Fibroblasts and Their Pathological Functions in the Fibrosis of Aortic Valve Sclerosis and Atherosclerosis


{kind=link}
Abstract
:1. Introduction
2. Role of Fibroblasts in Atherosclerosis
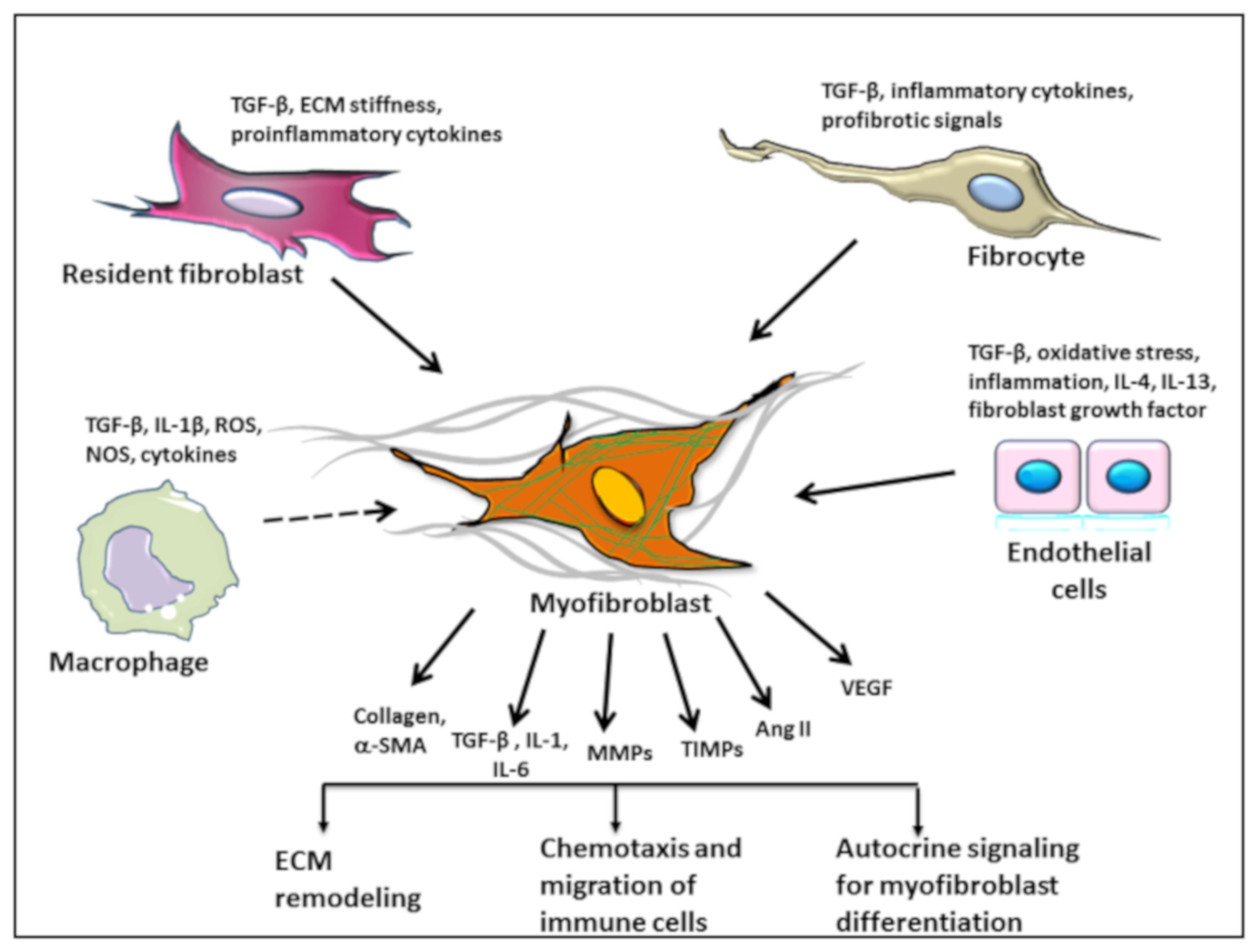
3. Role of Fibroblasts in AVS
4. Mechanisms of Fibrosis in AVS and Atherosclerosis
4.1. Transforming Growth Factor β
4.2. Renin-Angiotensin System
4.3. Immune Cells and Cytokines
5. Origin of Myofibroblasts
5.1. Macrophages and Monocytes
5.2. Endothelial to Mesenchymal Transition
5.3. Fibrocytes as the Source of Fibroblasts in AVS and Atherosclerosis
6. Clinical Implications of Fibrosis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Bobik, A. Transforming growth factor-betas and vascular disorders. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1712–1720. [Google Scholar] [CrossRef]
- Brokopp, C.E.; Schoenauer, R.; Richards, P.; Bauer, S.; Lohmann, C.; Emmert, M.Y.; Weber, B.; Winnik, S.; Aikawa, E.; Graves, K.; et al. Fibroblast activation protein is induced by inflammation and degrades type I collagen in thin-cap fibroatheromata. Eur. Heart J. 2011, 32, 2713–2722. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.D.; Shavelle, D.M.; Caulfield, M.T.; McDonald, T.O.; Olin-Lewis, K.; Otto, C.M.; Probstfield, J.L. Association of Angiotensin-Converting Enzyme With Low-Density Lipoprotein in Aortic Valvular Lesions and in Human Plasma. Circulation 2002, 106, 2224–2230. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.; Gabbiani, G.; Camenzind, E.; Bacchetta, M.; Virmani, R.; Bochaton-Piallat, M.-L. Phenotypic modulation of intima and media smooth muscle cells in fatal cases of coronary artery lesion. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 326–332. [Google Scholar] [CrossRef]
- Fina, L.; Molgaard, H.V.; Robertson, D.; Bradley, N.J.; Monaghan, P.; Delia, D.; Sutherland, D.R.; Baker, M.A.; Greaves, M.F. Expression of the CD34 gene in vascular endothelial cells. Blood 1990, 75, 2417–2426. [Google Scholar] [Green Version]
- Sartore, S.; Chiavegato, A.; Faggin, E.; Franch, R.; Puato, M.; Ausoni, S.; Pauletto, P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: From innocent bystander to active participant. Circ. Res. 2001, 89, 1111–1121. [Google Scholar] [CrossRef]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.-R.; D’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Weile, C.; Sondergaard, C.S.; Hindkjaer, J.; Kassem, M.; Falk, E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2696–2702. [Google Scholar] [CrossRef]
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260. [Google Scholar] [CrossRef]
- Paranya, G.; Vineberg, S.; Dvorin, E.; Kaushal, S.; Roth, S.J.; Rabkin, E.; Schoen, F.J.; Bischoff, J. Aortic valve endothelial cells undergo transforming growth factor-beta-mediated and non-transforming growth factor-beta-mediated transdifferentiation in vitro. Am. J. Pathol. 2001, 159, 1335–1343. [Google Scholar] [CrossRef]
- Rabkin, E.; Aikawa, M.; Stone, J.R.; Fukumoto, Y.; Libby, P.; Schoen, F.J. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 2001, 104, 2525–2532. [Google Scholar] [CrossRef]
- Shi, Y.; O’Brien, J.E.; Fard, A.; Zalewski, A. Transforming growth factor-beta 1 expression and myofibroblast formation during arterial repair. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1298–1305. [Google Scholar] [CrossRef]
- Muller, A.M.; Cronen, C.; Kupferwasser, L.I.; Oelert, H.; Muller, K.M.; Kirkpatrick, C.J. Expression of endothelial cell adhesion molecules on heart valves: Up-regulation in degeneration as well as acute endocarditis. J. Pathol. 2000, 191, 54–60. [Google Scholar] [CrossRef]
- Butcher, J.T.; Nerem, R.M. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: Effects of steady shear stress. Tissue Eng. 2006, 12, 905–915. [Google Scholar] [CrossRef]
- Taylor, P.M.; Allen, S.P.; Yacoub, M.H. Phenotypic and functional characterization of interstitial cells from human heart valves, pericardium and skin. J. Heart Valve Dis. 2000, 9, 150–158. [Google Scholar]
- Bairati, A.; DeBiasi, S. Presence of a smooth muscle system in aortic valve leaflets. Anat. Embryol. 1981, 161, 329–340. [Google Scholar] [CrossRef]
- Rabkin-Aikawa, E.; Farber, M.; Aikawa, M.; Schoen, F.J. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J. Heart Valve Dis. 2004, 13, 841–847. [Google Scholar]
- Stella, J.A.; Sacks, M.S. On the biaxial mechanical properties of the layers of the aortic valve leaflet. J. Biomech. Eng. 2007, 129, 757–766. [Google Scholar] [CrossRef]
- Volzke, H.; Haring, R.; Lorbeer, R.; Wallaschofski, H.; Reffelmann, T.; Empen, K.; Rettig, R.; John, U.; Felix, S.B.; Dorr, M. Heart valve sclerosis predicts all-cause and cardiovascular mortality. Atherosclerosis 2010, 209, 606–610. [Google Scholar] [CrossRef]
- Fuchs, C.; Mascherbauer, J.; Rosenhek, R.; Pernicka, E.; Klaar, U.; Scholten, C.; Heger, M.; Wollenek, G.; Czerny, M.; Maurer, G.; et al. Gender differences in clinical presentation and surgical outcome of aortic stenosis. Heart 2010, 96, 539–545. [Google Scholar] [CrossRef]
- Sritharen, Y.; Enriquez-Sarano, M.; Schaff, H.V.; Casaclang-Verzosa, G.; Miller, J.D. Pathophysiology of Aortic Valve Stenosis: Is It Both Fibrocalcific and Sex Specific? Physiology 2017, 32, 182–196. [Google Scholar] [CrossRef]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of Transforming Growth Factor β in Human Disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar] [CrossRef]
- Vaughan, M.B.; Howard, E.W.; Tomasek, J.J. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 2000, 257, 180–189. [Google Scholar] [CrossRef]
- Zhang, W.; Ou, J.; Inagaki, Y.; Greenwel, P.; Ramirez, F. Synergistic cooperation between Sp1 and Smad3/Smad4 mediates transforming growth factor β1 stimulation of α2(I)-collagen (COL1A2) transcription. J. Biol. Chem. 2000, 275, 39237–39245. [Google Scholar] [CrossRef]
- Mallat, Z.; Gojova, A.; Marchiol-Fournigault, C.; Esposito, B.; Kamate, C.; Merval, R.; Fradelizi, D.; Tedgui, A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 2001, 89, 930–934. [Google Scholar] [CrossRef]
- Grainger, D.J.; Kemp, P.R.; Metcalfe, J.C.; Liu, A.C.; Lawn, R.M.; Williams, N.R.; Grace, A.A.; Schofield, P.M.; Chauhan, A. The serum concentration of active transforming growth factor-beta is severely depressed in advanced atherosclerosis. Nat. Med. 1995, 1, 74–79. [Google Scholar] [CrossRef]
- Reifenberg, K.; Cheng, F.; Orning, C.; Crain, J.; Kupper, I.; Wiese, E.; Protschka, M.; Blessing, M.; Lackner, K.J.; Torzewski, M. Overexpression of TGF-ss1 in macrophages reduces and stabilizes atherosclerotic plaques in ApoE-deficient mice. PLoS ONE 2012, 7, e40990. [Google Scholar] [CrossRef]
- Frutkin, A.D.; Otsuka, G.; Stempien-Otero, A.; Sesti, C.; Du, L.; Jaffe, M.; Dichek, H.L.; Pennington, C.J.; Edwards, D.R.; Nieves-Cintrón, M.; et al. TGF-β1 Limits Plaque Growth, Stabilizes Plaque Structure, and Prevents Aortic Dilation in Apolipoprotein E-Null Mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1251–1257. [Google Scholar] [CrossRef]
- Zargham, R.; Pepin, J.; Thibault, G. Alpha8beta1 Integrin is up-regulated in the neointima concomitant with late luminal loss after balloon injury. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2007, 16, 212–220. [Google Scholar] [CrossRef]
- Helske, S.; Lindstedt, K.A.; Laine, M.; Mayranpaa, M.; Werkkala, K.; Lommi, J.; Turto, H.; Kupari, M.; Kovanen, P.T. Induction of local angiotensin II-producing systems in stenotic aortic valves. J. Am. Coll. Cardiol. 2004, 44, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.T.; Sun, Y.; Katwa, L.C. Myofibroblasts and local angiotensin II in rat cardiac tissue repair. Int. J. Biochem. Cell Biol. 1997, 29, 31–42. [Google Scholar] [CrossRef]
- Harrison, D.G.; Cai, H.; Landmesser, U.; Griendling, K.K. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2003, 4, 51–61. [Google Scholar] [CrossRef]
- Chen, K.; Chen, J.; Li, D.; Zhang, X.; Mehta, J.L. Angiotensin II Regulation of Collagen Type I Expression in Cardiac Fibroblasts. Hypertension 2004, 44, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Schiffrin, E.L. Signal Transduction Mechanisms Mediating the Physiological and Pathophysiological Actions of Angiotensin II in Vascular Smooth Muscle Cells. Pharmacol. Rev. 2000, 52, 639. [Google Scholar]
- Araujo, J.A.; Lusis, A.J. Atherosclerosis. In Encyclopedia of Endocrine Diseases; Martini, L., Ed.; Elsevier: New York, NY, USA, 2004; pp. 288–294. [Google Scholar] [CrossRef]
- Shen, W.-L.; Gao, P.-J.; Che, Z.-Q.; Ji, K.-D.; Yin, M.; Yan, C.; Berk, B.C.; Zhu, D.-L. NAD(P)H oxidase-derived reactive oxygen species regulate angiotensin-II induced adventitial fibroblast phenotypic differentiation. Biochem. Biophys. Res. Commun. 2006, 339, 337–343. [Google Scholar] [CrossRef]
- Gray, M.O.; Long, C.S.; Kalinyak, J.E.; Li, H.T.; Karliner, J.S. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc. Res. 1998, 40, 352–363. [Google Scholar] [CrossRef]
- Schultz Jel, J.; Witt, S.A.; Glascock, B.J.; Nieman, M.L.; Reiser, P.J.; Nix, S.L.; Kimball, T.R.; Doetschman, T. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Invest. 2002, 109, 787–796. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Probstfield, J.L.; Caulfield, M.T.; Nasir, K.; Takasu, J.; Shavelle, D.M.; Wu, A.H.; Zhao, X.Q.; Budoff, M.J. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch. Intern. Med. 2005, 165, 858–862. [Google Scholar] [CrossRef]
- Rosenhek, R.; Rader, F.; Loho, N.; Gabriel, H.; Heger, M.; Klaar, U.; Schemper, M.; Binder, T.; Maurer, G.; Baumgartner, H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004, 110, 1291–1295. [Google Scholar] [CrossRef]
- Liu, A.C.; Gotlieb, A.I. Transforming growth factor-beta regulates in vitro heart valve repair by activated valve interstitial cells. Am. J. Pathol. 2008, 173, 1275–1285. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1beta and the autoinflammatory diseases. N. Engl. J. Med. 2009, 360, 2467–2470. [Google Scholar] [CrossRef]
- Kaden, J.J.; Dempfle, C.-E.; Grobholz, R.; Tran, H.-T.; Kilic, R.; Sarikoc, A.; Brueckmann, M.; Vahl, C.; Hagl, S.; Haase, K.K.; et al. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis 2003, 170, 205–211. [Google Scholar] [CrossRef]
- Nadlonek, N.; Lee, J.H.; Reece, T.B.; Weyant, M.J.; Cleveland, J.C.; Meng, X.; Fullerton, D.A. Interleukin-1 Beta induces an inflammatory phenotype in human aortic valve interstitial cells through nuclear factor kappa Beta. Ann. Thorac. Surg. 2013, 96, 155–162. [Google Scholar] [CrossRef]
- Li, J.; Schwimmbeck, P.L.; Tschope, C.; Leschka, S.; Husmann, L.; Rutschow, S.; Reichenbach, F.; Noutsias, M.; Kobalz, U.; Poller, W.; et al. Collagen degradation in a murine myocarditis model: Relevance of matrix metalloproteinase in association with inflammatory induction. Cardiovasc. Res. 2002, 56, 235–247. [Google Scholar] [CrossRef]
- Lee, E.; Grodzinsky, A.J.; Libby, P.; Clinton, S.K.; Lark, M.W.; Lee, R.T. Human vascular smooth muscle cell-monocyte interactions and metalloproteinase secretion in culture. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2284–2289. [Google Scholar] [CrossRef]
- Meznarich, J.; Malchodi, L.; Helterline, D.; Ramsey, S.A.; Bertko, K.; Plummer, T.; Plawman, A.; Gold, E.; Stempien-Otero, A. Urokinase plasminogen activator induces pro-fibrotic/M2 phenotype in murine cardiac macrophages. PLoS ONE 2013, 8, e57837. [Google Scholar] [CrossRef]
- Hesse, M.; Modolell, M.; La Flamme, A.C.; Schito, M.; Fuentes, J.M.; Cheever, A.W.; Pearce, E.J.; Wynn, T.A. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: Granulomatous pathology is shaped by the pattern of L-arginine metabolism. J. Immunol. 2001, 167, 6533–6544. [Google Scholar] [CrossRef]
- Sullivan, D.E.; Ferris, M.; Nguyen, H.; Abboud, E.; Brody, A.R. TNF-alpha induces TGF-beta1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. J. Cell. Mol. Med. 2009, 13, 1866–1876. [Google Scholar] [CrossRef]
- Steinhauser, M.L.; Kunkel, S.L.; Hogaboam, C.M.; Evanoff, H.; Strieter, R.M.; Lukacs, N.W. Macrophage/fibroblast coculture induces macrophage inflammatory protein-1α production mediated by intercellular adhesion molecule-1 and oxygen radicals. J. Leukoc. Biol. 1998, 64, 636–641. [Google Scholar] [CrossRef]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef]
- Xu, K.; Zhou, T.; Huang, Y.; Chi, Q.; Shi, J.; Zhu, P.; Dong, N. Anthraquinone Emodin Inhibits Tumor Necrosis Factor Alpha-Induced Calcification of Human Aortic Valve Interstitial Cells via the NF-κB Pathway. Front. Pharmacol. 2018, 9, 1328. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Michael, L.H.; Entman, M.L. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc. Res. 2000, 48, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Haudek, S.B.; Xia, Y.; Huebener, P.; Lee, J.M.; Carlson, S.; Crawford, J.R.; Pilling, D.; Gomer, R.H.; Trial, J.; Frangogiannis, N.G.; et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18284–18289. [Google Scholar] [CrossRef] [Green Version]
- Pichler, M.; Rainer, P.P.; Schauer, S.; Hoefler, G. Cardiac fibrosis in human transplanted hearts is mainly driven by cells of intracardiac origin. J. Am. Coll. Cardiol. 2012, 59, 1008–1016. [Google Scholar] [CrossRef]
- Mollmann, H.; Nef, H.M.; Kostin, S.; Kalle, C.; Pilz, I.; Weber, M.; Schaper, J.; Hamm, C.W.; Elsasser, A. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc. Res. 2006, 71, 661–671. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723. [Google Scholar] [CrossRef]
- Leitinger, N.; Schulman, I.G. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1120–1126. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. A J. Virtual. Libr. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Meng, X.-M.; Wang, S.; Huang, X.-R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.-F.; Nikolic-Paterson, D.J.; Lan, H.-Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Jiang, H.; Pan, J.; Huang, X.-R.; Wang, Y.-C.; Huang, H.-F.; To, K.-F.; Nikolic-Paterson, D.J.; Lan, H.-Y.; Chen, J.-H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053. [Google Scholar] [CrossRef]
- Chu, P.-Y.; Mariani, J.; Finch, S.; McMullen, J.R.; Sadoshima, J.; Marshall, T.; Kaye, D.M. Bone marrow-derived cells contribute to fibrosis in the chronically failing heart. Am. J. Pathol. 2010, 176, 1735–1742. [Google Scholar] [CrossRef]
- Varga, J.; Jimenez, S.A. Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-beta. Biochem. Biophys. Res. Commun. 1986, 138, 974–980. [Google Scholar] [CrossRef]
- Overall, C.M.; Wrana, J.L.; Sodek, J. Independent regulation of collagenase, 72-kDa progelatinase, and metalloendoproteinase inhibitor expression in human fibroblasts by transforming growth factor-beta. J. Biol. Chem. 1989, 264, 1860–1869. [Google Scholar]
- Chen, S.J.; Yuan, W.; Mori, Y.; Levenson, A.; Trojanowska, M.; Varga, J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: Involvement of Smad 3. J. Investig. Dermatol. Symp. Proc. 1999, 112, 49–57. [Google Scholar] [CrossRef]
- Verrecchia, F.; Chu, M.L.; Mauviel, A. Identification of novel TGF-β/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 2001, 276, 17058–17062. [Google Scholar] [CrossRef]
- Nakajima, Y.; Yamagishi, T.; Hokari, S.; Nakamura, H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: Roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP). Anat. Rec. 2000, 258, 119–127. [Google Scholar] [CrossRef]
- Krenning, G.; Moonen, J.-R.A.J.; Van Luyn, M.J.A.; Harmsen, M.C. Vascular smooth muscle cells for use in vascular tissue engineering obtained by endothelial-to-mesenchymal transdifferentiation (EnMT) on collagen matrices. Biomaterials 2008, 29, 3703–3711. [Google Scholar] [CrossRef]
- Moonen, J.-R.A.J.; Krenning, G.; Brinker, M.G.L.; Koerts, J.A.; Van Luyn, M.J.A.; Harmsen, M.C. Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc. Res. 2010, 86, 506–515. [Google Scholar] [CrossRef] [Green Version]
- Dahal, S.; Huang, P.; Murray, B.T.; Mahler, G.J. Endothelial to mesenchymal transformation is induced by altered extracellular matrix in aortic valve endothelial cells. J. Biomed. Mater. Res. Part A 2017, 105, 2729–2741. [Google Scholar] [CrossRef]
- Zhong, A.; Mirzaei, Z.; Simmons, C.A. The Roles of Matrix Stiffness and ss-Catenin Signaling in Endothelial-to-Mesenchymal Transition of Aortic Valve Endothelial Cells. Cardiovasc. Eng. Technol. 2018, 9, 158–167. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef]
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; Paulis, A.d.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [Green Version]
- Kawabe, J.-I.; Hasebe, N. Role of the vasa vasorum and vascular resident stem cells in atherosclerosis. Biomed. Res. Int. 2014, 2014, 701571. [Google Scholar] [CrossRef]
- Van Meeteren, L.A.; Dijke, P.t. Regulation of endothelial cell plasticity by TGF-beta. Cell Tissue Res. 2012, 347, 177–186. [Google Scholar] [CrossRef]
- Feng, X.-H.; Derynck, R. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef]
- Cevallos, M.; Riha, G.M.; Wang, X.; Yang, H.; Yan, S.; Li, M.; Chai, H.; Yao, Q.; Chen, C. Cyclic strain induces expression of specific smooth muscle cell markers in human endothelial cells. Differ. Res. Biol. Divers. 2006, 74, 552–561. [Google Scholar] [CrossRef]
- Mahler, G.J.; Farrar, E.J.; Butcher, J.T. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 121–130. [Google Scholar] [CrossRef]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [CrossRef]
- Medbury, H.J.; Tarran, S.L.S.; Guiffre, A.K.; Williams, M.M.Y.; Lam, T.H.; Vicaretti, M.; Fletcher, J.P. Monocytes contribute to the atherosclerotic cap by transformation into fibrocytes. Int. Angiol. A J. Int. Union Angiol. 2008, 27, 114–123. [Google Scholar] [CrossRef]
- Sakai, N.; Wada, T.; Yokoyama, H.; Lipp, M.; Ueha, S.; Matsushima, K.; Kaneko, S. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 14098–14103. [Google Scholar] [CrossRef] [Green Version]
- Galligan, C.L.; Fish, E.N. The role of circulating fibrocytes in inflammation and autoimmunity. J. Leukoc. Biol. 2013, 93, 45–50. [Google Scholar] [CrossRef]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. The role of fibrocytes in fibrotic diseases of the lungs and heart. Fibrogenesis Tissue Repair 2011, 4, 2. [Google Scholar] [CrossRef]
- Egan, K.P.; Kim, J.-H.; Mohler, E.R.r.; Pignolo, R.J. Role for circulating osteogenic precursor cells in aortic valvular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2965–2971. [Google Scholar] [CrossRef]
- Baues, M.; Dasgupta, A.; Ehling, J.; Prakash, J.; Boor, P.; Tacke, F.; Kiessling, F.; Lammers, T. Fibrosis imaging: Current concepts and future directions. Adv. Drug Deliv. Rev. 2017, 121, 9–26. [Google Scholar] [CrossRef]
- Agarwal, I.; Arnold, A.; Glazer, N.L.; Barasch, E.; Djousse, L.; Fitzpatrick, A.L.; Gottdiener, J.S.; Ix, J.H.; Jensen, R.A.; Kizer, J.R.; et al. Fibrosis-related biomarkers and large and small vessel disease: The Cardiovascular Health Study. Atherosclerosis 2015, 239, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xu, M.; Wang, T.; Sun, J.; Sun, W.; Xu, B.; Huang, X.; Xu, Y.; Lu, J.; Li, X.; et al. Advanced fibrosis associates with atherosclerosis in subjects with nonalcoholic fatty liver disease. Atherosclerosis 2015, 241, 145–150. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, X.-Q.; Balu, N.; Neradilek, M.B.; Isquith, D.A.; Yamada, K.; Cantón, G.; Crouse, J.R., 3rd; Anderson, T.J.; Huston, J., 3rd; et al. Carotid Plaque Lipid Content and Fibrous Cap Status Predict Systemic CV Outcomes: The MRI Substudy in AIM-HIGH. JACC Cardiovasc. Imaging 2017, 10, 241–249. [Google Scholar] [CrossRef]
- Hellings, W.E.; Peeters, W.; Moll, F.L.; Piers, S.R.D.; Van Setten, J.; Van der Spek, P.J.; De Vries, J.-P.P.M.; Seldenrijk, K.A.; De Bruin, P.C.; Vink, A.; et al. Composition of carotid atherosclerotic plaque is associated with cardiovascular outcome: A prognostic study. Circulation 2010, 121, 1941–1950. [Google Scholar] [CrossRef]
- Milano, A.D.; Faggian, G.; Dodonov, M.; Golia, G.; Tomezzoli, A.; Bortolotti, U.; Mazzucco, A. Prognostic value of myocardial fibrosis in patients with severe aortic valve stenosis. J. Thorac. Cardiovasc. Surg. 2012, 144, 830–837. [Google Scholar] [CrossRef] [Green Version]
- Katbeh, A.; Ondrus, T.; Barbato, E.; Galderisi, M.; Trimarco, B.; Van Camp, G.; Vanderheyden, M.; Penicka, M. Imaging of Myocardial Fibrosis and Its Functional Correlates in Aortic Stenosis: A Review and Clinical Potential. Cardiology 2018, 141, 141–149. [Google Scholar] [CrossRef]
- Berry, J.M.; Le, V.; Rotter, D.; Battiprolu, P.K.; Grinsfelder, B.; Tannous, P.; Burchfield, J.S.; Czubryt, M.; Backs, J.; Olson, E.N.; et al. Reversibility of adverse, calcineurin-dependent cardiac remodeling. Circ. Res. 2011, 109, 407–417. [Google Scholar] [CrossRef]
- Patel, R.; Nagueh, S.F.; Tsybouleva, N.; Abdellatif, M.; Lutucuta, S.; Kopelen, H.A.; Quinones, M.A.; Zoghbi, W.A.; Entman, M.L.; Roberts, R.; et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2001, 104, 317–324. [Google Scholar] [CrossRef]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Forte, A.; Della Corte, A.; Feo, M.d.; Cerasuolo, F.; Cipollaro, M. Role of myofibroblasts in vascular remodelling: Focus on restenosis and aneurysm. Cardiovasc. Res. 2010, 88, 395–405. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Torzewski, M. Fibroblasts and Their Pathological Functions in the Fibrosis of Aortic Valve Sclerosis and Atherosclerosis. Biomolecules 2019, 9, 472. https://doi.org/10.3390/biom9090472
Singh S, Torzewski M. Fibroblasts and Their Pathological Functions in the Fibrosis of Aortic Valve Sclerosis and Atherosclerosis. Biomolecules. 2019; 9(9):472. https://doi.org/10.3390/biom9090472
Chicago/Turabian StyleSingh, Savita, and Michael Torzewski. 2019. "Fibroblasts and Their Pathological Functions in the Fibrosis of Aortic Valve Sclerosis and Atherosclerosis" Biomolecules 9, no. 9: 472. https://doi.org/10.3390/biom9090472
APA StyleSingh, S., & Torzewski, M. (2019). Fibroblasts and Their Pathological Functions in the Fibrosis of Aortic Valve Sclerosis and Atherosclerosis. Biomolecules, 9(9), 472. https://doi.org/10.3390/biom9090472