The Proteasome Lid Triggers COP9 Signalosome Activity during the Transition of Saccharomyces cerevisiae Cells into Quiescence

 and
and 
Abstract
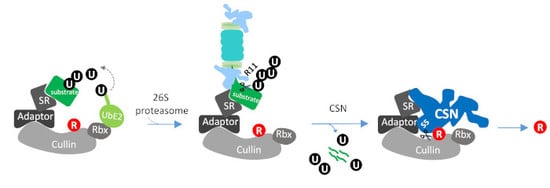
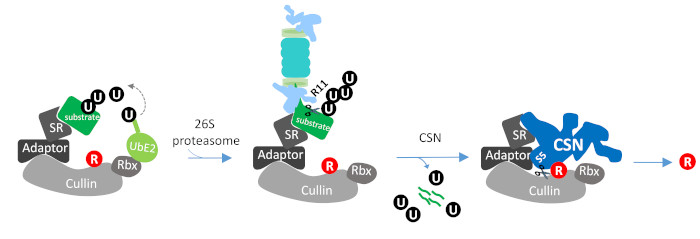
:
1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Growth Conditions
2.2. Vitality Test
2.3. Cell Harvest and Immunoblotting
2.4. Calmodulin-Based Affinity Purification
2.5. Histidine-Based Affinity Purification
2.6. Microscopy
2.6.1. Light Microscope Imaging
2.6.2. Confocal Imaging for Rpn5
2.6.3. Florescent Imaging
2.7. Antibodies
2.8. Inhibition of COP9 signalosome (CSN) Activity
3. Results and Discussion
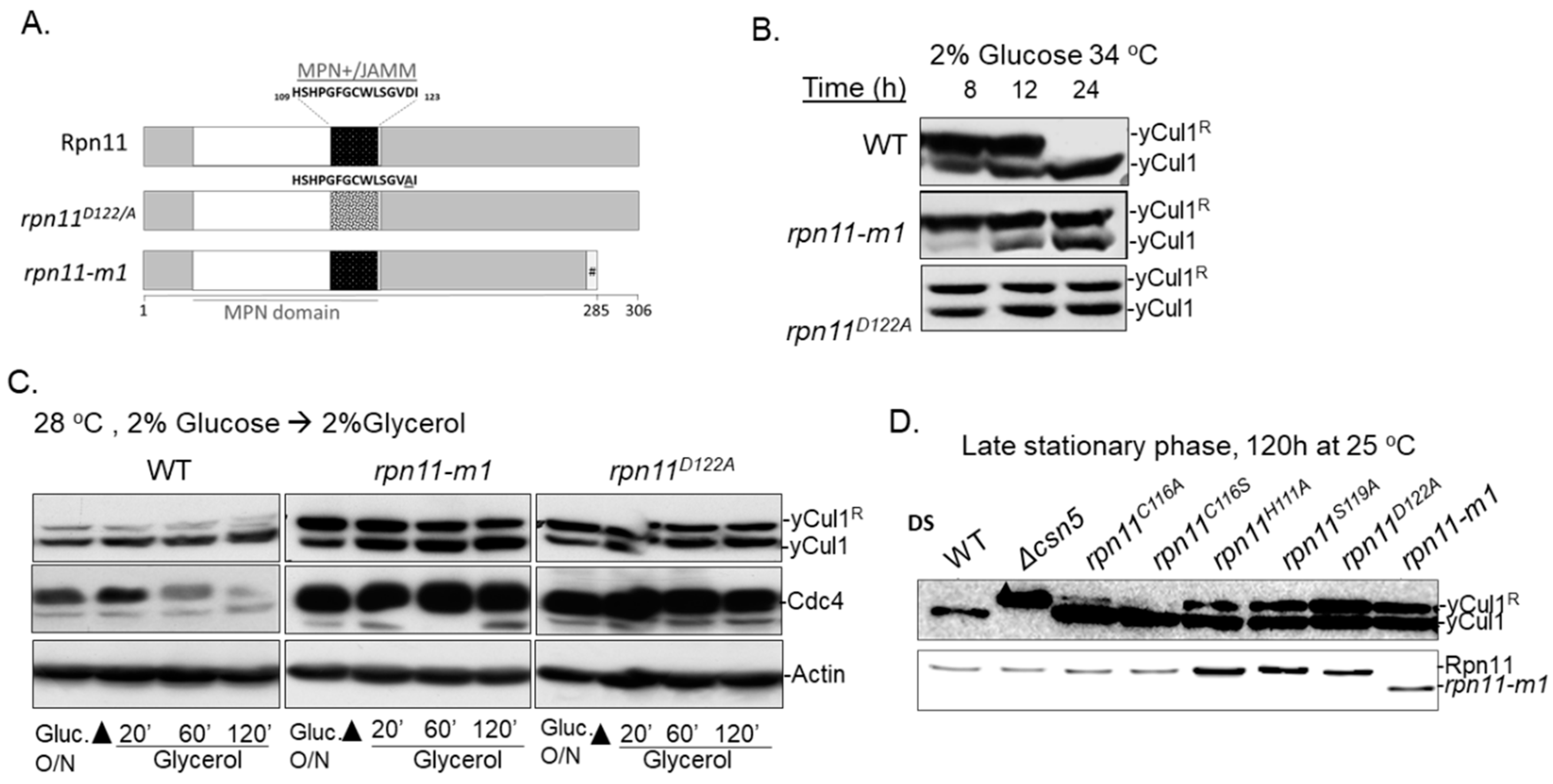
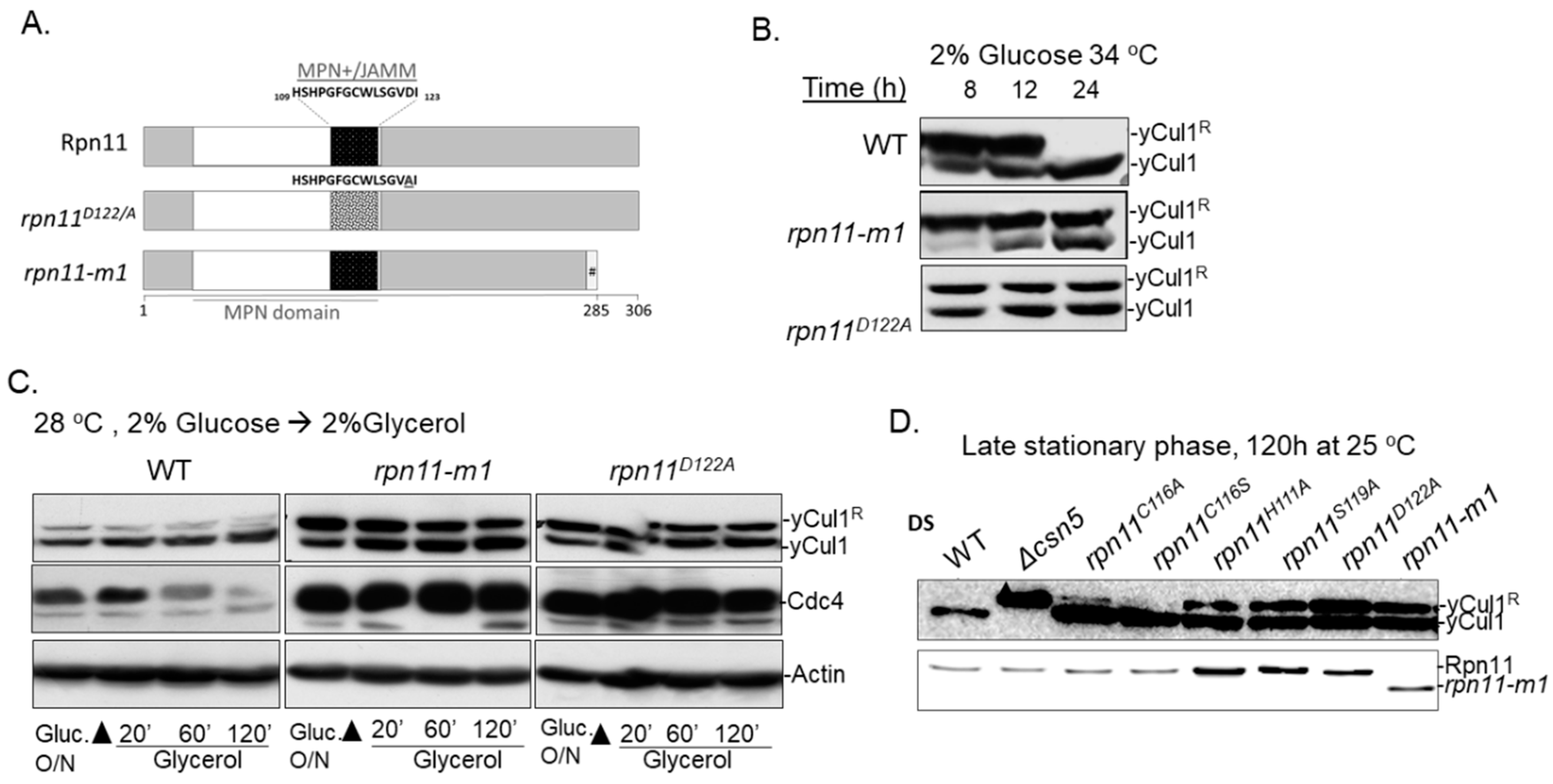
3.1. Distinct rpn11 Mutants Accumulate yCul1R
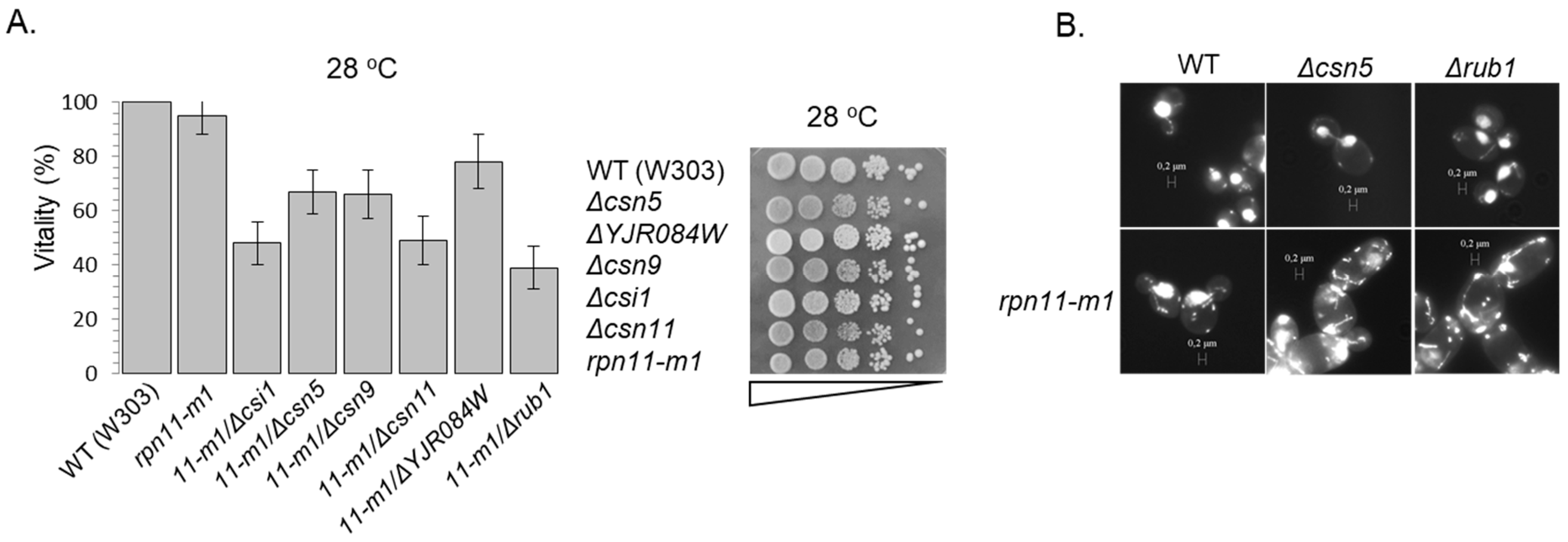
3.2. The Accumulation of yCul1R in rpn11 Mutants Is Not Associated with Cell Cycle Defects
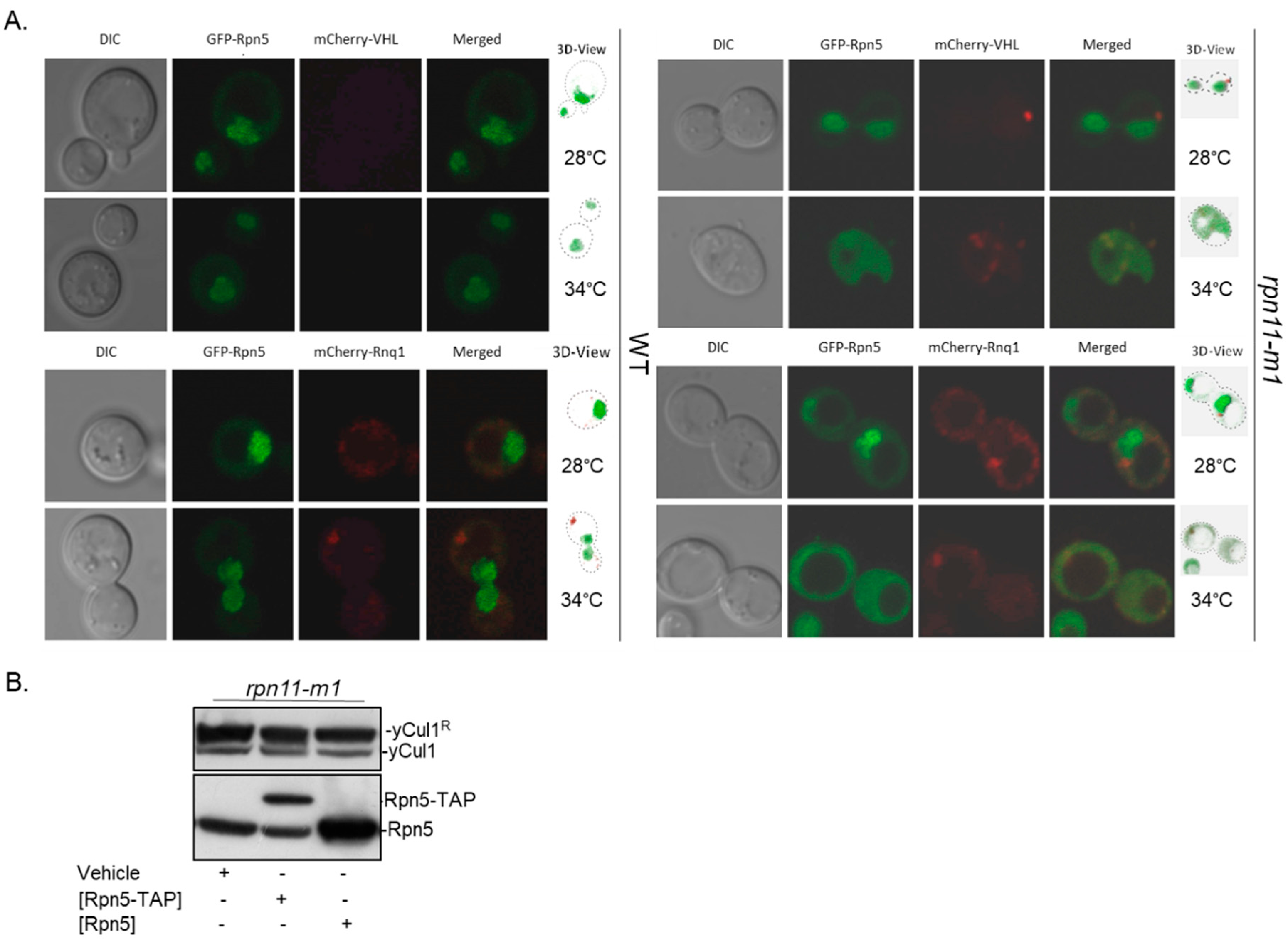
3.3. Rpn5 Availability Is Sufficient in rpn11-m1
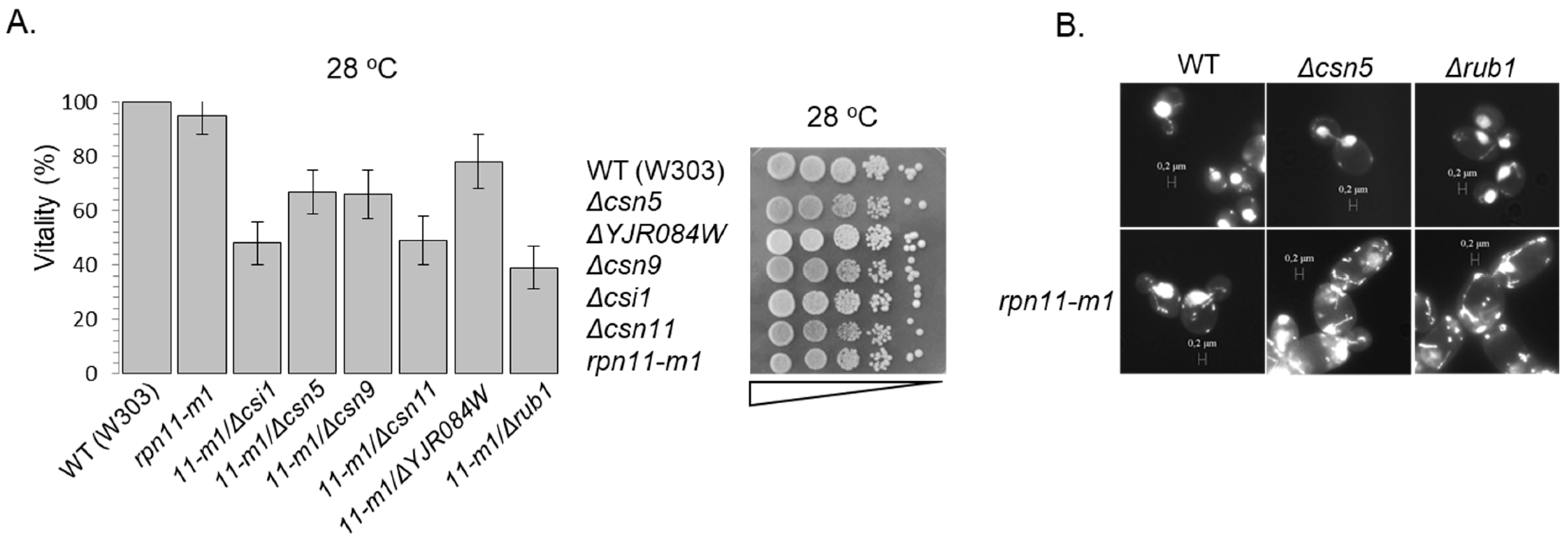
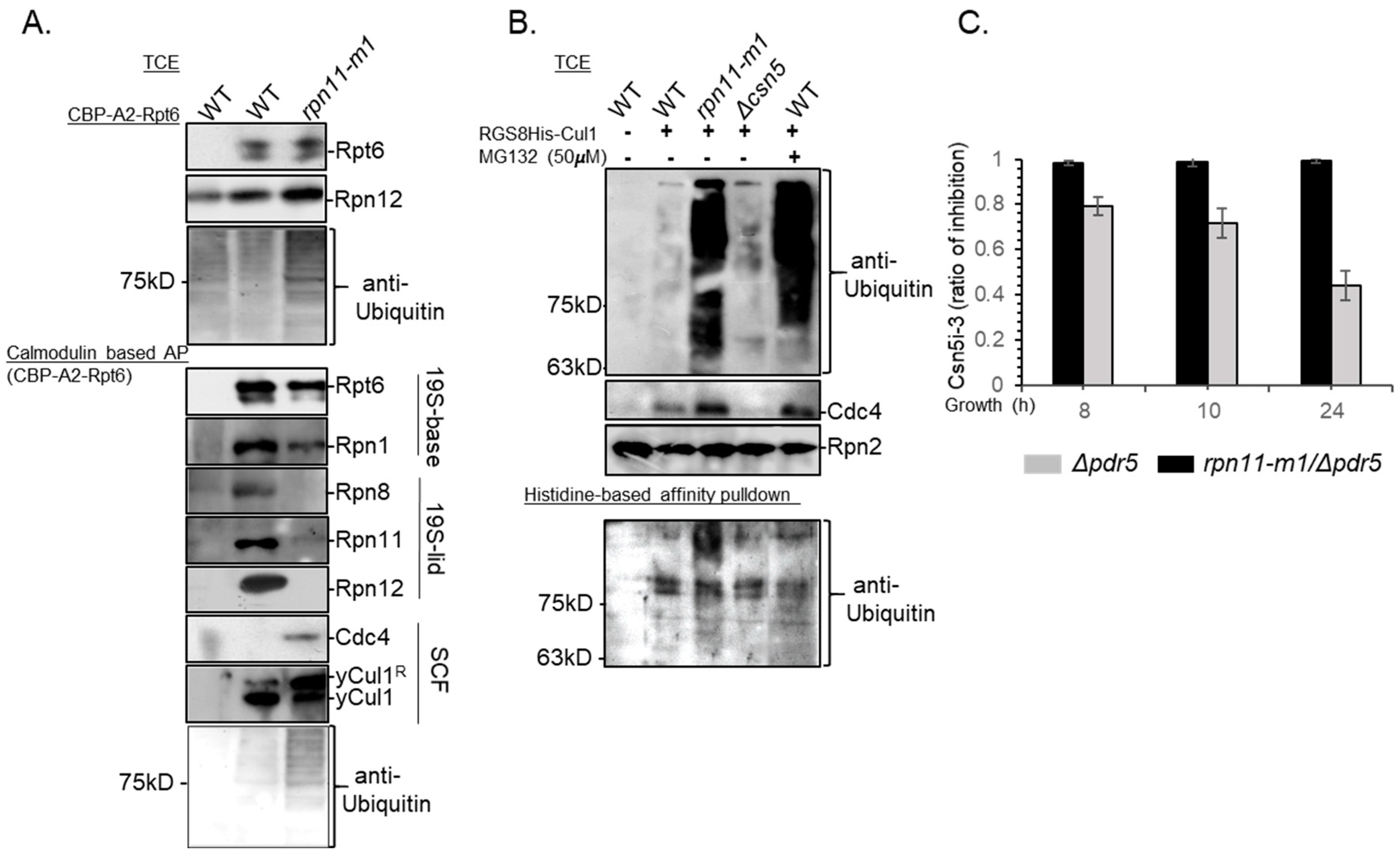
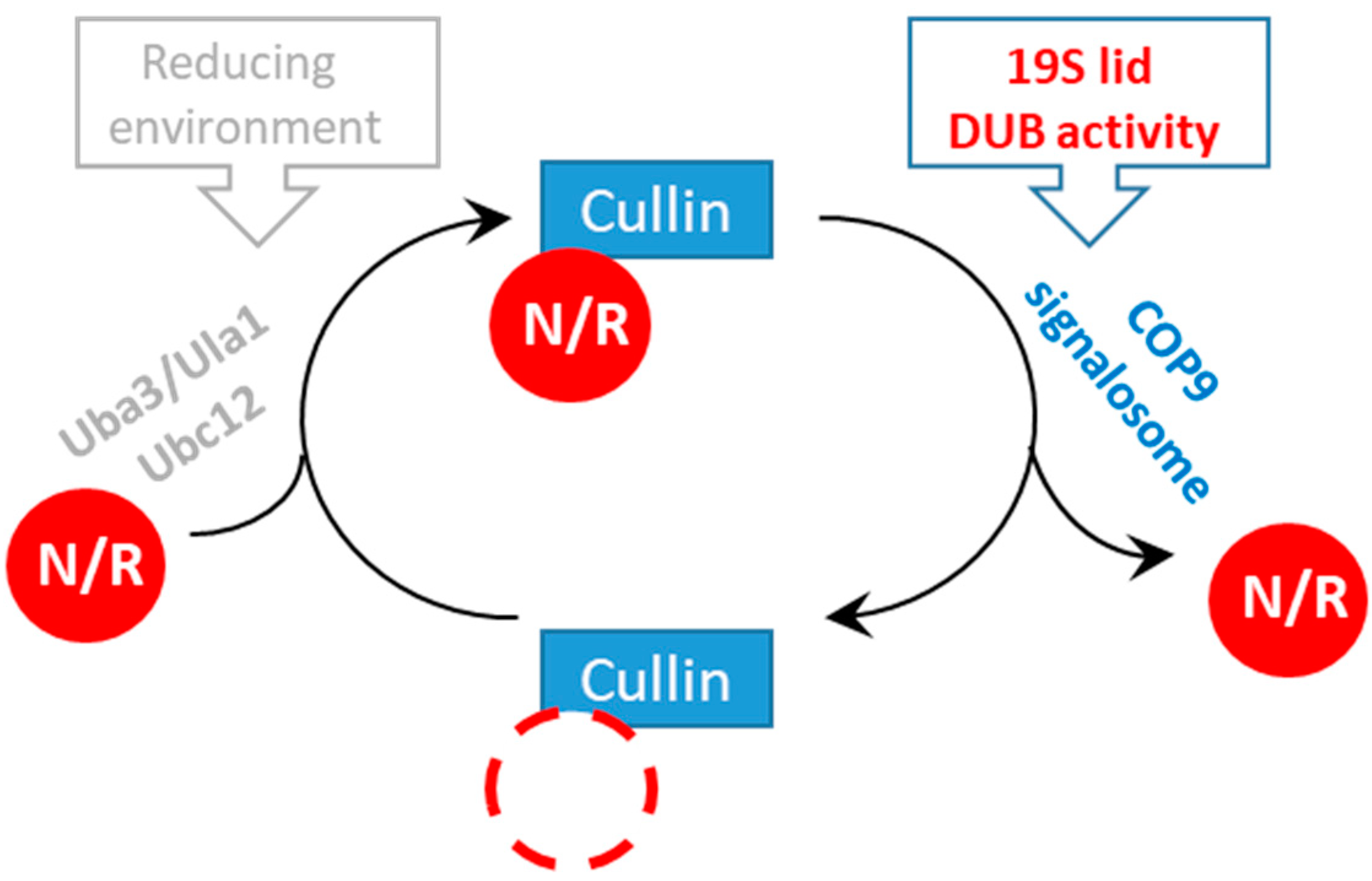
3.4. Rpn11-Mediated deubiquitinase Activity Authorizes COP9 signalosome-Mediated deNEDDylation of yCul1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van der Veen, A.G.; Ploegh, H.L. Ubiquitin-like proteins. Annu. Rev. Biochem. 2012, 81, 323–357. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A.; Varshavsky, A. The ubiquitin system. Nat. Med. 2000, 6, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Gelmann, E.P. The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. 2005, 23, 4776–4789. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Nezames, C.D.; Deng, X.W. The COP9 Signalosome: Its Regulation of Cullin-based E3 Ubiquitin Ligases and Role in Photomorphogenesis. Plant Physiol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Hotton, S.K.; Callis, J. Regulation of cullin RING ligases. Annu. Rev. Plant Biol. 2008, 59, 467–489. [Google Scholar] [CrossRef] [PubMed]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.M.; Correll, C.C.; Kaplan, K.B.; Deshaies, R.J. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 1997, 91, 221–230. [Google Scholar] [CrossRef]
- Skowyra, D.; Craig, K.L.; Tyers, M.; Elledge, S.J.; Harper, J.W. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 1997, 91, 209–219. [Google Scholar] [CrossRef]
- Verma, R.; Annan, R.; Huddleston, M.; Carr, S.; Reynard, G.; Deshaies, R.J. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 1997, 278, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.R.; Schwab, M.; Tyers, M. A hitchhiker’s guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta 2004, 1695, 133–170. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, W.; Rep, M. Lessons from fungal F-box proteins. Eukaryot. Cell 2009, 8, 677–695. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Mahon, C.; Krogan, N.J.; Craik, C.S.; Pick, E. Cullin E3 ligases and their rewiring by viral factors. Biomolecules 2014, 4, 897–930. [Google Scholar] [CrossRef]
- Whitby, F.G.; Xia, G.; Pickart, C.M.; Hill, C.P. Crystal Structure of the Human Ubiquitin-like Protein NEDD8 and Interactions with Ubiquitin Pathway Enzymes. J. Biol. Chem. 1998, 273, 34983–34991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.T.; Miller, D.W.; Mathew, R.; Cassell, R.; Holton, J.M.; Roussel, M.F.; Schulman, B.A. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat. Struct. Mol. Biol. 2004, 11, 927–935. [Google Scholar] [CrossRef]
- Calabrese, M.F.; Scott, D.C.; Duda, D.M.; Grace, C.R.; Kurinov, I.; Kriwacki, R.W.; Schulman, B.A. A RING E3-substrate complex poised for ubiquitin-like protein transfer: Structural insights into cullin-RING ligases. Nat. Struct. Mol. Biol. 2011, 18, 947–949. [Google Scholar] [CrossRef]
- Wei, N.; Serino, G.; Deng, X.W. The COP9 signalosome: More than a protease. Trends Biochem. Sci. 2008, 33, 592–600. [Google Scholar] [CrossRef]
- Dubiel, D.; Rockel, B.; Naumann, M.; Dubiel, W. Diversity of COP9 signalosome structures and functional consequences. FEBS Lett. 2015, 589, 2507–2513. [Google Scholar] [CrossRef] [Green Version]
- Lingaraju, G.M.; Bunker, R.D.; Cavadini, S.; Hess, D.; Hassiepen, U.; Renatus, M.; Fischer, E.S.; Thoma, N.H. Crystal structure of the human COP9 signalosome. Nature 2014, 512, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.S.; Scrima, A.; Bohm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Cavadini, S.; Fischer, E.S.; Bunker, R.D.; Potenza, A.; Lingaraju, G.M.; Goldie, K.N.; Mohamed, W.I.; Faty, M.; Petzold, G.; Beckwith, R.E.; et al. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 2016, 531, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Mosadeghi, R.; Reichermeier, K.M.; Winkler, M.; Schreiber, A.; Reitsma, J.M.; Zhang, Y.; Stengel, F.; Cao, J.; Kim, M.; Sweredoski, M.J.; et al. Structural and kinetic analysis of the COP9-Signalosome activation and the cullin-RING ubiquitin ligase deneddylation cycle. eLIFE 2016, 5. [Google Scholar] [CrossRef]
- Cope, G.A.; Suh, G.S.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of Predicted Metalloprotease Motif of Jab1/Csn5 in Cleavage of NEDD8 from CUL1. Science 2002, 298, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Maytal-Kivity, V.; Piran, R.; Pick, E.; Hofmann, K.; Glickman, M.H. COP9 signalosome components play a role in the mating pheromone response of S. cerevisiae. EMBO Rep. 2002, 12, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Rabut, G.; Le Dez, G.; Verma, R.; Makhnevych, T.; Knebel, A.; Kurz, T.; Boone, C.; Deshaies, R.J.; Peter, M. The TFIIH subunit Tfb3 regulates cullin neddylation. Mol. Cell 2011, 43, 488–495. [Google Scholar] [CrossRef]
- Zemla, A.; Thomas, Y.; Kedziora, S.; Knebel, A.; Wood, N.T.; Rabut, G.; Kurz, T. CSN-and CAND1-dependent remodelling of the budding yeast SCF complex. Nat. Commun. 2013, 4, 1641. [Google Scholar] [CrossRef]
- Scheel, H.; Hofmann, K. Prediction of a common structural scaffold for proteasome lid, COP9-signalosome and eIF3 complexes. BMC Bioinform. 2005, 6, 71. [Google Scholar] [CrossRef]
- Yu, Z.; Kleifeld, O.; Lande-Atir, A.; Bsoul, M.; Kleiman, M.; Krutauz, D.; Book, A.; Vierstra, R.D.; Hofmann, K.; Reis, N.; et al. Dual function of Rpn5 in two PCI complexes, the 26S proteasome and COP9 signalosome. Mol. Biol. Cell 2011, 22, 911–920. [Google Scholar] [CrossRef]
- Pick, E.; Golan, A.; Zimbler, J.Z.; Guo, L.; Sharaby, Y.; Tsuge, T.; Hofmann, K.; Wei, N. The Minimal Deneddylase Core of the COP9 Signalosome Excludes the Csn6 MPN(-) Domain. PLoS ONE 2012, 7, e43980. [Google Scholar] [CrossRef] [PubMed]
- Serino, G.; Pick, E. Duplication and familial promiscuity within the proteasome lid and COP9 signalosome kin complexes. Plant Sci. 2013, 203–204, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Pick, E.; Hofmann, K.; Glickman, M.H. PCI complexes: Beyond the proteasome, CSN, and eIF3 Troika. Mol. Cell 2009, 35, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Rubin, D.M.; Coux, O.; Wefes, I.; Pfeifer, G.; Cjeka, Z.; Baumeister, W.; Fried, V.A.; Finley, D. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 1998, 94, 615–623. [Google Scholar] [CrossRef]
- Ambroggio, X.I.; Rees, D.C.; Deshaies, R.J. JAMM: A Metalloprotease-Like Zinc Site in the Proteasome and Signalosome. PLoS Biol. 2004, 2, e2. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates III, J.R.; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 Metalloprotease in Deubiquitination and Degradation by the 26S Proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Maytal-Kivity, V.; Reis, N.; Hofmann, K.; Glickman, M.H. MPN+, a putative catalytic motif found in a subset of MPN domain proteins from eukaryotes and prokaryotes, is critical for Rpn11 function. BMC Biochem. 2002, 3, 28. [Google Scholar] [CrossRef]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef]
- Lyapina, S.; Cope, G.; Shevchenko, A.; Serino, G.; Tsuge, T.; Zhou, C.; Wolf, D.A.; Wei, N.; Shevchenko, A.; Deshaies, R.J. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science 2001, 292, 1382–1385. [Google Scholar] [CrossRef]
- Gray, J.V.; Petsko, G.A.; Johnston, G.C.; Ringe, D.; Singer, R.A.; Werner-Washburne, M. ”Sleeping beauty”: Quiescence in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2004, 68, 187–206. [Google Scholar] [CrossRef]
- Bramasole, L.; Sinha, A.; Gurevich, S.; Radzinski, M.; Klein, Y.; Panat, N.; Gefen, E.; Rinaldi, T.; Jimenez-Morales, D.; Johnson, J.; et al. Proteasome lid bridges mitochondrial stress with Cdc53/Cullin1 NEDDylation status. Redox Biol. 2019, 20, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Apodaca, J.; Davis, L.E.; Rao, H. Proteasome inhibition in wild-type yeast Saccharomyces cerevisiae cells. Biotechniques 2007, 42, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Livnat-Levanon, N.; Kleifeld, O.; Mansour, W.; Nakasone, M.A.; Castaneda, C.A.; Dixon, E.K.; Fushman, D.; Reis, N.; Pick, E.; et al. Base-CP proteasome can serve as a platform for stepwise lid formation. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Osmulski, P.A.; Gaczynska, M.; Glickman, M.H. The central unit within the 19S regulatory particle of the proteasome. Nat. Struct. Mol. Biol. 2008, 15, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Schlierf, A.; Altmann, E.; Quancard, J.; Jefferson, A.B.; Assenberg, R.; Renatus, M.; Jones, M.; Hassiepen, U.; Schaefer, M.; Kiffe, M.; et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat. Commun. 2016, 7, 13166. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Piatti, S.; Hofmann, L.; Frontali, L.; Delahodde, A.; Rinaldi, T. Analysis of the rpn11-m1 proteasomal mutant reveals connection between cell cycle and mitochondrial biogenesis. FEMS Yeast Res. 2011, 11, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Pick, E.; Gambadoro, A.; Zilli, S.; Maytal-Kivity, V.; Frontali, L.; Glickman, M.H. Participation of the proteasomal lid subunit Rpn11 in mitochondrial morphology and function is mapped to a distinct C-terminal domain. Biochem. J. 2004, 381, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Licursi, V.; Salvi, C.; De Cesare, V.; Rinaldi, T.; Mattei, B.; Fabbri, C.; Serino, G.; Bramasole, L.; Zimbler, J.Z.; Pick, E.; et al. The COP9 signalosome is involved in the regulation of lipid metabolism and of transition metals uptake in Saccharomyces cerevisiae. FEBS J. 2014, 281, 175–190. [Google Scholar] [CrossRef]
- Saunier, R.; Esposito, M.; Dassa, E.P.; Delahodde, A. Integrity of the Saccharomyces cerevisiae Rpn11 protein is critical for formation of proteasome storage granules (PSG) and survival in stationary phase. PLoS ONE 2013, 8, e70357. [Google Scholar] [CrossRef]
- Bai, C.; Sen, P.; Hofmann, K.; Ma, L.; Goebl, M.; Harper, J.W.; Elledge, S.J. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 1996, 86, 263–274. [Google Scholar] [CrossRef]
- Aviram, S.; Kornitzer, D. The ubiquitin ligase Hul5 promotes proteasomal processivity. Mol. Cell. Biol. 2010, 30, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Ricci, C.; Porro, D.; Bolotin-Fukuhara, M.; Frontali, L. A mutation in a novel yeast proteasomal gene produces a cell cycle arrest, overreplication of nuclear and mitochondrial DNA and an altered mitocondrial morphology. Mol. Biol. Cell 1998, 9, 2917–2931. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Hofmann, L.; Gambadoro, A.; Cossard, R.; Livnat-Levanon, N.; Glickman, M.H.; Frontali, L.; Delahodde, A. Dissection of the Carboxyl-Terminal Domain of the Proteasomal Subunit Rpn11 in Maintenance of Mitochondrial Structure and Function. Mol. Biol. Cell 2008, 19, 1022–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maytal-Kivity, V.; Pick, E.; Piran, R.; Hofmann, K.; Glickman, M.H. The COP9 signalosome-like complex in S. cerevisiae and links to other PCI complexes. Int. J. Biochem. Cell Biol. 2003, 35, 706–715. [Google Scholar] [CrossRef]
- Visintin, R.; Craig, K.; Hwang, E.S.; Prinz, S.; Tyers, M.; Amon, A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell 1998, 2, 709–718. [Google Scholar] [CrossRef]
- Laporte, D.; Salin, B.; Daignan-Fornier, B.; Sagot, I. Reversible cytoplasmic localization of the proteasome in quiescent yeast cells. J. Cell Biol. 2008, 181, 737–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, L.Z.; Hazan, R.; Breker, M.; Schuldiner, M.; Ben-Aroya, S. Formation and dissociation of proteasome storage granules are regulated by cytosolic pH. J. Cell Biol. 2013, 201, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Li, F.; Gemperline, D.C.; Book, A.J.; Vierstra, R.D. Autophagic Degradation of the 26S Proteasome Is Mediated by the Dual ATG8/Ubiquitin Receptor RPN10 in Arabidopsis. Mol. Cell 2015, 58, 1053–1066. [Google Scholar] [CrossRef]
- Peters, L.Z.; Karmon, O.; David-Kadoch, G.; Hazan, R.; Yu, T.; Glickman, M.H.; Ben-Aroya, S. The protein quality control machinery regulates its misassembled proteasome subunits. PLoS Genet. 2015, 11, e1005178. [Google Scholar] [CrossRef]
- Lehmann, A.; Niewienda, A.; Jechow, K.; Janek, K.; Enenkel, C. Ecm29 fulfils quality control functions in proteasome assembly. Mol. Cell 2010, 38, 879–888. [Google Scholar] [CrossRef]
- Verma, R.; Chen, S.; Feldman, R.; Schieltz, D.; Yates, J.; Dohmen, J.; Deshaies, R.J. Proteasomal Proteomics: Identification of Nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol. Biol. Cell 2000, 11, 3425–3439. [Google Scholar] [CrossRef]
- Bornstein, G.; Ganoth, D.; Hershko, A. Regulation of neddylation and deneddylation of cullin1 in SCFSkp2 ubiquitin ligase by F-box protein and substrate. Proc. Natl. Acad. Sci. USA 2006, 103, 11515–11520. [Google Scholar] [CrossRef]
- Enchev, R.I.; Scott, D.C.; da Fonseca, P.C.; Schreiber, A.; Monda, J.K.; Schulman, B.A.; Peter, M.; Morris, E.P. Structural Basis for a Reciprocal Regulation between SCF and CSN. Cell Rep. 2012. [Google Scholar] [CrossRef]
- Emberley, E.D.; Mosadeghi, R.; Deshaies, R.J. Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-F-box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism. J. Biol. Chem. 2012, 287, 29679–29689. [Google Scholar] [CrossRef]
- Toro, T.B.; Toth, J.I.; Petroski, M.D. The cyclomodulin cycle inhibiting factor (CIF) alters cullin neddylation dynamics. J. Biol. Chem. 2013, 288, 14716–14726. [Google Scholar] [CrossRef]
- Rinaldi, T.; Ricordy, R.; Bolotin-Fukuhara, M.; Frontali, L. Mitochondrial effects of the pleiotropic proteasomal mutation mpr1/rpn11: Uncoupling from cell cycle defects in extragenic revertants. Gene 2002, 286, 43–51. [Google Scholar] [CrossRef]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef]
- Bornstein, G.; Grossman, C. COP9-Signalosome deneddylase activity is enhanced by simultaneous neddylation: Insights into the regulation of an enzymatic protein complex. Cell Div. 2015, 10, 5. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Source | |
|---|---|---|---|
| EP25 | Csn5-TAP | GAL1p [CSN5-TAP] | Open Biosystems |
| EP53 | empty vector | Yeplac181 | |
| EP134 | CDC14-GFP | GAL1p [CDC14-GFP], Amp | [46] |
| EP149 | empty vector | pYes2 | |
| EP150 | pYC-RPN8 | pADH1, [RPN8], Amp | [47] |
| EP228 | mch-VHL | GAL1p [VHL-mCherry], Amp | [48] |
| EP229 | mch-Rnq1 | GAL1p [Rnq1-mCherry], Amp | [48] |
| EP204 | CBP -Rpt6 | RPT4p, [CBP-A2-RPT6-LEU2], Amp | [30] |
| EP234 | Rpn5-TAP | ADH1p [Rpn5-TAP] | Open Biosystems |
| EP235 | ScRpn5 | RPN10p [RPN5], Amp | [30] |
| M134 | RPN11 C116>A | YCPlac111, RPN11p [rpn11C116/A], Amp | [37] |
| M138 | RPN11 D116>S | YCPlac111, RPN11p [rpn11C116/S], Amp | [37] |
| M143 | RPN11 H111>A | YCPlac111, RPN11p [rpn11H111/A], Amp | [37] |
| M144 | RPN11 S119>A | YCPlac111, RPN11p [rpn11S119/A], Amp | [37] |
| M145 | RPN11 D122>A | YCPlac111, RPN11p [rpn11H122/A], Amp | [37] |
| Name | Strain | Genotype | Source |
|---|---|---|---|
| RC1 | Δcsn9 | W303: csn9:: G418 | This study |
| RC6 | rpn11-m1–Δcsi1 | W303ade2-1; can1-100; his3-11,15; leu2-3, trp1-1; ura3-1; GAL+; lys2, KanMX4::YMR025W | This study |
| RC13 | rpn11-m1–Δcsn9 | W303 ade2-1; can1-100; his3-11, 15; leu2-3, trp1-1; ura3-1; GAL+; lys2, KanMX4::YDR079C | This study |
| RC21 | ΔYJR084W | W303 YJR084W:: G418 | This study |
| RC22 | rpn11-m1–ΔYJR084W | W303 ade2-1; can1-100; his3-11,15; leu2-3, trp1-1; ura3-1; GAL+; lys2::ΔYJR084W | This study |
| RC25 | Δcsi1 | W303: csi1:: G418 | This study |
| YP61 | ∆ubp6 | BY4741 lys2-801 leu2-3, 2-112, ura3-52, his3-Δ200, trp1-1, Δubp6::HIS3 | Open Biosystems |
| YP76 | ∆ubp15 | BY4741 his3Δ1 leu2Δ0 ura3Δ0 met15Δ1, UBP::KanMX4 | Open Biosystems |
| YP77 | ∆ubp16 | BY4741; his3Δ1 leu2Δ0 ura3Δ0 met15Δ1, UBP::KanMX4 | Open Biosystems |
| YP86 | ∆ubp1 | BY4741 his3Δ1 leu2Δ0 ura3Δ0 met15Δ1, UBP::KanMX | EUROSCARF (Oberursel, Germany) |
| YP87 | ∆ubp2 | BY4741 his3D1; leu2D0; met15D0; ura3D0; YOR124c::kanMX4 | EUROSCARF (Oberursel, Germany) |
| YP89 | ∆ubp5 | BY4741 his3Δ1 leu2Δ0 ura3Δ0 met15Δ1, UBP::KanMX4 | Open Biosystems |
| YP90 | ∆ubp7 | BY4741 his3Δ1 leu2Δ0 ura3Δ0 met15Δ1, UBP::KanMX4 | Open Biosystems |
| YP91 | ∆ubp8 | BY4741 his3Δ1 leu2Δ0 ura3Δ0 met15Δ1, UBP::KanMX4 | Open Biosystems |
| YP92 | ∆ubp9 | BY4741 his3Δ1 leu2Δ0 ura3Δ0 met15Δ1, UBP::KanMX4 | Open Biosystems |
| YP94 | ∆ubp11 | BY4741 his3Δ1 leu2Δ0 ura3Δ0 met15Δ1, UBP::KanMX4 | Open Biosystems |
| YP207 | Δcsn11 | W303: csn11:: G418 | This study |
| YP212 | rpn11-m1–Δcsn5 | W303ade2-1; can1-100; his3-11,15; leu2-3, trp1-1; ura3-1; GAL+; lys2, KanMX4::YDL216C | This study |
| YP216 | rpn11-m1–Δcsn11 | W303 ade2-1; can1-100; his3-11,15; leu2-3, trp1-1; ura3-1; GAL+; lys2, KanMX4::YIL071C | This study |
| YP334 | W303 (parental) | Mat a, his3-200, ade2-101, leu21,ura3-52, lys2-801, trp162 | |
| YP335 | Δcsn5 | W303: Csn5:: KanMX4 | [49] |
| YP336 | Δrub1 | W303 his3ko1;leu2ko0;met15ko0;ura3ko0 YDR139c::kanMX4 | This study |
| YP337 | rpn11-m1 | W303 Mat a, his3_-200, ade2-101, leu2_1, ura3-52, lys2-801,trp1-62, YFR004W::rpn11-m1 | [47] |
| YP238 | rpn11-m1–Δcsn5 | W303 Mat a, his3_-200, ade2-101, leu2_1, ura3-52, lys2-801,trp1-62, YFR004W::rpn11-m1, Csn5:: KanMX4 | This study |
| YP339 | rpn11-m1–Δrub1 | W303 Mat a, his3_-200, ade2-101, leu2_1, ura3-52, lys2-801,trp1-62, YFR004W::rpn11-m1, YDR139c::kanMX4 | This study |
| YP444 | Rpn11-Rpn5-GFP | W303 MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 RPN11:3HA-KANMX6, RPN5:GFP(S65T)-TRP1 | [50] |
| YP445 | rpn11-m1-Rpn5-GFP | W303 MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 rpn11-m1:3HA-KANMX6, RPN5:GFP(S65T)-TRP1 | [50] |
| YP452 | cdc14-3 | BY4741 his3ko1;leu2ko0;lys2ko0;ura3ko0 | [46] |
| YP531 | cdc4-1 | W303 ura3-1,can1-100, gal+, leu2-3,112. trp1-1; ade2-1; his3-11,15; cdc4-1 | [51] |
| MY321 | rpn11D122/A | BY4741 his3ko1; leu2ko0; met15ko0; ura3ko0 YFR004W:kanMX4 with plasmid M145 | [37] |
| MY317 | rpn11C116/A | BY4741 his3ko1; leu2ko0; met15ko0; ura3ko0 YFR004W:kanMX4 with plasmid M134 | [37] |
| MY318 | rpn11C116/S | BY4741 his3ko1; leu2ko0; met15ko0; ura3ko0 YFR004W:kanMX4 with plasmid M138 | [37] |
| MY319 | rpn11H111/A | BY4741 his3ko1; leu2ko0; met15ko0; ura3ko0 YFR004W:kanMX4 with plasmid M143 | [37] |
| MY320 | rpn11S119/A | BY4741 his3ko1; leu2ko0; met15ko0; ura3ko0 YFR004W:kanMX4 with plasmid M144 | [37] |
| MY1021 | Δpdr5 | W303 ura3-1; can1-100; GAL+leu3,112,trp1-1; ade2-1; his3-11,15; pdr5::hisG | [52] |
| MY1424 | rpn11-m1–Δpdr5 | W303 ura3-1; can1-100; GAL+leu3,112,trp1-1; ade2-1; his3-11,15; pdr5::hisG; YFR004W::rpn11-m1 | This study |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bramasole, L.; Sinha, A.; Harshuk, D.; Cirigliano, A.; Sylvia, G.; Yu, Z.; Carmeli, R.L.; Glickman, M.H.; Rinaldi, T.; Pick, E. The Proteasome Lid Triggers COP9 Signalosome Activity during the Transition of Saccharomyces cerevisiae Cells into Quiescence. Biomolecules 2019, 9, 449. https://doi.org/10.3390/biom9090449
Bramasole L, Sinha A, Harshuk D, Cirigliano A, Sylvia G, Yu Z, Carmeli RL, Glickman MH, Rinaldi T, Pick E. The Proteasome Lid Triggers COP9 Signalosome Activity during the Transition of Saccharomyces cerevisiae Cells into Quiescence. Biomolecules. 2019; 9(9):449. https://doi.org/10.3390/biom9090449
Chicago/Turabian StyleBramasole, Laylan, Abhishek Sinha, Dana Harshuk, Angela Cirigliano, Gurevich Sylvia, Zanlin Yu, Rinat Lift Carmeli, Michael H. Glickman, Teresa Rinaldi, and Elah Pick. 2019. "The Proteasome Lid Triggers COP9 Signalosome Activity during the Transition of Saccharomyces cerevisiae Cells into Quiescence" Biomolecules 9, no. 9: 449. https://doi.org/10.3390/biom9090449
APA StyleBramasole, L., Sinha, A., Harshuk, D., Cirigliano, A., Sylvia, G., Yu, Z., Carmeli, R. L., Glickman, M. H., Rinaldi, T., & Pick, E. (2019). The Proteasome Lid Triggers COP9 Signalosome Activity during the Transition of Saccharomyces cerevisiae Cells into Quiescence. Biomolecules, 9(9), 449. https://doi.org/10.3390/biom9090449