The Early Phase of β2m Aggregation: An Integrative Computational Study Framed on the D76N Mutant and the ΔN6 Variant


 ,
,
Abstract
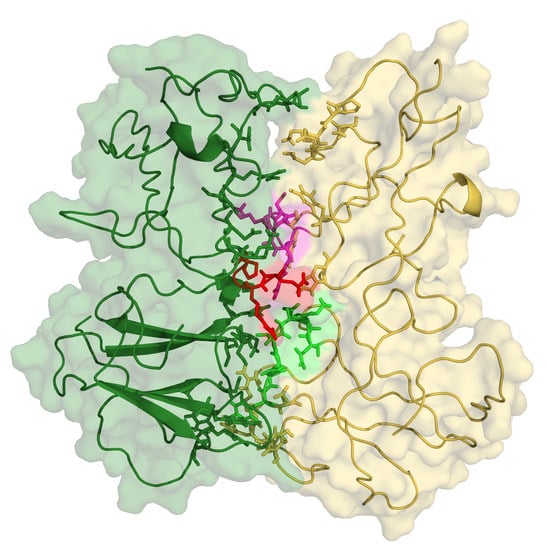
:
1. Introduction
2. Materials and Methods
2.1. Overview of the Methodological Approach
2.2. Monte Carlo Ensemble Docking
3. Results and Discussion
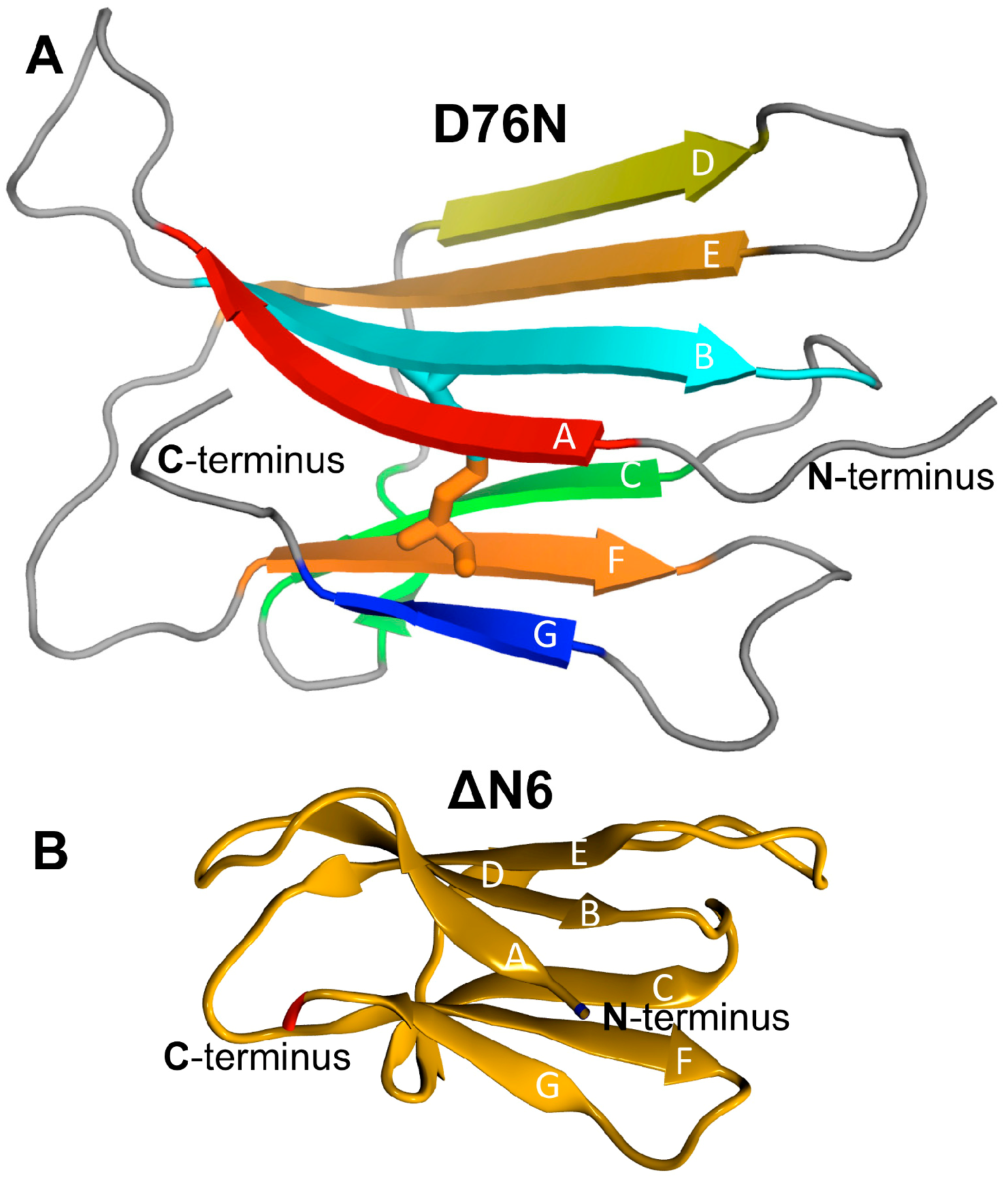
3.1. Model Systems
3.2. Dimer Stability under Different pH Conditions
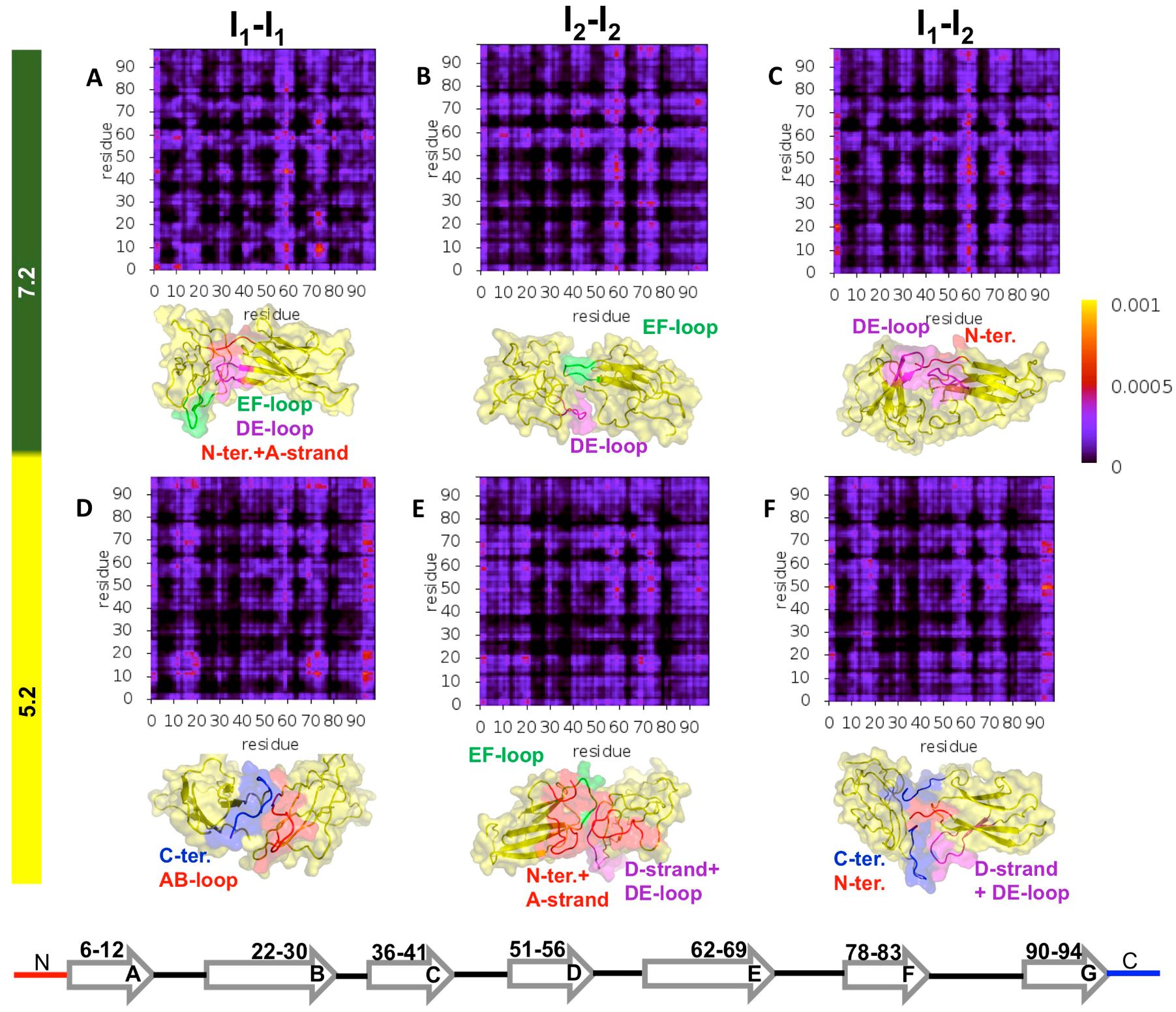
3.3. Structure of D76N Dimers under Different pH Conditions and Dimerization Hot Spots
3.4. Structure of ΔN6 Dimers under Different pH Conditions and Dimerization Hot Spots
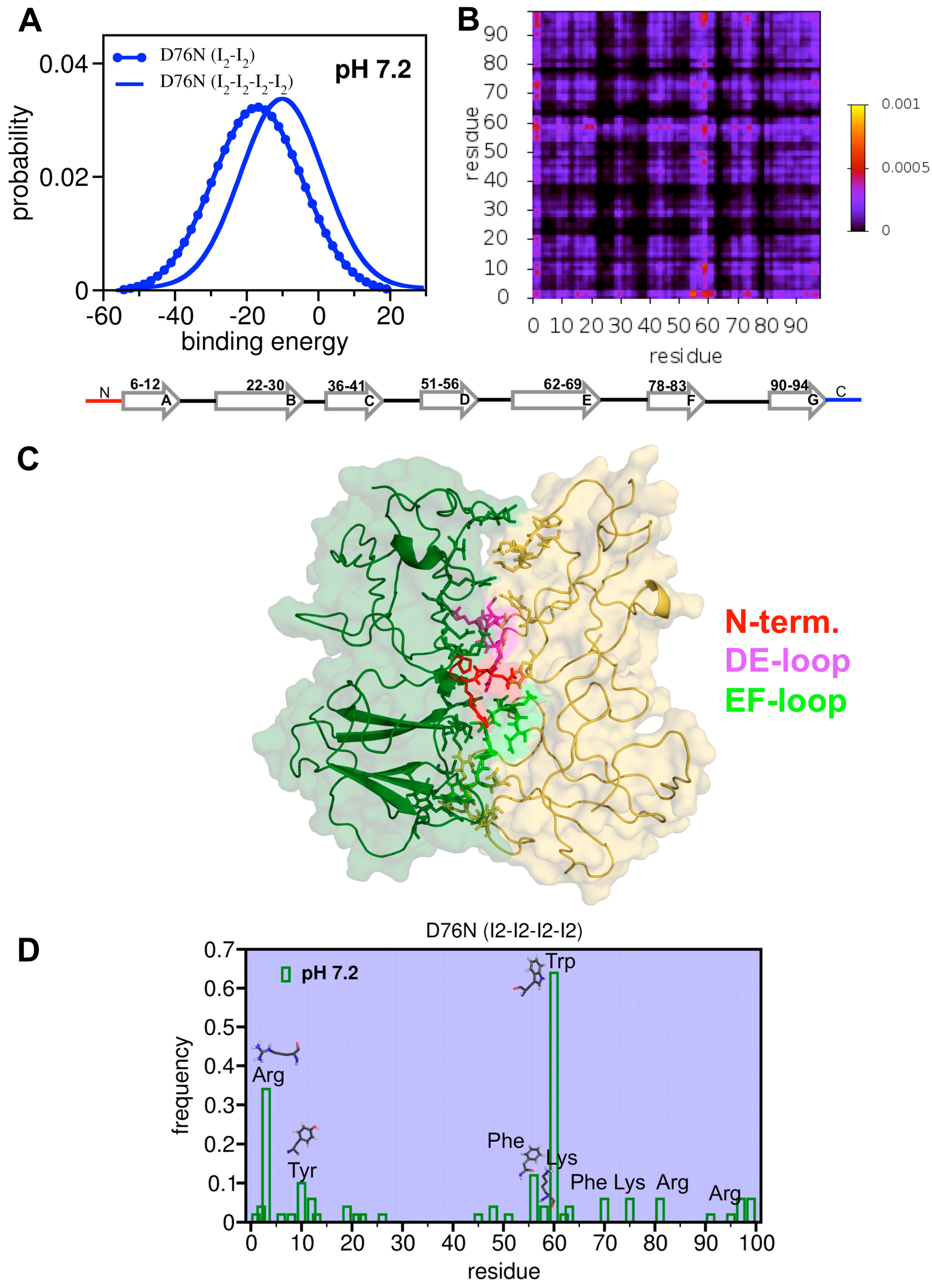
3.5. First Glimpses into the Tetrameritation Phase of D76N
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dovidchenko, N.V.; Leonova, E.I.; Galzitskaya, O.V. Mechanisms of amyloid fibril formation. Biochem. Biokhimiia 2014, 79, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Corazza, A.; Bellotti, V. Pathological Self-Aggregation of b2-Microglobulin: A Challenge for Protein Biophysics. In Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease; Harris, J.R., Ed.; Springer: Dordrecht, The Netherlands, 2012; Volume 65. [Google Scholar]
- Chong, S.H.; Hong, J.; Lim, S.; Cho, S.; Lee, J.; Ham, S. Structural and Thermodynamic Characteristics of Amyloidogenic Intermediates of beta-2-Microglobulin. Sci. Rep. 2015, 5, 13631. [Google Scholar] [CrossRef] [PubMed]
- Camilloni, C.; Sala, B.M.; Sormanni, P.; Porcari, R.; Corazza, A.; De Rosa, M.; Zanini, S.; Barbiroli, A.; Esposito, G.; Bolognesi, M.; et al. Rational design of mutations that change the aggregation rate of a protein while maintaining its native structure and stability. Sci. Rep. 2016, 6, 25559. [Google Scholar] [CrossRef] [PubMed]
- Gumral, D.; Fogolari, F.; Corazza, A.; Viglino, P.; Giorgetti, S.; Stoppini, M.; Bellotti, V.; Esposito, G. Reduction of conformational mobility and aggregation in W60G beta2-microglobulin: Assessment by 15N NMR relaxation. Magn. Reson. Chem. MRC 2013, 51, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Estacio, S.G.; Shakhnovich, E.I.; Faisca, P.F. Assessing the effect of loop mutations in the folding space of beta2-microglobulin with molecular dynamics simulations. Int. J. Mol. Sci. 2013, 14, 17256–17278. [Google Scholar] [CrossRef] [PubMed]
- Narang, S.S.; Shuaib, S.; Goyal, D.; Goyal, B. Assessing the effect of D59P mutation in the DE loop region in amyloid aggregation propensity of beta2-microglobulin: A molecular dynamics simulation study. J. Cell. Biochem. 2018, 119, 782–792. [Google Scholar] [CrossRef]
- Natalello, A.; Relini, A.; Penco, A.; Halabelianw, L.; Bolognesi, M.; Doglia, S.M.; Ricagno, S. Wild type beta-2 microglobulin and DE loop mutants display a common fibrillar architecture. PLoS ONE 2015, 10, e0122449. [Google Scholar] [CrossRef]
- Santambrogio, C.; Ricagno, S.; Colombo, M.; Barbiroli, A.; Bonomi, F.; Bellotti, V.; Bolognesi, M.; Grandori, R. DE-loop mutations affect beta2 microglobulin stability, oligomerization, and the low-pH unfolded form. Protein Sci. A Publ. Protein Soc. 2010, 19, 1386–1394. [Google Scholar] [CrossRef]
- Esposito, G.; Ricagno, S.; Corazza, A.; Rennella, E.; Gumral, D.; Mimmi, M.C.; Betto, E.; Pucillo, C.E.; Fogolari, F.; Viglino, P.; et al. The controlling roles of Trp60 and Trp95 in beta2-microglobulin function, folding and amyloid aggregation properties. J. Mol. Biol. 2008, 378, 887–897. [Google Scholar] [CrossRef]
- Ricagno, S.; Colombo, M.; de Rosa, M.; Sangiovanni, E.; Giorgetti, S.; Raimondi, S.; Bellotti, V.; Bolognesi, M. DE loop mutations affect beta2-microglobulin stability and amyloid aggregation. Biochem. Biophys. Res. Commun. 2008, 377, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Ricagno, S.; Raimondi, S.; Giorgetti, S.; Bellotti, V.; Bolognesi, M. Human beta-2 microglobulin W60V mutant structure: Implications for stability and amyloid aggregation. Biochem. Biophys. Res. Commun. 2009, 380, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Kihara, M.; Chatani, E.; Iwata, K.; Yamamoto, K.; Matsuura, T.; Nakagawa, A.; Naiki, H.; Goto, Y. Conformation of amyloid fibrils of beta2-microglobulin probed by tryptophan mutagenesis. J. Biol. Chem. 2006, 281, 31061–31069. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.F.; Eakin, C.M.; Wang, J.M.; Miranker, A.D. A regulatable switch mediates self-association in an immunoglobulin fold. Nat. Struct. Mol. Biol. 2008, 15, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Blaho, D.V.; Miranker, A.D. Delineating the conformational elements responsible for Cu(2+)-induced oligomerization of beta-2 microglobulin. Biochemistry 2009, 48, 6610–6617. [Google Scholar] [CrossRef]
- Rosano, C.; Zuccotti, S.; Mangione, P.; Giorgetti, S.; Bellotti, V.; Pettirossi, F.; Corazza, A.; Viglino, P.; Esposito, G.; Bolognesi, M. beta2-microglobulin H31Y variant 3D structure highlights the protein natural propensity towards intermolecular aggregation. J. Mol. Biol. 2004, 335, 1051–1064. [Google Scholar] [CrossRef]
- Esposito, G.; Corazza, A.; Viglino, P.; Verdone, G.; Pettirossi, F.; Fogolari, F.; Makek, A.; Giorgetti, S.; Mangione, P.; Stoppini, M.; et al. Solution structure of beta(2)-microglobulin and insights into fibrillogenesis. Biochim. Biophys. Acta 2005, 1753, 76–84. [Google Scholar] [CrossRef]
- Eakin, C.M.; Berman, A.J.; Miranker, A.D. A native to amyloidogenic transition regulated by a backbone trigger. Nat. Struct. Mol. Biol. 2006, 13, 202–208. [Google Scholar] [CrossRef]
- Ma, B.; Nussinov, R. Molecular dynamics simulations of the unfolding of beta(2)-microglobulin and its variants. Protein Eng. 2003, 16, 561–575. [Google Scholar] [CrossRef]
- Heegaard, N.H.; Jorgensen, T.J.; Cheng, L.; Schou, C.; Nissen, M.H.; Trapp, O. Interconverting conformations of variants of the human amyloidogenic protein beta2-microglobulin quantitatively characterized by dynamic capillary electrophoresis and computer simulation. Anal. Chem. 2006, 78, 3667–3673. [Google Scholar] [CrossRef]
- Heegaard, N.H.; Jorgensen, T.J.; Rozlosnik, N.; Corlin, D.B.; Pedersen, J.S.; Tempesta, A.G.; Roepstorff, P.; Bauer, R.; Nissen, M.H. Unfolding, aggregation, and seeded amyloid formation of lysine-58-cleaved beta 2-microglobulin. Biochemistry 2005, 44, 4397–4407. [Google Scholar] [CrossRef]
- Corlin, D.B.; Sen, J.W.; Ladefoged, S.; Lund, G.B.; Nissen, M.H.; Heegaard, N.H. Quantification of cleaved beta2-microglobulin in serum from patients undergoing chronic hemodialysis. Clin. Chem. 2005, 51, 1177–1184. [Google Scholar] [CrossRef]
- Heegaard, N.H.; Roepstorff, P.; Melberg, S.G.; Nissen, M.H. Cleaved beta 2-microglobulin partially attains a conformation that has amyloidogenic features. J. Biol. Chem. 2002, 277, 11184–11189. [Google Scholar] [CrossRef]
- Estacio, S.G.; Krobath, H.; Vila-Vicosa, D.; Machuqueiro, M.; Shakhnovich, E.I.; Faisca, P.F. A simulated intermediate state for folding and aggregation provides insights into DeltaN6 beta2-microglobulin amyloidogenic behavior. PLoS Comput. Biol. 2014, 10, e1003606. [Google Scholar] [CrossRef]
- Fang, P.-S.; Zhao, J.-H.; Liu, H.-L.; Liu, K.-T.; Chen, J.-T.; Tsai, W.-B.; Lin, H.-Y.; Fang, H.-W.; Ho, Y. Molecular dynamics simulations to investigate the relationship between the structural stability and amyloidogenesis of the wild-type and N-terminal hexapeptide deletion ΔN6 β2-microglobulin. Mol. Simul. 2009, 35, 755–765. [Google Scholar] [CrossRef]
- Hall, Z.; Schmidt, C.; Politis, A. Uncovering the Early Assembly Mechanism for Amyloidogenic beta2-Microglobulin Using Cross-linking and Native Mass Spectrometry. J. Biol. Chem. 2016, 291, 4626–4637. [Google Scholar] [CrossRef]
- Esposito, G.; Michelutti, R.; Verdone, G.; Viglino, P.; Hernandez, H.; Robinson, C.V.; Amoresano, A.; Dal Piaz, F.; Monti, M.; Pucci, P.; et al. Removal of the N-terminal hexapeptide from human beta2-microglobulin facilitates protein aggregation and fibril formation. Protein Sci. A Publ. Protein Soc. 2000, 9, 831–845. [Google Scholar] [CrossRef]
- Eichner, T.; Kalverda, A.P.; Thompson, G.S.; Homans, S.W.; Radford, S.E. Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 2011, 41, 161–172. [Google Scholar] [CrossRef]
- Domanska, K.; Vanderhaegen, S.; Srinivasan, V.; Pardon, E.; Dupeux, F.; Marquez, J.A.; Giorgetti, S.; Stoppini, M.; Wyns, L.; Bellotti, V.; et al. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic beta2-microglobulin variant. Proc. Natl. Acad. Sci. USA 2011, 108, 1314–1319. [Google Scholar] [CrossRef]
- Bellotti, V.; Gallieni, M.; Giorgetti, S.; Brancaccio, D. Dynamic of beta(2)-microglobulin fibril formation and reabsorption: The role of proteolysis. Semin. Dial. 2001, 14, 117–122. [Google Scholar] [CrossRef]
- Mangione, P.P.; Esposito, G.; Relini, A.; Raimondi, S.; Porcari, R.; Giorgetti, S.; Corazza, A.; Fogolari, F.; Penco, A.; Goto, Y.; et al. Structure, folding dynamics, and amyloidogenesis of D76N beta2-microglobulin: Roles of shear flow, hydrophobic surfaces, and alpha-crystallin. J. Biol. Chem. 2013, 288, 30917–30930. [Google Scholar] [CrossRef]
- Loureiro, R.J.S.; Vila-Vicosa, D.; Machuqueiro, M.; Shakhnovich, E.I.; Faisca, P.F.N. A tale of two tails: The importance of unstructured termini in the aggregation pathway of beta2-microglobulin. Proteins 2017. [Google Scholar] [CrossRef]
- Le Marchand, T.; de Rosa, M.; Salvi, N.; Sala, B.M.; Andreas, L.B.; Barbet-Massin, E.; Sormanni, P.; Barbiroli, A.; Porcari, R.; Sousa Mota, C.; et al. Conformational dynamics in crystals reveal the molecular bases for D76N beta-2 microglobulin aggregation propensity. Nat. Commun. 2018, 9, 1658. [Google Scholar] [CrossRef]
- Cohen, S.I.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef]
- Rennella, E.; Cutuil, T.; Schanda, P.; Ayala, I.; Gabel, F.; Forge, V.; Corazza, A.; Esposito, G.; Brutscher, B. Oligomeric states along the folding pathways of beta2-microglobulin: Kinetics, thermodynamics, and structure. J. Mol. Biol. 2013, 425, 2722–2736. [Google Scholar] [CrossRef]
- Eichner, T.; Radford, S.E. A generic mechanism of beta2-microglobulin amyloid assembly at neutral pH involving a specific proline switch. J. Mol. Biol. 2009, 386, 1312–1326. [Google Scholar] [CrossRef]
- Fabian, H.; Gast, K.; Laue, M.; Misselwitz, R.; Uchanska-Ziegler, B.; Ziegler, A.; Naumann, D. Early stages of misfolding and association of beta2-microglobulin: Insights from infrared spectroscopy and dynamic light scattering. Biochemistry 2008, 47, 6895–6906. [Google Scholar] [CrossRef]
- Halabelian, L.; Relini, A.; Barbiroli, A.; Penco, A.; Bolognesi, M.; Ricagno, S. A covalent homodimer probing early oligomers along amyloid aggregation. Sci. Rep. 2015, 5, 14651. [Google Scholar] [CrossRef] [Green Version]
- Antwi, K.; Mahar, M.; Srikanth, R.; Olbris, M.R.; Tyson, J.F.; Vachet, R.W. Cu(II) organizes beta-2-microglobulin oligomers but is released upon amyloid formation. Protein Sci. A Publ. Protein Soc. 2008, 17, 748–759. [Google Scholar] [CrossRef]
- White, H.E.; Hodgkinson, J.L.; Jahn, T.R.; Cohen-Krausz, S.; Gosal, W.S.; Muller, S.; Orlova, E.V.; Radford, S.E.; Saibil, H.R. Globular tetramers of beta(2)-microglobulin assemble into elaborate amyloid fibrils. J. Mol. Biol. 2009, 389, 48–57. [Google Scholar] [CrossRef]
- Estacio, S.G.; Fernandes, C.S.; Krobath, H.; Faisca, P.F.; Shakhnovich, E.I. Robustness of atomistic Go models in predicting native-like folding intermediates. J. Chem. Phys. 2012, 137, 085102. [Google Scholar] [CrossRef]
- Vila-Vicosa, D.; Campos, S.R.; Baptista, A.M.; Machuqueiro, M. Reversibility of prion misfolding: Insights from constant-pH molecular dynamics simulations. J. Chem. Phys. B 2012, 116, 8812–8821. [Google Scholar] [CrossRef]
- Krobath, H.; Estacio, S.G.; Faisca, P.F.; Shakhnovich, E.I. Identification of a conserved aggregation-prone intermediate state in the folding pathways of Spc-SH3 amyloidogenic variants. J. Mol. Biol. 2012, 422, 705–722. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Kinoshita, K.; Nakamura, H. Analyses of homo-oligomer interfaces of proteins from the complementarity of molecular surface, electrostatic potential and hydrophobicity. Protein Eng. Des. Sel. PEDS 2006, 19, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Thornton, J.M. Protein-protein interactions: A review of protein dimer structures. Prog. Biophys. Mol. Biol. 1995, 63, 31–65. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef]
- Urbanc, B.; Borreguero, J.M.; Cruz, L.; Stanley, H.E. Ab initio discrete molecular dynamics approach to protein folding and aggregation. Methods Enzymol. 2006, 412, 314–338. [Google Scholar] [CrossRef]
- Urbanc, B.; Cruz, L.; Yun, S.; Buldyrev, S.V.; Bitan, G.; Teplow, D.B.; Stanley, H.E. In silico study of amyloid beta-protein folding and oligomerization. Proc. Natl. Acad. Sci. USA 2004, 101, 17345–17350. [Google Scholar] [CrossRef]
- Baker, E.N.; Hubbard, R.E. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 1984, 44, 97–179. [Google Scholar] [CrossRef]
- Kortemme, T.; Morozov, A.V.; Baker, D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J. Mol. Biol. 2003, 326, 1239–1259. [Google Scholar] [CrossRef]
- Ding, F.; Buldyrev, S.V.; Dokholyan, N.V. Folding Trp-cage to NMR resolution native structure using a coarse-grained protein model. Biophys. J. 2005, 88, 147–155. [Google Scholar] [CrossRef]
- Xu, D.; Tsai, C.J.; Nussinov, R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997, 10, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, D.; de Groot, B.L. Atomic contacts in protein structures. A detailed analysis of atomic radii, packing, and overlaps. Proteins 2007, 68, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.; Urbanc, B.; Cruz, L.; Bitan, G.; Teplow, D.B.; Stanley, H.E. Role of electrostatic interactions in amyloid beta-protein (A beta) oligomer formation: A discrete molecular dynamics study. Biophys. J. 2007, 92, 4064–4077. [Google Scholar] [CrossRef]
- Sheu, S.Y.; Yang, D.Y.; Selzle, H.L.; Schlag, E.W. Energetics of hydrogen bonds in peptides. Proc. Natl. Acad. Sci. USA 2003, 100, 12683–12687. [Google Scholar] [CrossRef] [Green Version]
- Brändén, C.I.; Tooze, J. Introduction to Protein Structure; Garland Pub.: Spokane, WA, USA, 1999. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Teixeira, V.H.; Cunha, C.A.; Machuqueiro, M.; Oliveira, A.S.; Victor, B.L.; Soares, C.M.; Baptista, A.M. On the use of different dielectric constants for computing individual and pairwise terms in poisson-boltzmann studies of protein ionization equilibrium. J. Phys. Chem. B 2005, 109, 14691–14706. [Google Scholar] [CrossRef]
- Gitlin, I.; Carbeck, J.D.; Whitesides, G.M. Why are proteins charged? Networks of charge-charge interactions in proteins measured by charge ladders and capillary electrophoresis. Angew. Chem. Int. Ed. Engl. 2006, 45, 3022–3060. [Google Scholar] [CrossRef]
- Luisi, D.L.; Snow, C.D.; Lin, J.J.; Hendsch, Z.S.; Tidor, B.; Raleigh, D.P. Surface salt bridges, double-mutant cycles, and protein stability: An experimental and computational analysis of the interaction of the Asp 23 side chain with the N-terminus of the N-terminal domain of the ribosomal protein l9. Biochemistry 2003, 42, 7050–7060. [Google Scholar] [CrossRef]
- Biedermann, F.; Schneider, H.J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Higueruelo, A.P.; Blundell, T.L. Comprehensive, atomic-level characterization of structurally characterized protein-protein interactions: The PICCOLO database. BMC Bioinform. 2011, 12, 313. [Google Scholar] [CrossRef]
- Cummings, M.D.; Hart, T.N.; Read, R.J. Atomic solvation parameters in the analysis of protein-protein docking results. Protein Sci. A Publ. Protein Soc. 1995, 4, 2087–2099. [Google Scholar] [CrossRef] [Green Version]
- Fauchere, J.-L.; Pliska, V. Hydrophobic parameters pi of amino-acid side chains from the partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem. 1983, 18, 369–375. [Google Scholar]
- Lesser, G.J.; Rose, G.D. Hydrophobicity of amino acid subgroups in proteins. Proteins 1990, 8, 6–13. [Google Scholar] [CrossRef]
- Becker, J.W.; Reeke, G.N., Jr. Three-dimensional structure of beta 2-microglobulin. Proc. Natl. Acad. Sci. USA 1985, 82, 4225–4229. [Google Scholar] [CrossRef]
- Valleix, S.; Gillmore, J.D.; Bridoux, F.; Mangione, P.P.; Dogan, A.; Nedelec, B.; Boimard, M.; Touchard, G.; Goujon, J.M.; Lacombe, C.; et al. Hereditary systemic amyloidosis due to Asp76Asn variant beta2-microglobulin. N. Engl. J. Med. 2012, 366, 2276–2283. [Google Scholar] [CrossRef]
- De Rosa, M.; Barbiroli, A.; Giorgetti, S.; Mangione, P.P.; Bolognesi, M.; Ricagno, S. Decoding the Structural Bases of D76N ß2-Microglobulin High Amyloidogenicity through Crystallography and Asn-Scan Mutagenesis. PLoS ONE 2015, 10, e0144061. [Google Scholar] [CrossRef]
- Karamanos, T.K.; Kalverda, A.P.; Thompson, G.S.; Radford, S.E. Visualization of transient protein-protein interactions that promote or inhibit amyloid assembly. Mol. Cell 2014, 55, 214–226. [Google Scholar] [CrossRef]
- Colombo, M.; de Rosa, M.; Bellotti, V.; Ricagno, S.; Bolognesi, M. A recurrent D-strand association interface is observed in beta-2 microglobulin oligomers. FEBS J. 2012, 279, 1131–1143. [Google Scholar] [CrossRef]
- Mendoza, V.L.; Baron-Rodriguez, M.A.; Blanco, C.; Vachet, R.W. Structural insights into the pre-amyloid tetramer of beta-2-microglobulin from covalent labeling and mass spectrometry. Biochemistry 2011, 50, 6711–6722. [Google Scholar] [CrossRef]
- Jadoul, M.; Garbar, C.; Noel, H.; Sennesael, J.; Vanholder, R.; Bernaert, P.; Rorive, G.; Hanique, G.; van Ypersele de Strihou, C. Histological prevalence of beta 2-microglobulin amyloidosis in hemodialysis: A prospective post-mortem study. Kidney Int. 1997, 51, 1928–1932. [Google Scholar] [CrossRef]
- Morgan, C.J.; Gelfand, M.; Atreya, C.; Miranker, A.D. Kidney dialysis-associated amyloidosis: A molecular role for copper in fiber formation. J. Mol. Biol. 2001, 309, 339–345. [Google Scholar] [CrossRef]
- Piazza, R.; Pierno, M.; Iacopini, S.; Mangione, P.; Esposito, G.; Bellotti, V. Micro-heterogeneity and aggregation in beta2-microglobulin solutions: Effects of temperature, pH, and conformational variant addition. Eur. Biophys. J. EBJ 2006, 35, 439–445. [Google Scholar] [CrossRef]
- Pal-Gabor, H.; Gombos, L.; Micsonai, A.; Kovacs, E.; Petrik, E.; Kovacs, J.; Graf, L.; Fidy, J.; Naiki, H.; Goto, Y.; et al. Mechanism of lysophosphatidic acid-induced amyloid fibril formation of beta(2)-microglobulin in vitro under physiological conditions. Biochemistry 2009, 48, 5689–5699. [Google Scholar] [CrossRef]
- Hasegawa, K.; Tsutsumi-Yasuhara, S.; Ookoshi, T.; Ohhashi, Y.; Kimura, H.; Takahashi, N.; Yoshida, H.; Miyazaki, R.; Goto, Y.; Naiki, H. Growth of beta(2)-microglobulin-related amyloid fibrils by non-esterified fatty acids at a neutral pH. Biochem. J. 2008, 416, 307–315. [Google Scholar] [CrossRef]
- Relini, A.; De Stefano, S.; Torrassa, S.; Cavalleri, O.; Rolandi, R.; Gliozzi, A.; Giorgetti, S.; Raimondi, S.; Marchese, L.; Verga, L.; et al. Heparin strongly enhances the formation of beta2-microglobulin amyloid fibrils in the presence of type I collagen. J. Biol. Chem. 2008, 283, 4912–4920. [Google Scholar] [CrossRef]
- Motomiya, Y.; Ando, Y.; Haraoka, K.; Sun, X.; Morita, H.; Amano, I.; Uchimura, T.; Maruyama, I. Studies on unfolded β2-microglobulin at C-terminal in dialysis-related amyloidosis. Kidney Int. 2005, 67, 314–320. [Google Scholar] [CrossRef]
- Mukaiyama, A.; Nakamura, T.; Makabe, K.; Maki, K.; Goto, Y.; Kuwajima, K. The molten globule of beta(2)-microglobulin accumulated at pH 4 and its role in protein folding. J. Mol. Biol. 2013, 425, 273–291. [Google Scholar] [CrossRef]
- Verdone, G.; Corazza, A.; Viglino, P.; Pettirossi, F.; Giorgetti, S.; Mangione, P.; Andreola, A.; Stoppini, M.; Bellotti, V.; Esposito, G. The solution structure of human beta2-microglobulin reveals the prodromes of its amyloid transition. Protein Sci. A Publ. Protein Soc. 2002, 11, 487–499. [Google Scholar] [CrossRef]
- McParland, V.J.; Kad, N.M.; Kalverda, A.P.; Brown, A.; Kirwin-Jones, P.; Hunter, M.G.; Sunde, M.; Radford, S.E. Partially unfolded states of beta(2)-microglobulin and amyloid formation in vitro. Biochemistry 2000, 39, 8735–8746. [Google Scholar] [CrossRef]
- McParland, V.J.; Kalverda, A.P.; Homans, S.W.; Radford, S.E. Structural properties of an amyloid precursor of beta(2)-microglobulin. Nat. Struct. Biol. 2002, 9, 326–331. [Google Scholar] [CrossRef]
- Corazza, A.; Pettirossi, F.; Viglino, P.; Verdone, G.; Garcia, J.; Dumy, P.; Giorgetti, S.; Mangione, P.; Raimondi, S.; Stoppini, M.; et al. Properties of some variants of human beta2-microglobulin and amyloidogenesis. J. Biol. Chem. 2004, 279, 9176–9189. [Google Scholar] [CrossRef]
- Mompean, M.; Chakrabartty, A.; Buratti, E.; Laurents, D.V. Electrostatic Repulsion Governs TDP-43 C-terminal Domain Aggregation. PLoS Biol. 2016, 14, e1002447. [Google Scholar] [CrossRef]
- Zou, Y.; Sun, Y.; Zhu, Y.; Ma, B.; Nussinov, R.; Zhang, Q. Critical Nucleus Structure and Aggregation Mechanism of the C-terminal Fragment of Copper-Zinc Superoxide Dismutase Protein. ACS Chem. Neurosci. 2016, 7, 286–296. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury, L.P., Jr. The C-terminus of the beta protein is critical in amyloidogenesis. Ann. N. Y. Acad. Sci. 1993, 695, 144–148. [Google Scholar] [CrossRef]
- Zheng, X.; Jia, L.; Hu, B.; Sun, Y.; Zhang, Y.; Gao, X.; Deng, T.; Bao, S.; Xu, L.; Zhou, J. The C-terminal amyloidogenic peptide contributes to self-assembly of Avibirnavirus viral protease. Sci. Rep. 2015, 5, 14794. [Google Scholar] [CrossRef] [Green Version]
- Patino, M.M.; Liu, J.J.; Glover, J.R.; Lindquist, S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996, 273, 622–626. [Google Scholar] [CrossRef]
- Beland, M.; Roucou, X. The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J. Neurochem. 2012, 120, 853–868. [Google Scholar] [CrossRef]
- Smaoui, M.R.; Mazza-Anthony, C.; Waldispuhl, J. Investigating Mutations to Reduce Huntingtin Aggregation by Increasing Htt-N-Terminal Stability and Weakening Interactions with PolyQ Domain. Comput. Math. Methods Med. 2016, 2016, 6247867. [Google Scholar] [CrossRef]
- Baias, M.; Smith, P.E.; Shen, K.; Joachimiak, L.A.; Zerko, S.; Kozminski, W.; Frydman, J.; Frydman, L. Structure and Dynamics of the Huntingtin Exon-1 N-Terminus: A Solution NMR Perspective. J. Am. Chem. Soc. 2017, 139, 1168–1176. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SC | SN/O | SN+ | SO− | SS |
|---|---|---|---|---|
| 18 ± 2 | −7 ± 3 | −34 ± 4 | −20 ± 8 | 18 ± 6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
J. S. Loureiro, R.; Vila-Viçosa, D.; Machuqueiro, M.; Shakhnovich, E.I.; F. N. Faísca, P. The Early Phase of β2m Aggregation: An Integrative Computational Study Framed on the D76N Mutant and the ΔN6 Variant. Biomolecules 2019, 9, 366. https://doi.org/10.3390/biom9080366
J. S. Loureiro R, Vila-Viçosa D, Machuqueiro M, Shakhnovich EI, F. N. Faísca P. The Early Phase of β2m Aggregation: An Integrative Computational Study Framed on the D76N Mutant and the ΔN6 Variant. Biomolecules. 2019; 9(8):366. https://doi.org/10.3390/biom9080366
Chicago/Turabian StyleJ. S. Loureiro, Rui, Diogo Vila-Viçosa, Miguel Machuqueiro, Eugene I. Shakhnovich, and Patrícia F. N. Faísca. 2019. "The Early Phase of β2m Aggregation: An Integrative Computational Study Framed on the D76N Mutant and the ΔN6 Variant" Biomolecules 9, no. 8: 366. https://doi.org/10.3390/biom9080366