Protein Engineering of Multi-Modular Transcription Factor Alcohol Dehydrogenase Repressor 1 (Adr1p), a Tool for Dissecting In Vitro Transcription Activation


Abstract
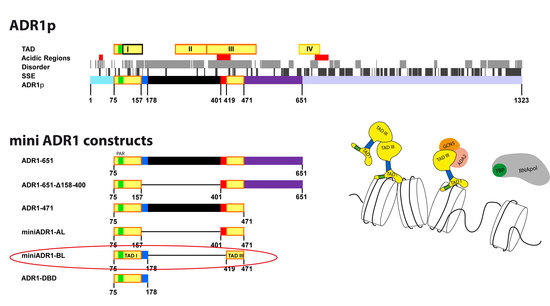
:
1. Introduction
2. Materials and Methods
2.1. Design of the Minimal Regulatory Units
2.2. Cloning and Expression
2.2.1. Cloning
2.2.2. Expression in Escherichia coli
2.2.3. Expression in Komagataella pastoris
2.3. Purification of miniAdr1 Constructs
2.3.1. Purification of His-tagged miniAdr1 Constructs
2.3.2. Purification of Untagged miniAdr1 Constructs
2.4. Functional Characterization
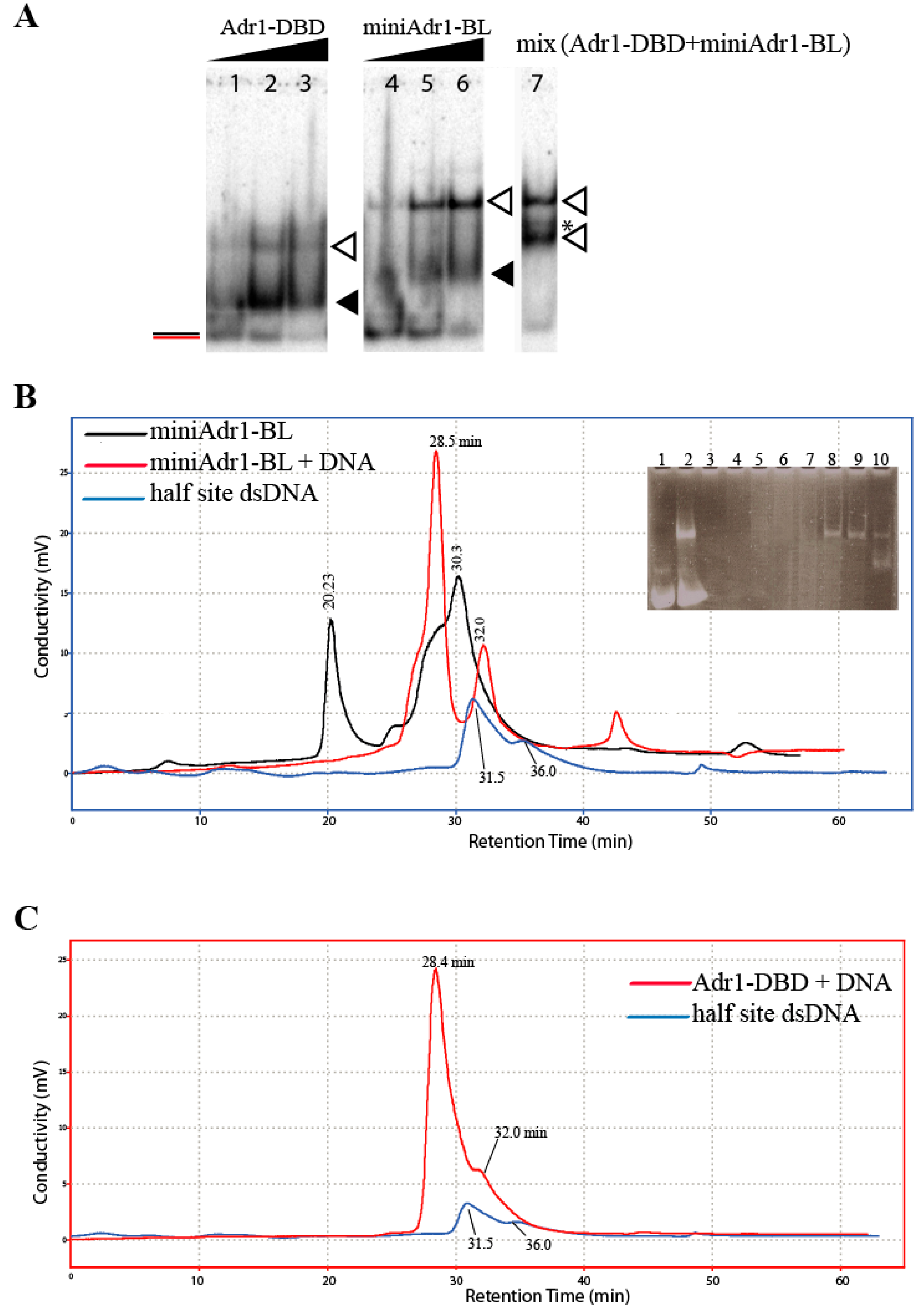
2.4.1. DNA-Binding Assays
2.4.2. Quaternary Assembly by HPLC
3. Results
3.1. Design of the Minimal Regulatory Units
3.2. Cloning, Expression, and Purification of miniARD1 Constructs in Escherichia coli and Komagtaella pastoris
3.3. Functional Characterization of miniAdr1 Constructs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Denis, C.L.; Ciriacy, M.; Young, E.T. A positive regulatory gene is required for accumulation of the functional messenger RNA for the glucose-repressible alcohol dehydrogenase from Saccharomyces cerevisiae. J. Mol. Biol. 1981, 148, 355–368. [Google Scholar] [CrossRef]
- Bemis, L.T.; Denis, C.L. Identification of functional regions in the yeast transcriptional activator Adr1. Mol. Cell. Biol. 1988, 8, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.A.; Rapatz, W.G.; Spevak, W.; Ruis, H. The Saccharomyces cerevisiae Adr1 gene is a positive regulator of transcription of genes encoding peroxisomal proteins. Mol. Cell. Biol. 1991, 11, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Young, E.T. Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 2011, 189, 705–736. [Google Scholar] [CrossRef] [PubMed]
- Wierman, M.B.; Maqani, N.; Strickler, E.; Li, M.; Smith, J.S. Caloric Restriction Extends Yeast Chronological Life Span by Optimizing the Snf1 (AMPK) Signaling Pathway. Mol Cell Biol. 2017, 37, 13. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, H.; Hartshorne, T.A.; Young, E.T. Regulation of expression and activity of the yeast transcription factor Adr1. Mol. Cell. Biol. 1988, 8, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.E.; Young, E.T. cAMP-dependent phosphorylation and inactivation of yeast transcription factor Adr1 does not affect DNA binding. Proc. Natl. Acad. Sci. USA 1990, 87, 4098–4102. [Google Scholar] [CrossRef]
- Verdone, L.; Cesari, F.; Denis, C.L.; Di Mauro, E.; Caserta, M. Factors affecting Saccharomyces cerevisiae ADH2 chromatin remodelling and transcription. J. Biol. Chem. 1997, 272, 30828–30834. [Google Scholar] [CrossRef]
- Abate, G.; Bastonini, E.; Braun, K.A.; Verdone, L.; Young, E.T.; Caserta, M. Snf1/AMPK regulates Gcn5 occupancy, H3 acetylation and chromatin remodelling at S. cerevisiae ADY2 promoter. Biochim. Biophys. Acta 2012, 1819, 419–427. [Google Scholar] [CrossRef]
- Panetta, G.; Buttinelli, M.; Flaus, A.; Richmond, T.J.; Rhodes, D. Differential nucleosome positioning on Xenopus oocyte and somatic 5 S RNA genes determines both TFIIIA and H1 binding: A mechanism for selective H1 repression. J. Mol. Biol. 1998, 282, 683–697. [Google Scholar] [CrossRef]
- Klug, A. The discovery of zinc fingers and their development for practical applications in gene regulation and genome manipulation. Rev. Biophys. 2010, 43, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thukral, S.K.; Eisen, A.; Young, E.T. Two monomers of yeast transcription factor Adr1 bind a palindromic sequence symmetrically to activate ADH2 expression. Mol. Cell. Biol. 1991, 11, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Drew, H.R.; Travers, A. DNA bending and its relation to nucleosome positioning. J. Mol. Biol. 1985, 186, 773–790. [Google Scholar] [CrossRef]
- Battistini, F.; Hunter, C.A.; Moore, I.K.; Widom, J. Structure-based identification of new high-affinity nucleosome binding sequences. J. Mol. Biol. 2012, 420, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K.; Segal, E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 2013, 20, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Di Marcotullio, L.; Buttinelli, M.; Costanzo, G.; Di Mauro, E.; Negri, R. Changing nucleosome positions in vivo through modification of the DNA rotational information. Biochem. J. 1998, 333, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Buttinelli, M.; Minnock, A.; Panetta, G.; Waring, M.; Travers, A. The exocyclic groups of DNA modulate the affinity and positioning of the histone octamer. Proc. Natl. Acad. Sci. USA 1998, 95, 8544–8549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negri, R.; Buttinelli, M.; Panetta, G.; De Arcangelis, V.; Di Mauro, E.; Travers, A. Sequence dependence of translational positioning of core nucleosomes. J. Mol. Biol. 2001, 307, 307–987. [Google Scholar] [CrossRef] [PubMed]
- Cook, W.J.; Chase, D.; Audino, D.C.; Denis, C.L. Dissection of the Adr1 protein reveals multiple, functionally redundant activation domains interspersed with inhibitory regions: Evidence for a repressor binding to the Adr1c region. Mol. Cell. Biol. 1994. [Google Scholar] [CrossRef]
- Hintze, S.; Engelhardt, M.; van Diepen, L.; Witt, E.; Schüller, H.J. Multiple Taf subunits of TFIID interact with Ino2 activation domains and contribute to expression of genes required for yeast phospholipid biosynthesis. Mol. Microbiol. 2017, 106, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Chiang, Y.C.; Komarnitsky, F.; Chase, D.; Denis, C.L. Adr1 activation domains contact the histone acetyl transferase GCN5 and the core transcriptional factor TFIIB. J. Biol. Chem. 1996, 271, 32359–32365. [Google Scholar] [CrossRef] [PubMed]
- Thukral, S.K.; Tavianini, M.A.; Blumberg, H.; Young, E.T. Localization of a minimal binding domain and activation regions in the yeast regulatory protein Adr1. Mol. Cell. Biol. 1989, 9, 2360–2369. [Google Scholar] [CrossRef] [PubMed]
- Bowers, P.M.; Schaufler, L.E.; Klevit, R.E. A folding transition and novel zinc finger accessory domain in the transcription factor Adr1. Nat. Struct. Biol. 1999, 6, 478–485. [Google Scholar] [PubMed]
- Bernstein, B.E.; Hoffman, R.C.; Horvath, S.; Herriott, J.R.; Klevit, R.E. Structure of a histidine-X4-histidine zinc finger domain: Insights into Adr1-UAS1 protein-DNA recognition. Biochemistry 1994, 33, 4460–4470. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, O.B.; Hedman, R.; Marino, J.; Wickles, S.; Bischoff, L.; Johansson, M.; Müller-Lucks, A.; Trovato, F.; Puglisi, J.D.; O’Brien, E.P.; et al. Cotranslational Protein Folding inside the Ribosome Exit Tunnel. Cell Rep. 2015, 12, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Nagai, S.; Davis, R.E.; Mattei, P.J.; Eagen, K.P.; Kornberg, R.D. Chromatin potentiates transcription. Proc. Natl. Acad. Sci. USA 2017, 114, 1536–1541. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.; Minneci, F.; Nugent, T.C.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef]
- De Beer, T.A.; Berka, K.; Thornton, J.M.; Laskowski, R.A. PDBsum additions. Nucleic Acids Res. 2014, 42, D292–D296. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protocols. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, D.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Mao, B.; Aramini, J.M.; Montelione, G.T. Assessment of template-based protein structure predictions in CASP10. Proteins 2014, 82, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyy, B.; Kryshtafovych, A.; Moult, J.; Tramontano, A.; Fidelis, K. Assessment of protein disorder region predictions in CASP10. Proteins 2014, 82, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, E.; Kendrew, S.G.; Caserta, M. Two distinct nucleosome alterations characterize chromatin remodelling at the Saccharomyces cerevisiae ADH2 promoter. J. Biol. Chem. 2000, 275, 7612–7618. [Google Scholar] [CrossRef] [PubMed]
- Weidner, M.; Taupp, M.; Hallam, S.J. Expression of recombinant proteins in the methylotrophic yeast Pichia pastoris. J. Vis. Exp. 2010, 25. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyre, D.E.; Klevit, R.E. A disorder-to-order transition coupled to DNA binding in the essential zinc-finger DNA-binding domain of yeast Adr1. J. Mol. Biol. 1998, 279, 929–943. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J. Biol. Chem. 2016, 291, 6714–6722. [Google Scholar] [CrossRef] [Green Version]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Schaufler, L.E.; Klevit, R.E. Mechanism of DNA binding by the Adr1 zinc finger transcription factor as determined by SPR. J. Mol. Biol. 2003, 329, 931–939. [Google Scholar] [CrossRef]
- Haase, S.B.; Wittenberg, C. Topology and control of the cell-cycle-regulated transcriptional circuitry. Genetics. 2014, 196, 65–90. [Google Scholar] [CrossRef] [PubMed]
- Lieleg, C.; Krietenstein, N.; Walker, M.; Korber, P. Nucleosome positioning in yeasts: methods, maps, and mechanisms. Chromosoma 2015, 124, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Rudnizky, S.; Malik, O.; Bavly, A.; Pnueli, L.; Melamed, P.; Kaplan, A. Nucleosome mobility and the regulation of gene expression: Insights from single-molecule studies. Protein Sci. 2017, 26, 1266–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
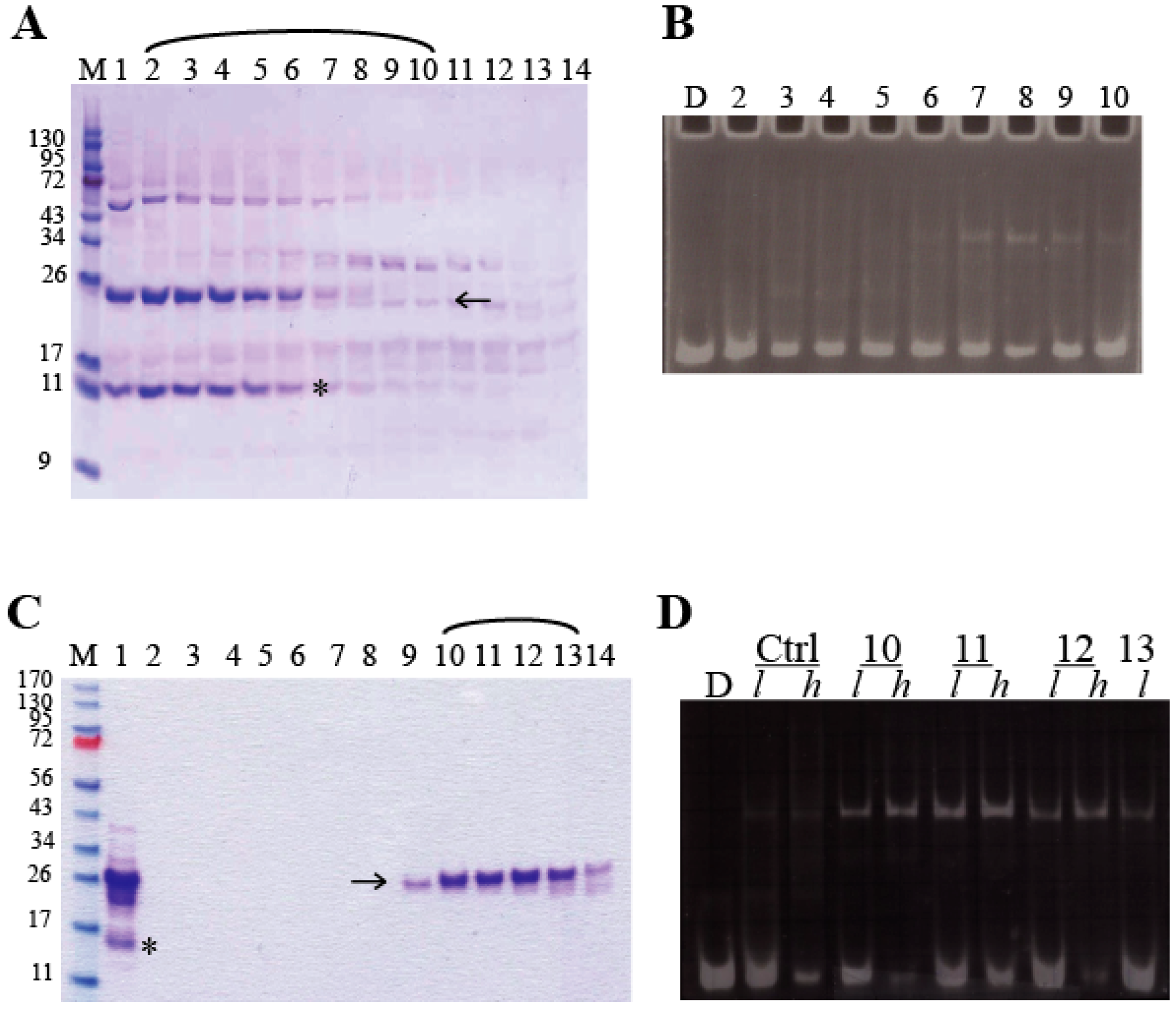
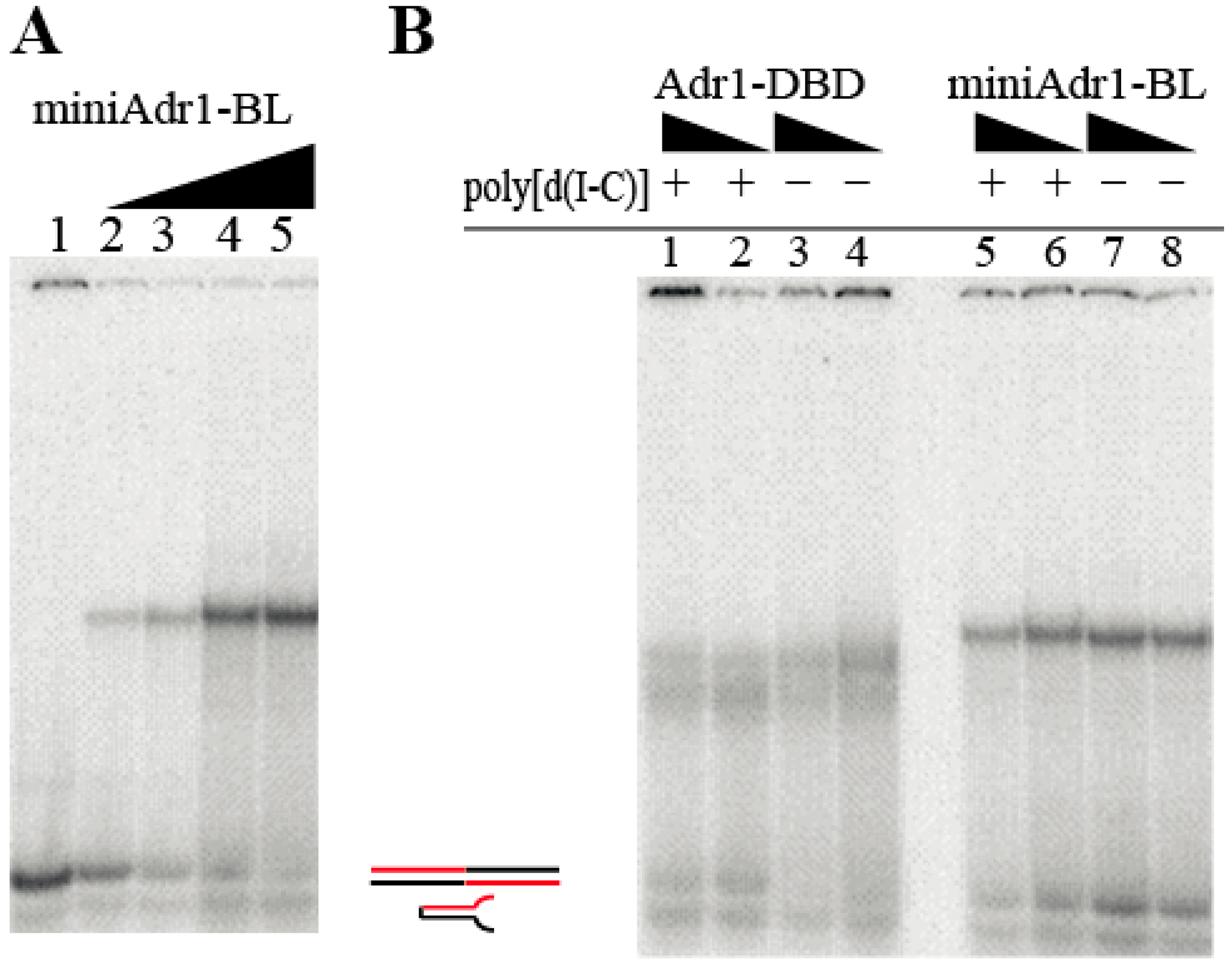

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Escherichia coli (His-tag) | Komagataella pastoris (His-tag) | K. pastoris (no tag) | |
|---|---|---|---|
| Adr1-651 | Insoluble (inclusion bodies, IB) | - | - |
| Adr1-651_Δ158–400 | Insoluble (IB) | - | - |
| Adr1-471 | Insoluble (IB) | insoluble | - |
| MiniAdr1-AL | <1 mg/L culture | <1 mg/L | 1 mg/L unstable1 |
| MiniAdr1-BL | <1 mg/L | <1 mg/L | 3 mg/L |
| Adr1-DBD | - | - | 1 mg/L unstable1 |
| HisAdr1-165 | ~1 mg/L unstable 1 | - | - |
| Oligonucleotide Name | Sequence |
|---|---|
| Palindromic | 5′–CCGGCCTCTCCAACTTA·TAAGTTGGAGAGG–3′ |
| Half site “a” | 5′–TCCGGGCATCTCCAACTTA–3′ |
| Half site “b” | 5′–CTAAGTTGGAGATGCCCGG–3′ |
| Composition of the Translated Proteins with Respect to the Full Length Primary Structure | Linker Sequence | |
|---|---|---|
| miniAdr1-AL | (75–157)–(401–471) | 401–SWTVAIDNNSNNNKVSDN–418 |
| miniAdr1-BL | (75–178)–(419–471) | 158–NLGETISHTKKVSRTITKAR–178 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buttinelli, M.; Panetta, G.; Bucci, A.; Frascaria, D.; Morea, V.; Miele, A.E. Protein Engineering of Multi-Modular Transcription Factor Alcohol Dehydrogenase Repressor 1 (Adr1p), a Tool for Dissecting In Vitro Transcription Activation. Biomolecules 2019, 9, 497. https://doi.org/10.3390/biom9090497
Buttinelli M, Panetta G, Bucci A, Frascaria D, Morea V, Miele AE. Protein Engineering of Multi-Modular Transcription Factor Alcohol Dehydrogenase Repressor 1 (Adr1p), a Tool for Dissecting In Vitro Transcription Activation. Biomolecules. 2019; 9(9):497. https://doi.org/10.3390/biom9090497
Chicago/Turabian StyleButtinelli, Memmo, Gianna Panetta, Ambra Bucci, Daniele Frascaria, Veronica Morea, and Adriana Erica Miele. 2019. "Protein Engineering of Multi-Modular Transcription Factor Alcohol Dehydrogenase Repressor 1 (Adr1p), a Tool for Dissecting In Vitro Transcription Activation" Biomolecules 9, no. 9: 497. https://doi.org/10.3390/biom9090497