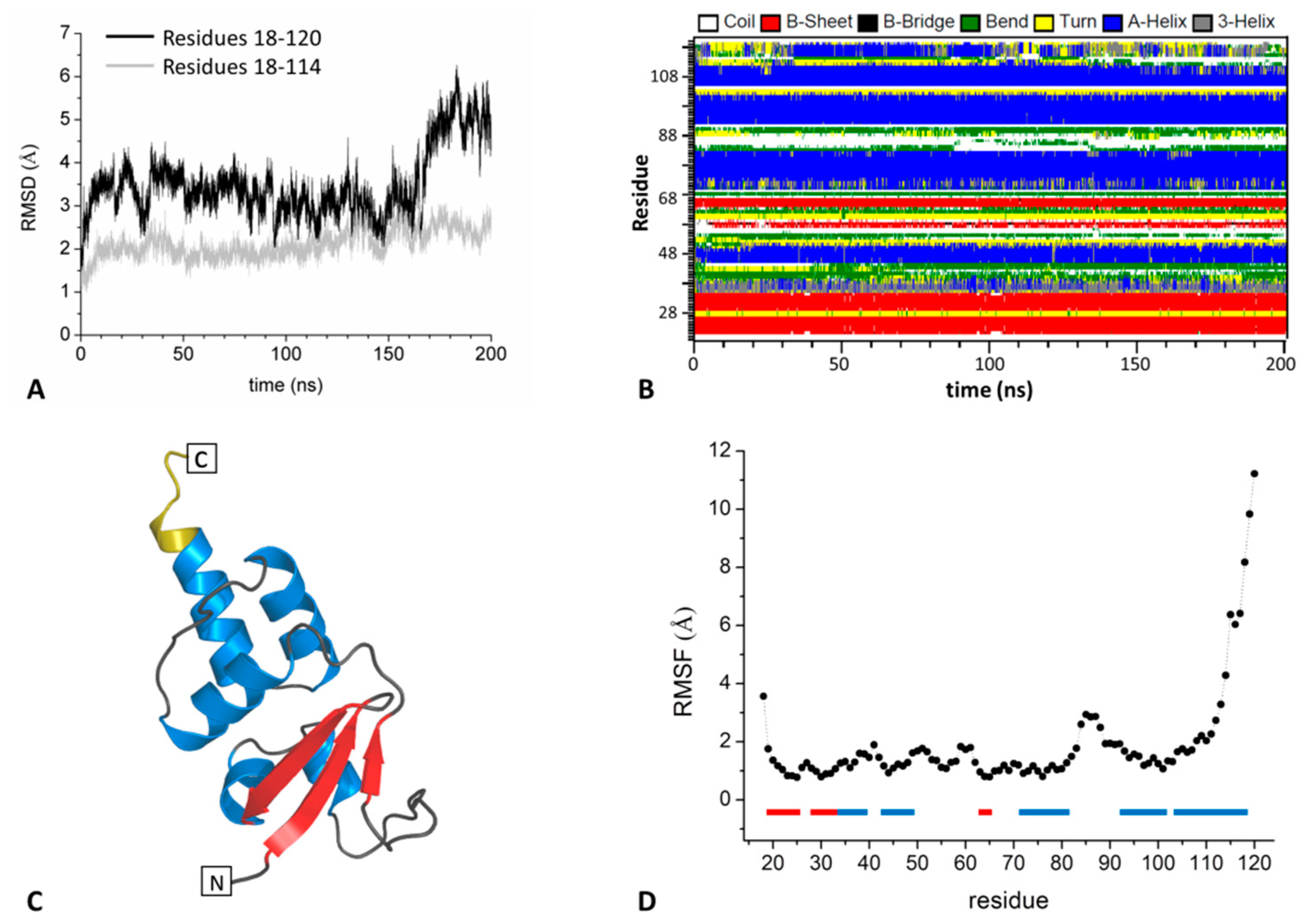
2.1.1. The BTB Domain of SHKBP1
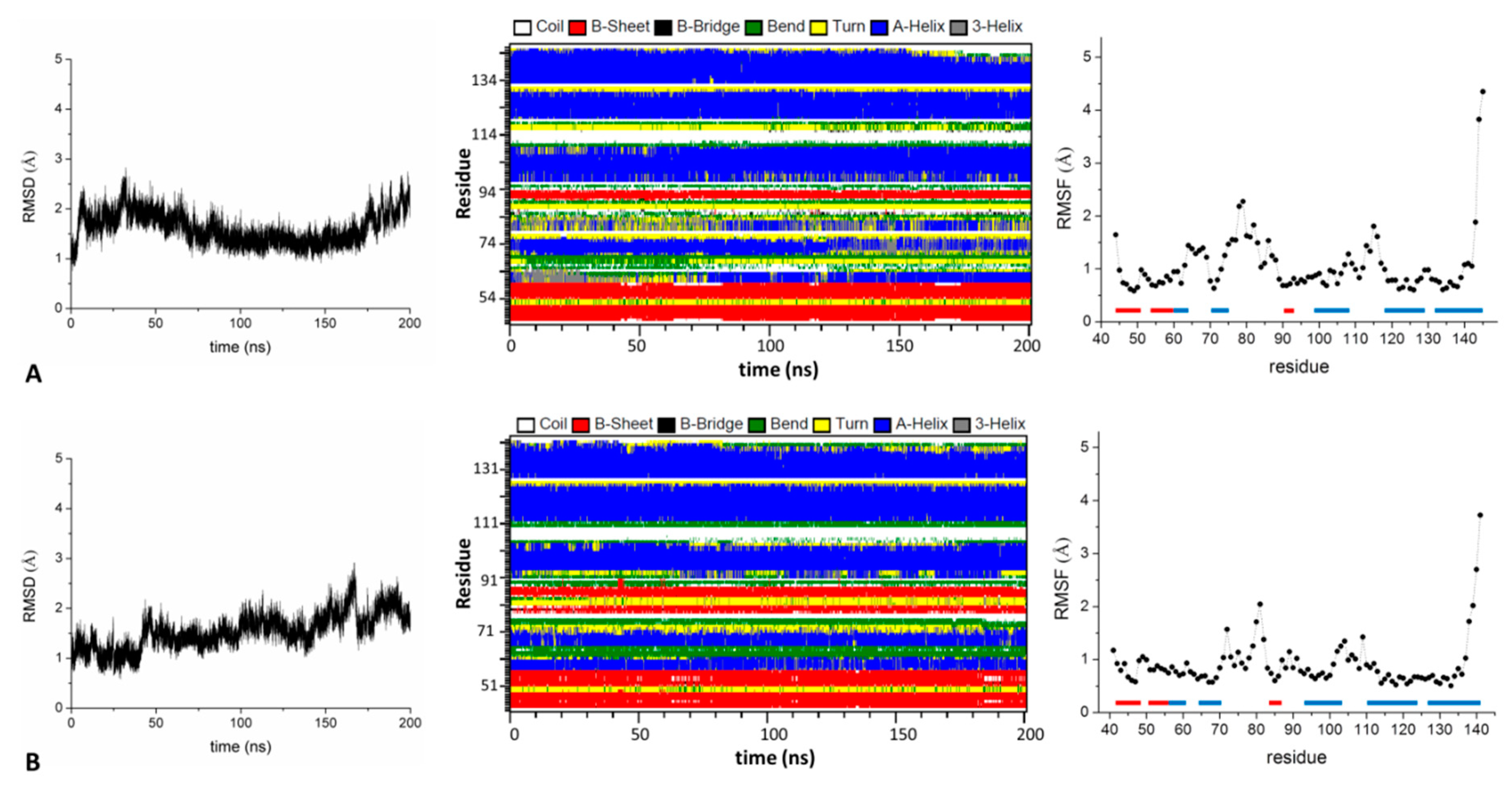
2.1.2. The BTB Domain of KCTD1, KCTD5, KCTD9, KCTD10, KCTD13, KCTD16, and KCTD17
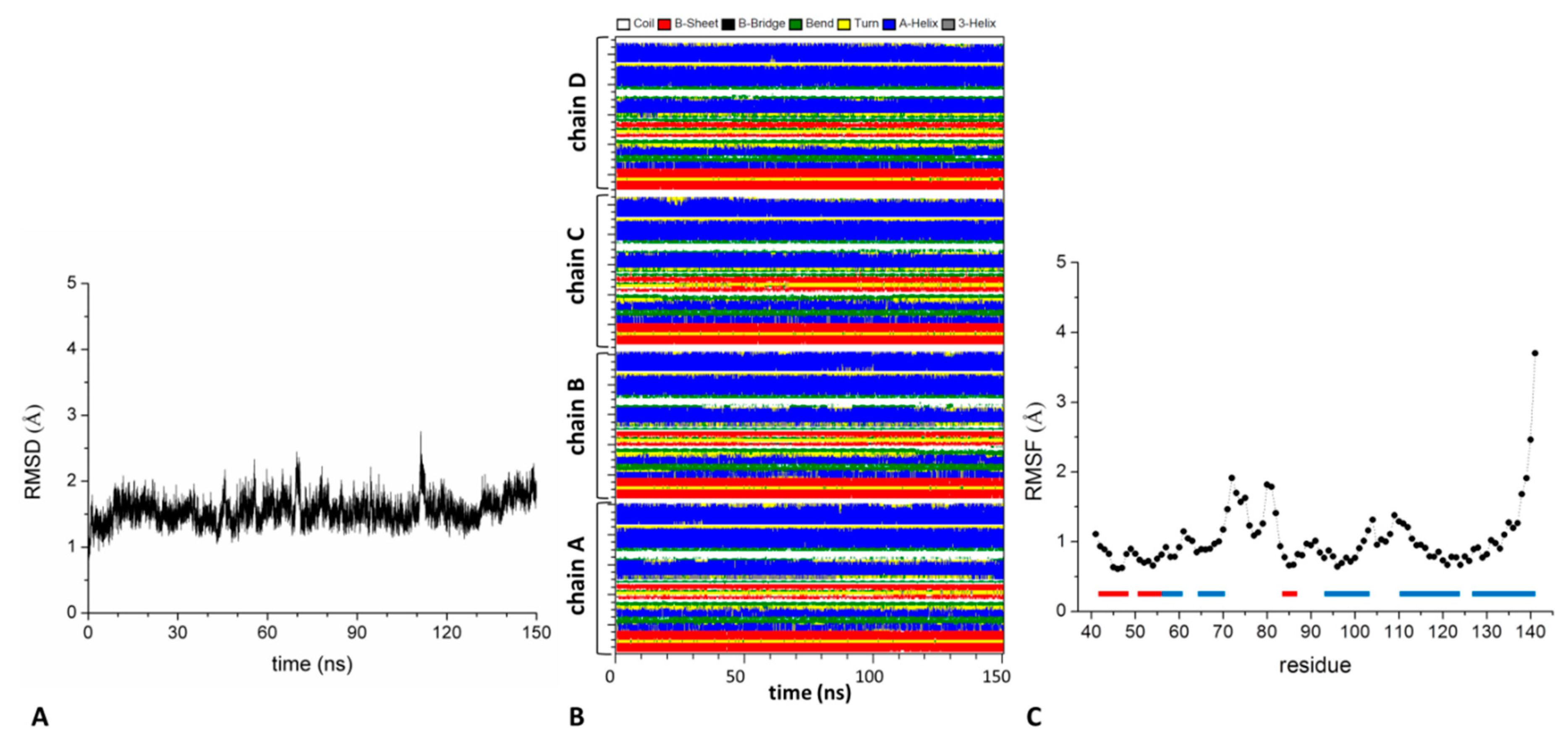
2.2. Analysis of the Structure and Stability of the KCTD-BTB Domains in Tetrameric and Dimeric States
2.3. Analysis of the Structure and Stability of the KCTD-BTB Domains in Pentameric States
2.3.1. Close States (KCTD1BTB, KCTD17BTB, and KCTD9BTB)
2.3.2. Open States (KCTD1BTB and KCTD16BTB)
2.3.3. Mixed Open Pentamer Formed by KCTD12BTB and KCTD16BTB
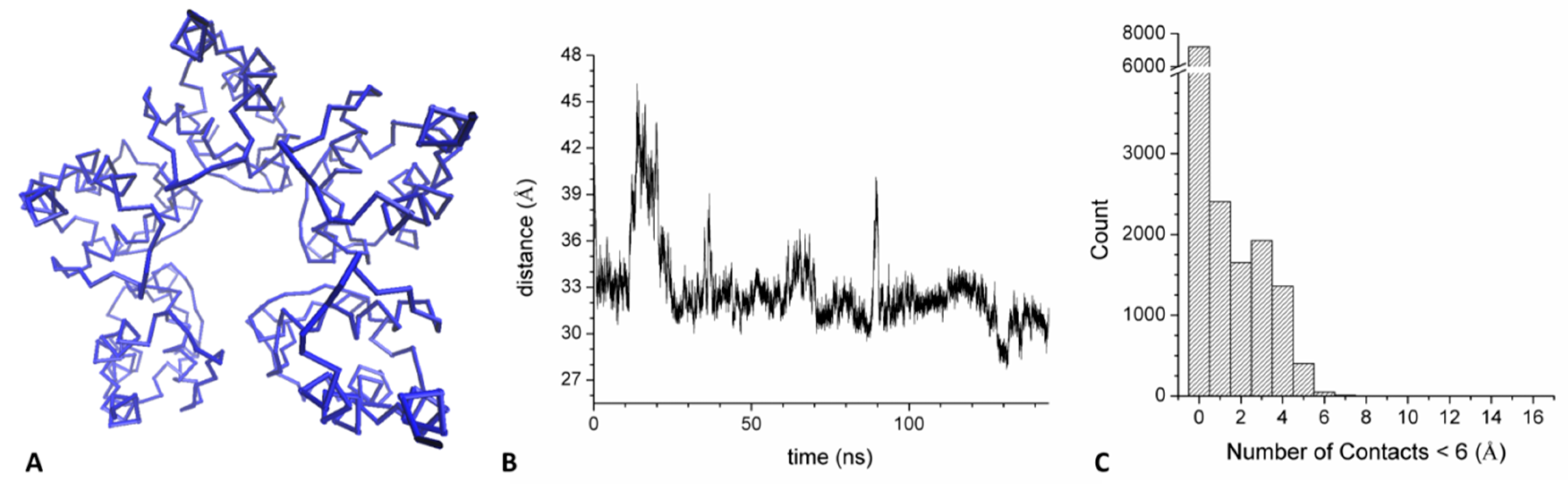
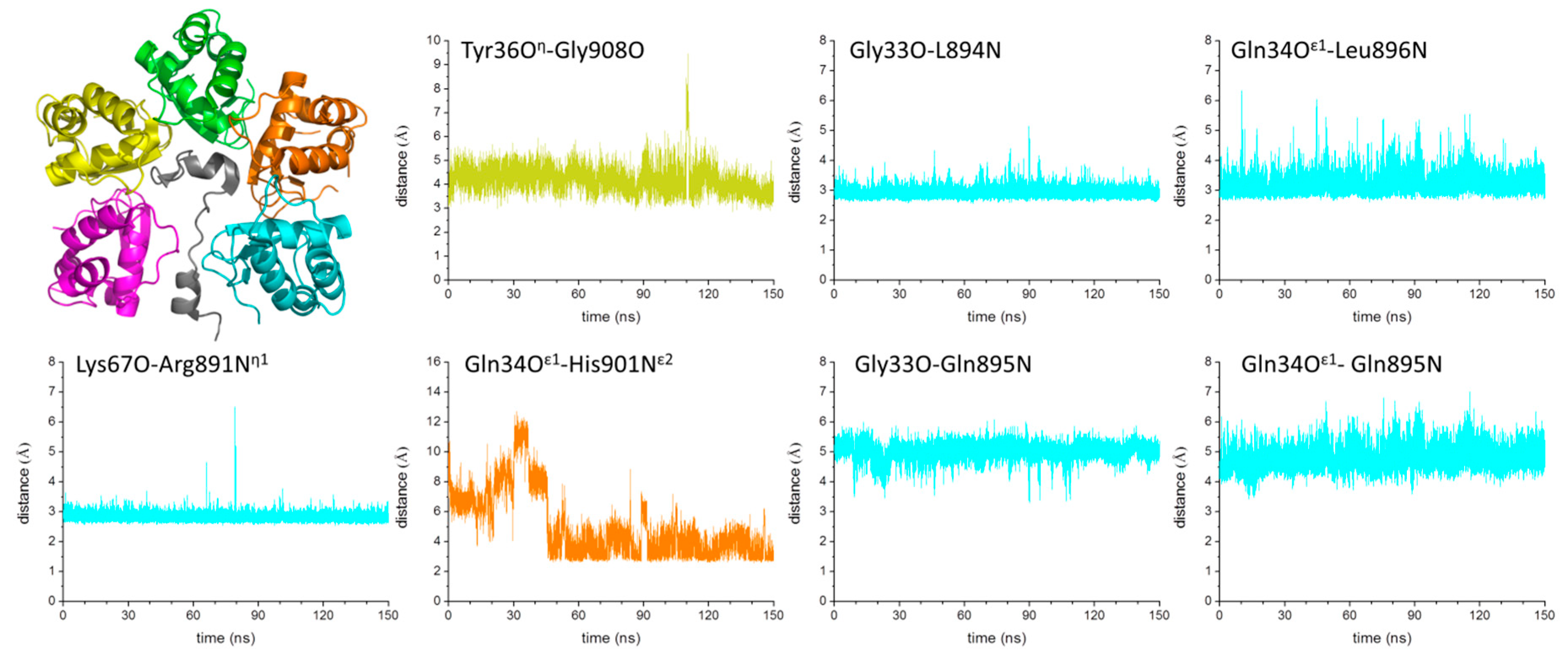
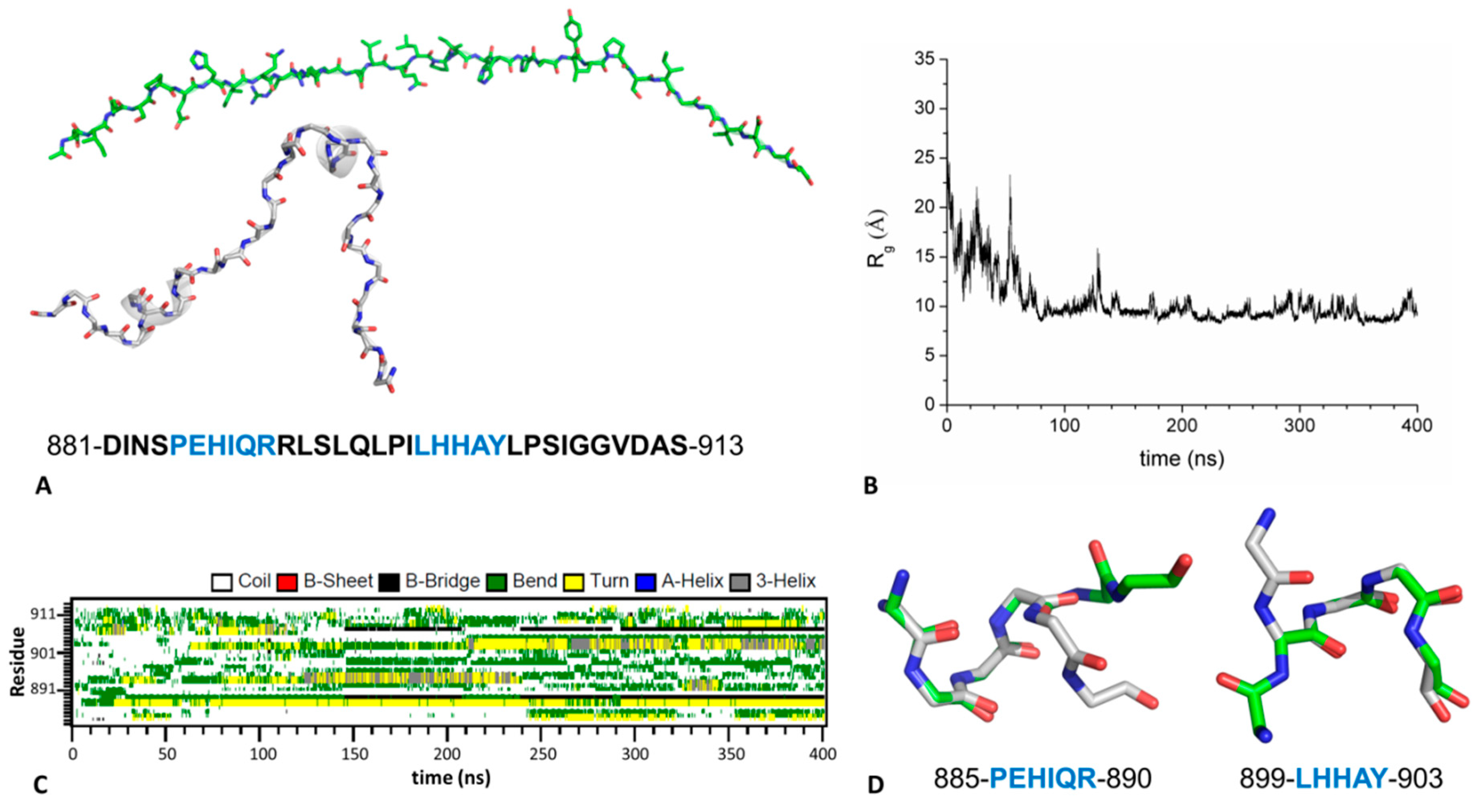
2.4. Mechanism of GABAB2R Recognition by KCTD16
3. Discussion
4. Material and Methods
4.1. Notations and System
4.2. MD Protocol
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Z.; Xiang, Y.; Sun, G. The KCTD family of proteins: Structure, function, disease relevance. Cell Biosci. 2013, 3, 45. [Google Scholar] [CrossRef]
- Skoblov, M.; Marakhonov, A.; Marakasova, E.; Guskova, A.; Chandhoke, V.; Birerdinc, A.; Baranova, A. Protein partners of KCTD proteins provide insights about their functional roles in cell differentiation and vertebrate development. BioEssays 2013, 35, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin Mediates Degradation of RhoA through Evolutionarily Conserved BTB Adaptors to Control Actin Cytoskeleton Structure and Cell Movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Aouacheria, A.; Lionnard, L.; Metz, K.A.; Soane, L.; Kamiya, A.; Hardwick, J.M. KCTD: A new gene family involved in neurodevelopmental and neuropsychiatric disorders. CNS Neurosci. Ther. 2019, 25, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, M.; Blomen, V.A.; Nieuwenhuis, J.; Stickel, E.; Raaben, M.; Bleijerveld, O.B.; Altelaar, A.F.M.; Jae, L.T.; Brummelkamp, T.R. Genetic wiring maps of single-cell protein states reveal an off-switch for GPCR signalling. Nature 2017, 546, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3–RENKCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nature 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Van Bogaert, P.; Azizieh, R.; Desir, J.; Aeby, A.; De Meirleir, L.; Laes, J.-F.; Christiaens, F.; Abramowicz, M.J. Mutation of a potassium channel-related gene in progressive myoclonic epilepsy. Ann. Neurol. 2007, 61, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Pirone, L.; Smaldone, G.; Esposito, C.; Balasco, N.; Petoukhov, M.V.; Spilotros, A.; Svergun, D.I.; Di Gaetano, S.; Vitagliano, L.; Pedone, E.M. Proteins involved in sleep homeostasis: Biophysical characterization of INC and its partners. Biochimie 2016, 131, 106–114. [Google Scholar] [CrossRef]
- Li, Q.; Kellner, D.A.; Hatch, H.A.M.; Yumita, T.; Sanchez, S.; Machold, R.P.; Frank, C.A.; Stavropoulos, N. Conserved properties of Drosophila Insomniac link sleep regulation and synaptic function. PLoS Genet. 2017, 13, e1006815. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffenberger, C.; Allada, R. Cul3 and the BTB Adaptor Insomniac Are Key Regulators of Sleep Homeostasis and a Dopamine Arousal Pathway in Drosophila. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Schwenk, J.; Metz, M.; Zolles, G.; Turecek, R.; Fritzius, T.; Bildl, W.; Tarusawa, E.; Kulik, A.; Unger, A.; Ivankova, K.; et al. Native GABAB receptors are heteromultimers with a family of auxiliary subunits. Nature 2010, 465, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Escamilla, C.O.; Filonova, I.; Walker, A.K.; Xuan, Z.X.; Holehonnur, R.; Espinosa, F.; Liu, S.; Thyme, S.B.; López-García, I.A.; Mendoza, D.B.; et al. Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 2017, 551, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Golzio, C.; Willer, J.; Talkowski, M.E.; Oh, E.C.; Taniguchi, Y.; Jacquemont, S.; Reymond, A.; Sun, M.; Sawa, A.; Gusella, J.F.; et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012, 485, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, G.; Pirone, L.; Capolupo, A.; Vitagliano, L.; Monti, M.C.; Di Gaetano, S.; Pedone, E. The essential player in adipogenesis GRP78 is a novel KCTD15 interactor. Int. J. Boil. Macromol. 2018, 115, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Maekawa, M.; Kawai, K.; Nakayama, J.; Araki, N.; Semba, K.; Taguchi, T.; Kamei, Y.; Takada, Y.; Higashiyama, S. Cullin-3/KCTD10 E3 complex is essential for Rac1 activation through RhoB degradation in human epidermal growth factor receptor 2-positive breast cancer cells. Cancer Sci. 2019, 110, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Marneros, A.G.; Beck, A.E.; Turner, E.H.; McMillin, M.J.; Edwards, M.J.; Field, M.; Sobreira, N.L.D.M.; Perez, A.B.A.; Fortes, J.A.; Lampe, A.K.; et al. Mutations in KCTD1 Cause Scalp-Ear-Nipple Syndrome. Am. J. Hum. Genet. 2013, 92, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Smaldone, G.; Balasco, N.; Pirone, L.; Caruso, D.; Di Gaetano, S.; Pedone, E.M.; Vitagliano, L. Molecular basis of the scalp-ear-nipple syndrome unraveled by the characterization of disease-causing KCTD1 mutants. Sci. Rep. 2019, 9, 10519. [Google Scholar] [CrossRef]
- Stogios, P.J.; Downs, G.S.; Jauhal, J.J.S.; Nandra, S.K.; Privé, G.G. Sequence and structural analysis of BTB domain proteins. Genome Boil. 2005, 6, R82. [Google Scholar] [CrossRef]
- Balasco, N.; Pirone, L.; Smaldone, G.; Di Gaetano, S.; Esposito, L.; Pedone, E.M.; Vitagliano, L. Molecular recognition of Cullin3 by KCTDs: Insights from experimental and computational investigations. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1289–1298. [Google Scholar] [CrossRef]
- Smaldone, G.; Pirone, L.; Balasco, N.; Di Gaetano, S.; Pedone, E.M.; Vitagliano, L. Cullin 3 Recognition Is Not a Universal Property among KCTD Proteins. PLoS ONE 2015, 10, e0126808. [Google Scholar] [CrossRef]
- Pinkas, D.M.; Sanvitale, C.E.; Bufton, J.C.; Sorrell, F.J.; Solcan, N.; Chalk, R.; Doutch, J.; Bullock, A.N. Structural complexity in the KCTD family of Cullin3-dependent E3 ubiquitin ligases. Biochem. J. 2017, 474, 3747–3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarelli, V.E.; Dawid, I.B. Inhibition of neural crest formation by Kctd15 involves regulation of transcription factor AP-2. Proc. Natl. Acad. Sci. USA 2013, 110, 2870–2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dementieva, I.S.; Tereshko, V.; McCrossan, Z.A.; Solomaha, E.; Araki, D.; Xu, C.; Grigorieff, N.; Goldstein, S.A.N. Pentameric Assembly of Potassium Channel Tetramerization Domain-Containing Protein 5 (KCTD5). J. Mol. Boil. 2009, 387, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, G.; Pirone, L.; Pedone, E.; Marlovits, T.; Vitagliano, L.; Ciccarelli, L. The BTB domains of the potassium channel tetramerization domain proteins prevalently assume pentameric states. FEBS Lett. 2016, 590, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Barone, D.; Balasco, N.; Vitagliano, L. KCTD5 is endowed with large, functionally relevant, interdomain motions. J. Biomol. Struct. Dyn. 2016, 34, 1725–1735. [Google Scholar] [CrossRef]
- Zheng, S.; Abreu, N.; Levitz, J.; Kruse, A.C. Structural basis for KCTD-mediated rapid desensitization of GABAB signalling. Nature 2019, 567, 127–131. [Google Scholar] [CrossRef]
- Ji, A.X.; Chu, A.; Nielsen, T.K.; Benlekbir, S.; Rubinstein, J.L.; Privé, G.G. Structural Insights into KCTD Protein Assembly and Cullin3 Recognition. J. Mol. Boil. 2016, 428, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Zuo, H.; Glaaser, I.; Zhao, Y.; Kurinov, I.; Mosyak, L.; Wang, H.; Liu, J.; Park, J.; Frangaj, A.; Sturchler, E.; et al. Structural basis for auxiliary subunit KCTD16 regulation of the GABAB receptor. Proc. Natl. Acad. Sci. USA 2019, 116, 8370–8379. [Google Scholar] [CrossRef]
- De Paola, I.; Pirone, L.; Palmieri, M.; Balasco, N.; Esposito, L.; Russo, L.; Mazzà, D.; Di Marcotullio, L.; Di Gaetano, S.; Malgieri, G.; et al. Cullin3—BTB Interface: A Novel Target for Stapled Peptides. PLoS ONE 2015, 10, e0121149. [Google Scholar] [CrossRef]
- Fritzius, T.; Turecek, R.; Seddik, R.; Kobayashi, H.; Tiao, J.; Rem, P.D.; Metz, M.; Kralikova, M.; Bouvier, M.; Gassmann, M.; et al. KCTD Hetero-oligomers Confer Unique Kinetic Properties on Hippocampal GABAB Receptor-Induced K+ Currents. J. Neurosci. 2017, 37, 1162–1175. [Google Scholar] [CrossRef]
- Correale, S.; Esposito, C.; Pirone, L.; Vitagliano, L.; Di Gaetano, S.; Pedone, E.M. A biophysical characterization of the folded domains of KCTD12: insights into interaction with the GABAB2receptor. J. Mol. Recognit. 2013, 26, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.; Prive, G. The BACK domain in BTB-kelch proteins. Trends Biochem. Sci. 2004, 29, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Zollman, S.; Godt, D.; Prive, G.G.; Couderc, J.L.; Laski, F.A. The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in Drosophila. Proc. Natl. Acad. Sci. USA 1994, 91, 10717–10721. [Google Scholar] [CrossRef] [PubMed]
- Bixby, K.A.; Nanao, M.H.; Shen, N.V.; Kreusch, A.; Bellamy, H.; Pfaffinger, P.J.; Choe, S. Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+ channels. Nat. Struct. Mol. Biol. 1999, 6, 38–43. [Google Scholar]
- De Smaele, E.; Di Marcotullio, L.; Moretti, M.; Pelloni, M.; Occhione, M.A.; Infante, P.; Cucchi, D.; Greco, A.; Pietrosanti, L.; Todorovic, J.; et al. Identification and Characterization of KCASH2 and KCASH3, 2 Novel Cullin3 Adaptors Suppressing Histone Deacetylase and Hedgehog Activity in Medulloblastoma. Neoplasia 2011, 13, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Balasco, N.; Barone, D.; Iaccarino, E.; Sandomenico, A.; De Simone, A.; Ruvo, M.; Vitagliano, L. Intrinsic structural versatility of the highly conserved 412–423 epitope of the Hepatitis C Virus E2 protein. Int. J. Boil. Macromol. 2018, 116, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ma, B.; Tsai, C.J.; Sinha, N.; Nussinov, R. Folding and binding cascades: Dynamic landscapes and population shifts. Protein Sci. 2000, 9, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Amadei, A.; Ceruso, M.A.; Di Nola, A. On the convergence of the conformational coordinates basis set obtained by the essential dynamics analysis of proteins’ molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 1999, 36, 419–424. [Google Scholar] [CrossRef]
- Besker, N.; Molina, R.; Marcaida, M.J.; Montoya, G.; Prieto, J.; D’Abramo, M. Key Players in I-DmoI Endonuclease Catalysis Revealed from Structure and Dynamics. ACS Chem. Boil. 2016, 11, 1401–1407. [Google Scholar]
- Merlino, A.; Vitagliano, L.; Ceruso, M.A.; Mazzarella, L. Subtle functional collective motions in pancreatic-like ribonucleases: From ribonuclease A to angiogenin. Proteins Struct. Funct. Bioinform. 2003, 53, 101–110. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Timescale (ns) | Box Dimensions (nm3) | No. of Water Molecules | RMSIP a |
|---|---|---|---|---|
| SHKBP1BTB monomer | 200 | 5.24 × 5.20 × 7.02 | 5770 | 0.78 |
| KCTD1BTB monomer | 200 | 6.43 × 5.22 × 4.96 | 4924 | 0.80 |
| KCTD5BTB monomer | 200 | 4.83 × 5.56 × 6.39 | 5158 | 0.72 |
| KCTD9BTB monomer | 200 | 5.15 × 4.91 × 6.61 | 4985 | 0.68 |
| KCTD10BTB monomer | 200 | 5.23 × 5.00 × 6.69 | 5284 | 0.82 |
| KCTD13BTB monomer | 200 | 6.89 × 5.25 × 5.16 | 5589 | 0.78 |
| KCTD16BTB monomer | 200 | 6.43 × 6.05 × 5.44 | 6318 | 0.72 |
| KCTD17BTB monomer | 200 | 5.95 × 5.23 × 6.53 | 6245 | 0.77 |
| KCTD13BTB tetramer | 150 | 7.73 × 7.46 × 9.07 | 15618 | 0.65 |
| KCTD10BTB tetramer | 150 | 7.67 × 9.06 × 7.23 | 14570 | 0.79 |
| KCTD13BTB dimer | 150 | 7.32 × 6.84 × 6.51 | 9735 | 0.78 |
| KCTD1BTB close pentamer | 150 | 8.58 × 6.87 × 8.38 | 13845 | 0.60 |
| KCTD9BTB close pentamer | 150 | 8.93 × 9.16 × 6.87 | 15988 | 0.49 |
| KCTD17BTB close pentamer | 150 | 8.73 × 9.09 × 8.13 | 18724 | 0.60 |
| KCTD1BTB open pentamer | 200 | 7.65 × 9.93 × 8.47 | 18280 | 0.67 |
| KCTD16BTB open pentamer | 200 | 7.87 × 10.41 × 8.99 | 21809 | 0.72 |
| KCTD12-16BTB open pentamer | 150 | 7.83 × 9.79 × 8.99 | 19886 | 0.73 |
| KCTD16BTB-GABAB2R complex | 150 | 7.84 × 9.62 × 9.07 | 19624 | 0.66 |
| GABAB2R peptide | 400 | 11.15 × 6.10 × 4.77 | 10509 | 0.73 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balasco, N.; Smaldone, G.; Vitagliano, L. The Structural Versatility of the BTB Domains of KCTD Proteins and Their Recognition of the GABAB Receptor. Biomolecules 2019, 9, 323. https://doi.org/10.3390/biom9080323
Balasco N, Smaldone G, Vitagliano L. The Structural Versatility of the BTB Domains of KCTD Proteins and Their Recognition of the GABAB Receptor. Biomolecules. 2019; 9(8):323. https://doi.org/10.3390/biom9080323
Chicago/Turabian StyleBalasco, Nicole, Giovanni Smaldone, and Luigi Vitagliano. 2019. "The Structural Versatility of the BTB Domains of KCTD Proteins and Their Recognition of the GABAB Receptor" Biomolecules 9, no. 8: 323. https://doi.org/10.3390/biom9080323
APA StyleBalasco, N., Smaldone, G., & Vitagliano, L. (2019). The Structural Versatility of the BTB Domains of KCTD Proteins and Their Recognition of the GABAB Receptor. Biomolecules, 9(8), 323. https://doi.org/10.3390/biom9080323