
Quantitative Proteomics Reveal Region-Specific Alterations in Neuroserpin-Deficient Mouse Brain and Retina: Insights into Serpini1 Function

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
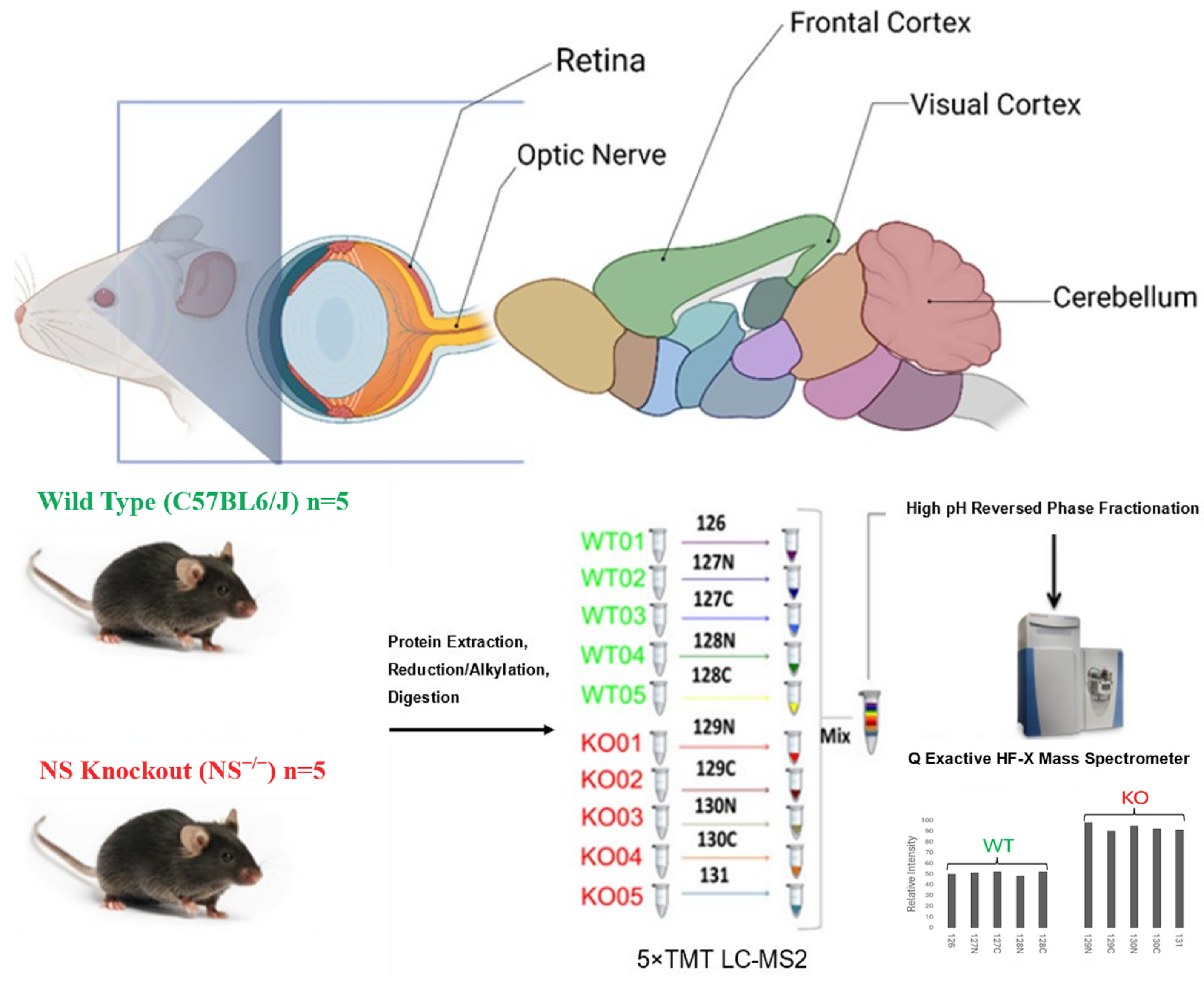
2.1. Animals
2.2. Protein Sample Preparation
2.3. Tandem Mass Tag Labelling of Peptides
2.4. Liquid Chromatography-Tandem Mass Spectrometry
2.5. Database Search, Peptide Quantification and Statistical Analysis
2.6. Normalization of Multiplexed Quantitative Proteomics Data and Bioinformatic Analysis
2.7. Protein Validation by Western Blotting
2.8. Statistical Analysis
3. Results
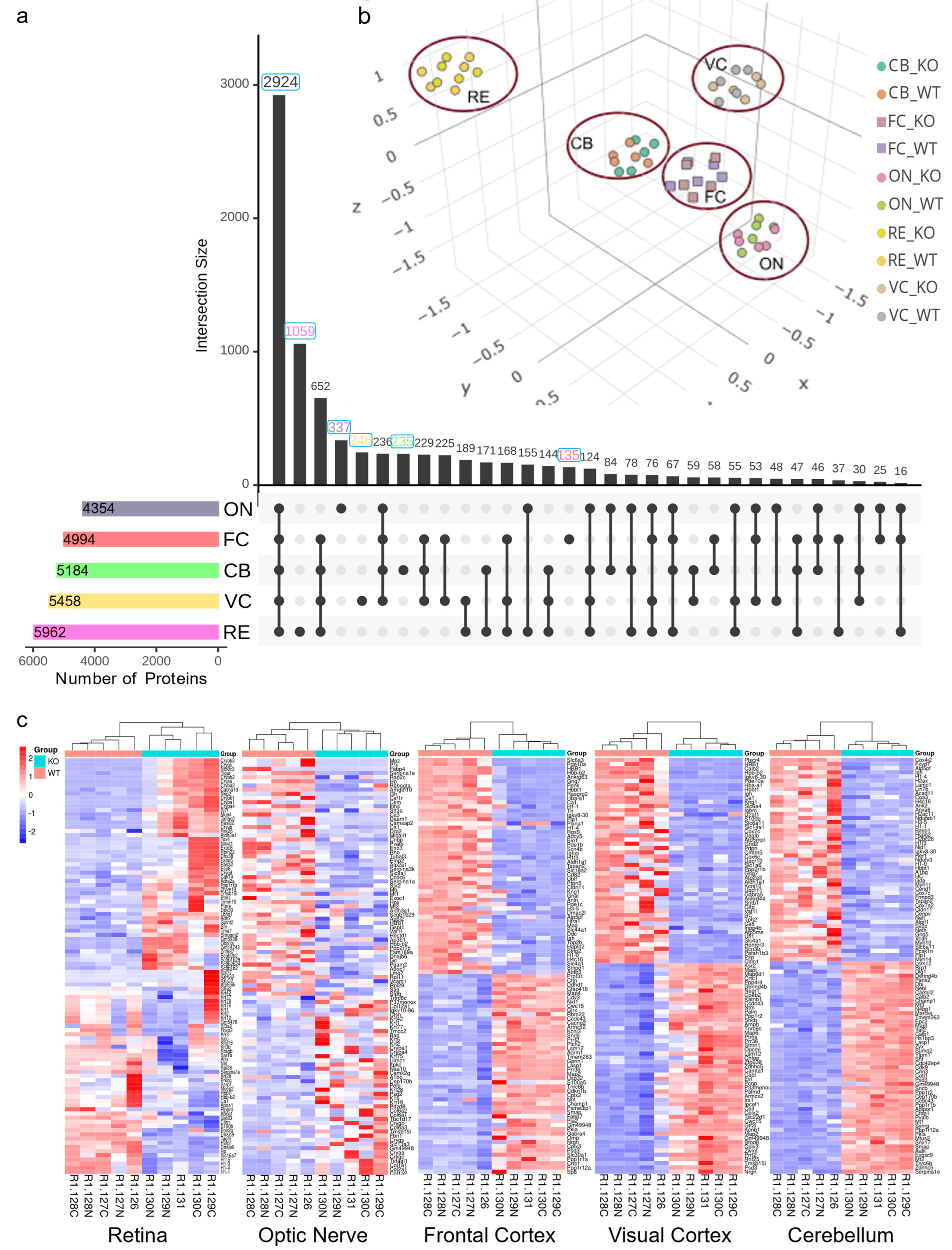
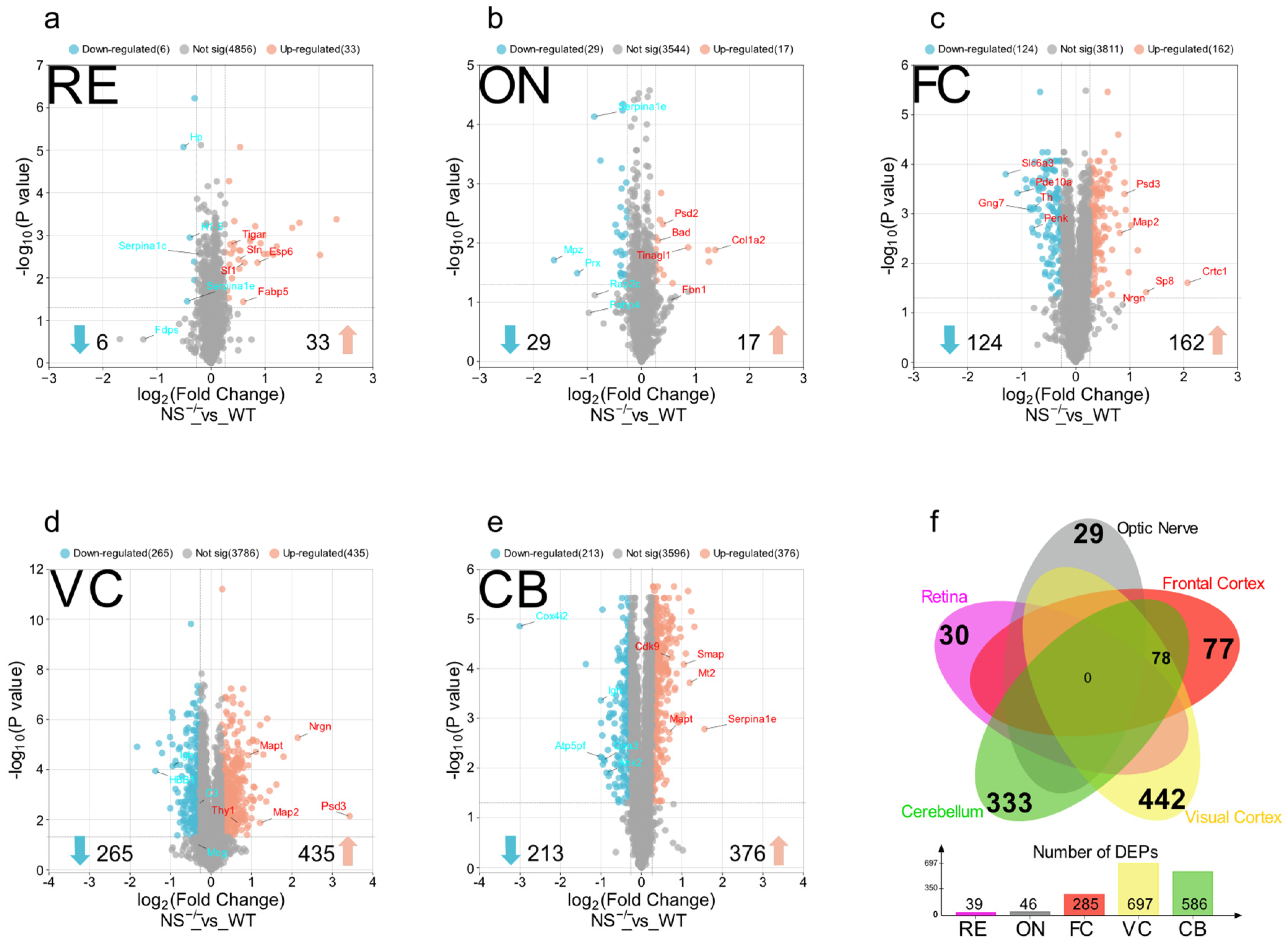
3.1. Differential Regulation of the Proteome in the Different Brain Regions of Neuroserpin-Deficient Mice
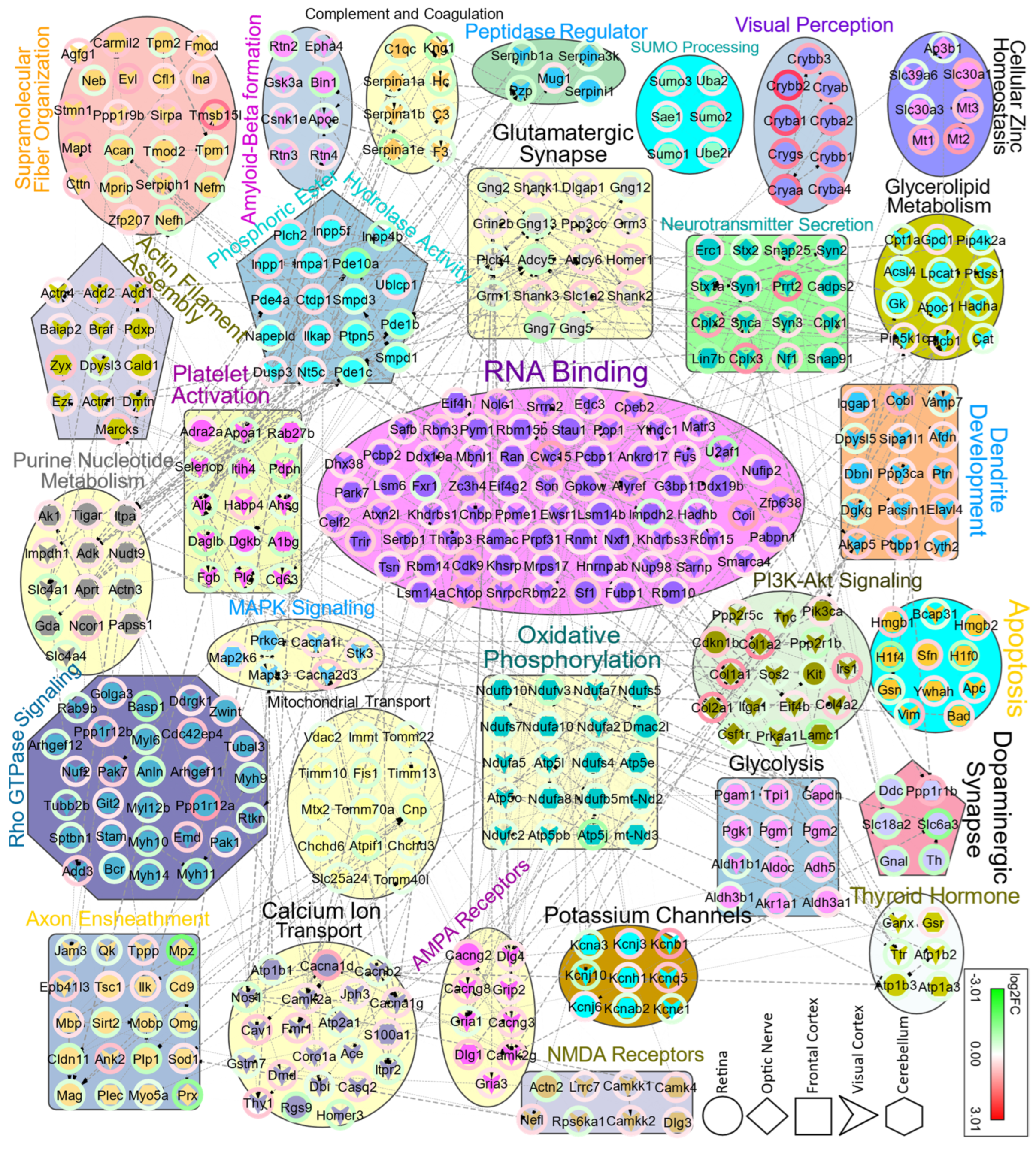
3.2. Functional Pathway and Protein–Protein Interaction Analysis Identify Region-Specific Pathways Affected in the Brain and Retina of NS−/− Mice
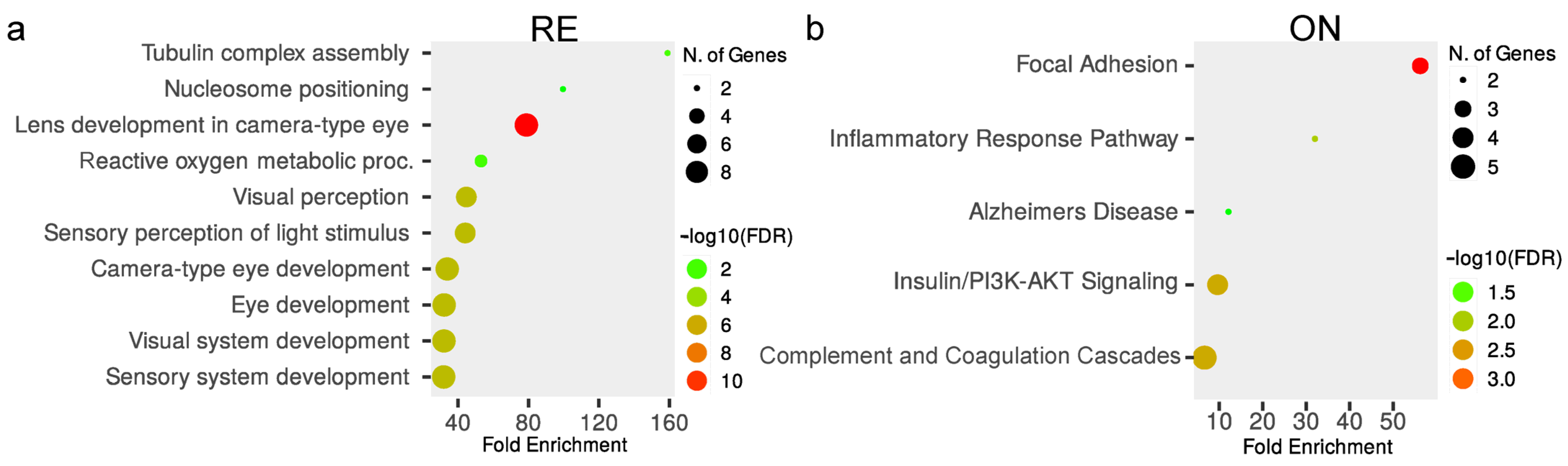
3.3. Nucleosome Positioning and Visual Perception Are Affected in the Retina of Knockout Mice
3.4. Optic Nerve Shows Dysregulation of Focal Adhesion and Complement/Coagulation Cascades in the Absence of Serpini1
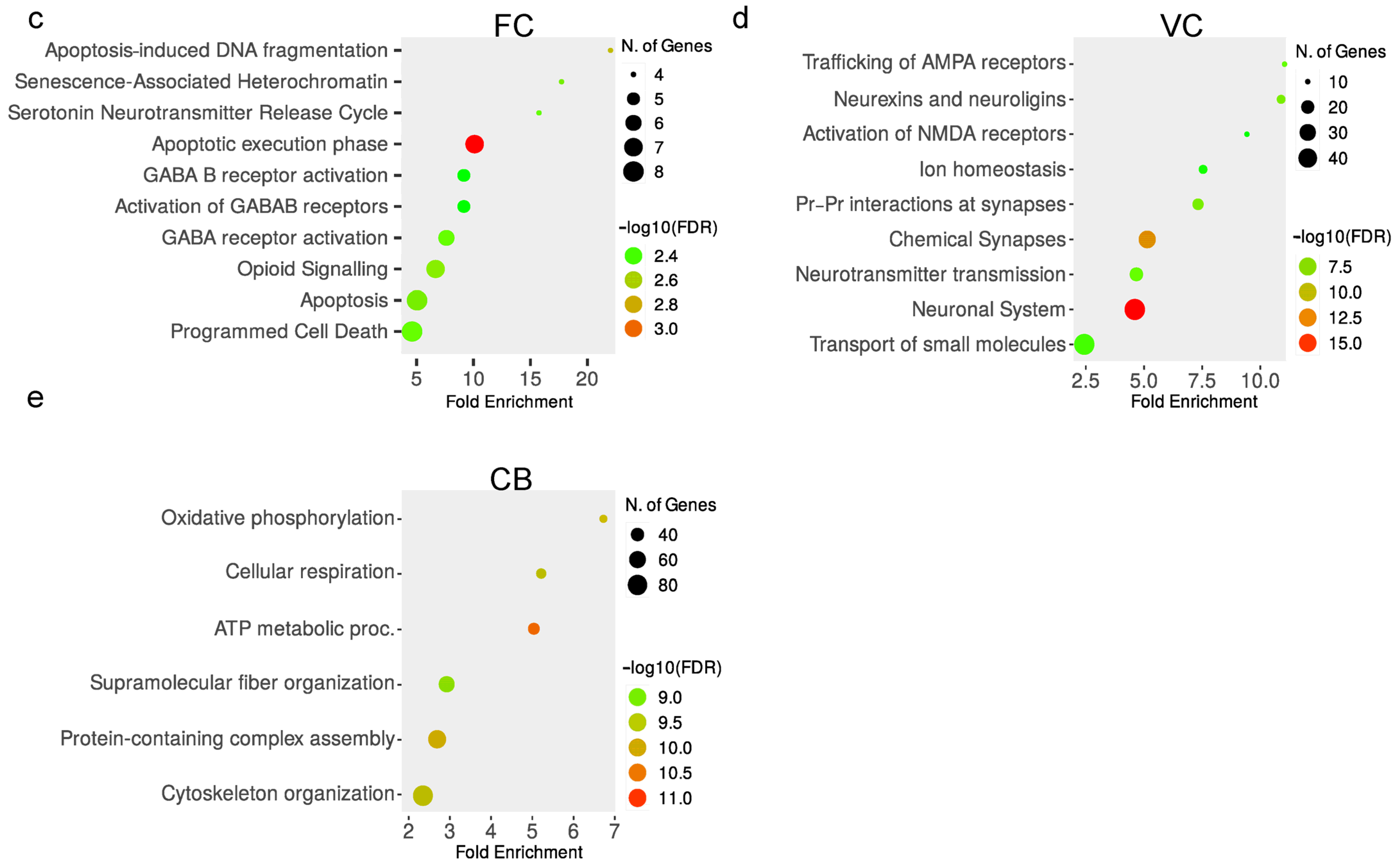
3.5. Activation of GABA Receptors and Apoptosis in the Frontal Cortex of NS−/− Mice
3.6. Enrichment of Glutamate Receptor Activation and Neurexins/Neuroligins in the Visual Cortex
3.7. Oxidative Phosphorylation and Supramolecular Fiber Organization Are Affected in the Cerebellum of NS−/− Mice
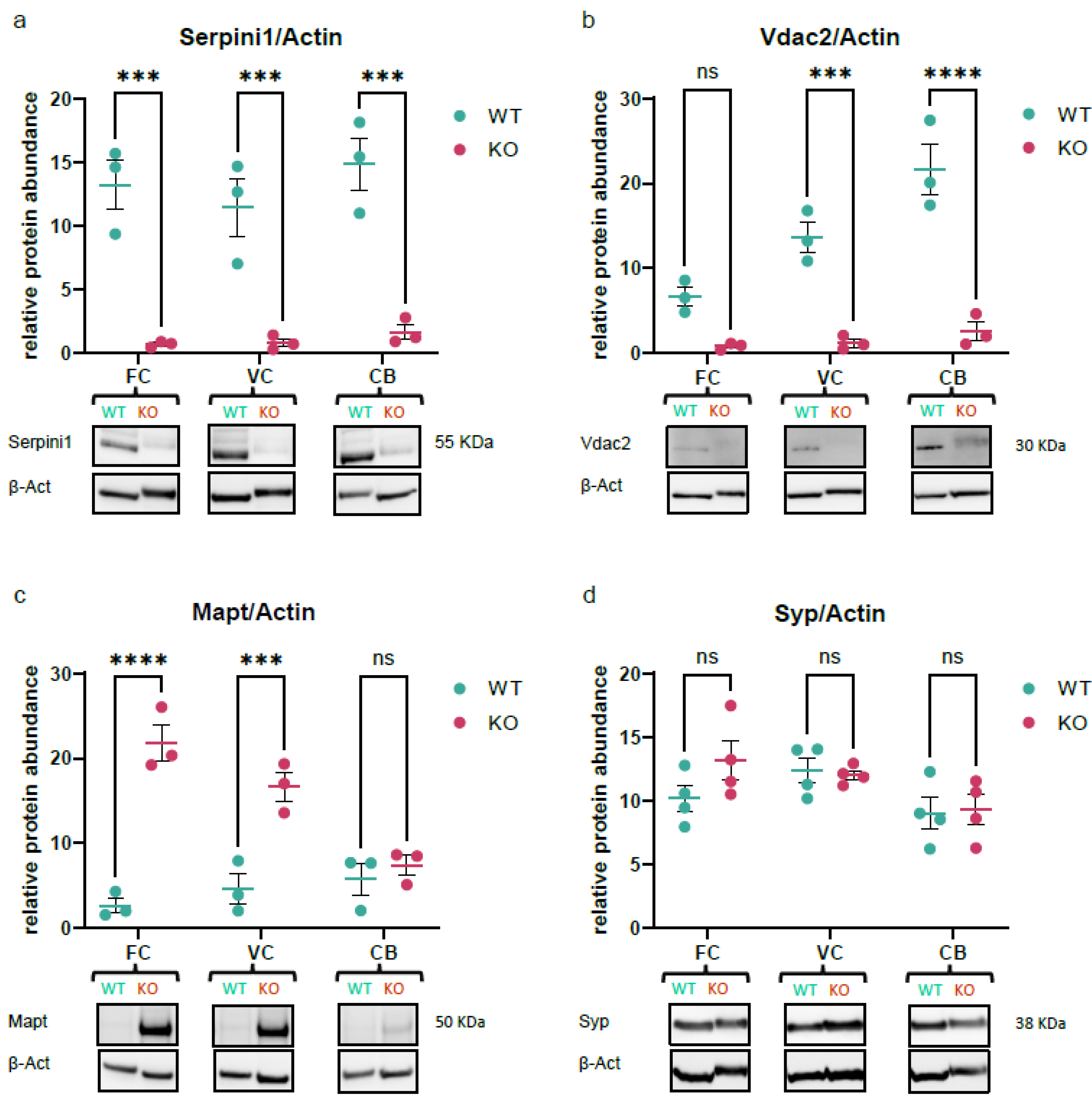
3.8. Validation of Quantitative MS Data Using Western Blotting
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 301–307. [Google Scholar]
- Patterson, C. World Alzheimer Report 2018; Alzheimer’s Disease International (ADI): London, UK, 2018. [Google Scholar]
- Temple, J.B.; McDonald, P.F. Population ageing and the labour force: 2000–2015 and 2015–2030. Australas. J. Ageing 2017, 36, 264–270. [Google Scholar] [CrossRef]
- Knickman, J.R.; Snell, E.K. The 2030 problem: Caring for aging baby boomers. Health Serv. Res. 2002, 37, 849–884. [Google Scholar] [CrossRef]
- James, B.D.; Bennett, D.A. Causes and Patterns of Dementia: An Update in the Era of Redefining Alzheimer’s Disease. Annu. Rev. Public Health 2019, 40, 65–84. [Google Scholar] [CrossRef]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef]
- Zádori, D.; Klivényi, P.; Szalárdy, L.; Fülöp, F.; Toldi, J.; Vécsei, L. Mitochondrial disturbances, excitotoxicity, neuroinflammation and kynurenines: Novel therapeutic strategies for neurodegenerative disorders. J. Neurol. Sci. 2012, 322, 187–191. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- Ahmad Bhat, S.; Amjad Kamal, M.; Sastry Yarla, N.; Md Ashraf, G. Synopsis on managment strategies for neurodegenerative disorders: Challenges from bench to bedside in successful drug discovery and development. Curr. Top. Med. Chem. 2017, 17, 1371–1378. [Google Scholar] [CrossRef]
- Galliciotti, G.; Sonderegger, P. Neuroserpin. Front. Biosci. 2006, 11, 33–45. [Google Scholar] [CrossRef]
- Hastings, G.A.; Coleman, T.A.; Haudenschild, C.C.; Stefansson, S.; Smith, E.P.; Barthlow, R.; Cherry, S.; Sandkvist, M.; Lawrence, D.A. Neuroserpin, a brain-associated inhibitor of tissue plasminogen activator is localized primarily in neurons. Implications for the regulation of motor learning and neuronal survival. J. Biol. Chem. 1997, 272, 33062–33067. [Google Scholar] [CrossRef]
- Yepes, M.; Lawrence, D.A. Neuroserpin: A selective inhibitor of tissue-type plasminogen activator in the central nervous system. Thromb. Haemost. 2004, 91, 457–464. [Google Scholar] [CrossRef]
- Gelderblom, M.; Neumann, M.; Ludewig, P.; Bernreuther, C.; Krasemann, S.; Arunachalam, P.; Gerloff, C.; Glatzel, M.; Magnus, T. Deficiency in serine protease inhibitor neuroserpin exacerbates ischemic brain injury by increased postischemic inflammation. PLoS ONE 2013, 8, e63118. [Google Scholar] [CrossRef]
- Reumann, R.; Vierk, R.; Zhou, L.; Gries, F.; Kraus, V.; Mienert, J.; Romswinkel, E.; Morellini, F.; Ferrer, I.; Nicolini, C.; et al. The serine protease inhibitor neuroserpin is required for normal synaptic plasticity and regulates learning and social behavior. Learn. Mem. 2017, 24, 650–659. [Google Scholar] [CrossRef]
- Lee, T.W.; Coates, L.C.; Birch, N.P. Neuroserpin regulates N-cadherin-mediated cell adhesion independently of its activity as an inhibitor of tissue plasminogen activator. J. Neurosci. Res. 2008, 86, 1243–1253. [Google Scholar] [CrossRef]
- Hakak, Y.; Walker, J.R.; Li, C.; Wong, W.H.; Davis, K.L.; Buxbaum, J.D.; Haroutunian, V.; Fienberg, A.A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4746–4751. [Google Scholar] [CrossRef]
- Fabbro, S.; Seeds, N.W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 2009, 109, 303–315. [Google Scholar] [CrossRef]
- Gupta, V.; Mirzaei, M.; Gupta, V.B.; Chitranshi, N.; Dheer, Y.; Vander Wall, R.; Abbasi, M.; You, Y.; Chung, R.; Graham, S. Glaucoma is associated with plasmin proteolytic activation mediated through oxidative inactivation of neuroserpin. Sci. Rep. 2017, 7, 8412. [Google Scholar] [CrossRef]
- Zattoni, M.; Mearelli, M.; Vanni, S.; Colini Baldeschi, A.; Tran, T.H.; Ferracin, C.; Catania, M.; Moda, F.; Di Fede, G.; Giaccone, G. Serpin signatures in prion and Alzheimer’s diseases. Mol. Neurobiol. 2022, 59, 3778–3799. [Google Scholar] [CrossRef]
- Chitranshi, N.; Rajput, R.; Godinez, A.; Pushpitha, K.; Mirzaei, M.; Basavarajappa, D.; Gupta, V.; Sharma, S.; You, Y.; Galliciotti, G. Neuroserpin gene therapy inhibits retinal ganglion cell apoptosis and promotes functional preservation in glaucoma. Mol. Ther. 2023, 31, 2056–2076. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Seger, C.; Salzmann, L. After another decade: LC–MS/MS became routine in clinical diagnostics. Clin. Biochem. 2020, 82, 2–11. [Google Scholar] [CrossRef]
- Dayon, L.; Affolter, M. Progress and pitfalls of using isobaric mass tags for proteome profiling. Expert. Rev. Proteom. 2020, 17, 149–161. [Google Scholar] [CrossRef]
- O’Connell, J.D.; Paulo, J.A.; O’Brien, J.J.; Gygi, S.P. Proteome-wide evaluation of two common protein quantification methods. J. Proteome Res. 2018, 17, 1934–1942. [Google Scholar] [CrossRef]
- Banfi, C.; Baetta, R.; Gianazza, E.; Tremoli, E. Technological advances and proteomic applications in drug discovery and target deconvolution: Identification of the pleiotropic effects of statins. Drug Discov. Today 2017, 22, 848–869. [Google Scholar] [CrossRef]
- Ning, M.; Lopez, M.; Cao, J.; Buonanno, F.S.; Lo, E.H. Application of proteomics to cerebrovascular disease. Electrophoresis 2012, 33, 3582–3597. [Google Scholar] [CrossRef]
- Rayaprolu, S.; Higginbotham, L.; Bagchi, P.; Watson, C.M.; Zhang, T.; Levey, A.I.; Rangaraju, S.; Seyfried, N.T. Systems-based proteomics to resolve the biology of Alzheimer’s disease beyond amyloid and tau. Neuropsychopharmacology 2021, 46, 98–115. [Google Scholar] [CrossRef]
- Hall, B.; Limaye, A.; Kulkarni, A.B. Overview: Generation of gene knockout mice. Curr. Protoc. Cell Biol. 2009, 44, 19.12.1–19.12.17. [Google Scholar] [CrossRef]
- Wong, H.H.-W.; Chou, C.Y.C.; Watt, A.J.; Sjöström, P.J. Comparing mouse and human brains. eLife 2023, 12, e90017. [Google Scholar] [CrossRef]
- Loomba, S.; Straehle, J.; Gangadharan, V.; Heike, N.; Khalifa, A.; Motta, A.; Ju, N.; Sievers, M.; Gempt, J.; Meyer, H.S.; et al. Connectomic comparison of mouse and human cortex. Science 2022, 377, eabo0924. [Google Scholar] [CrossRef]
- Kement, D.; Reumann, R.; Schostak, K.; Voß, H.; Douceau, S.; Dottermusch, M.; Schweizer, M.; Schlüter, H.; Vivien, D.; Glatzel, M. Neuroserpin is strongly expressed in the developing and adult mouse neocortex but its absence does not perturb cortical lamination and synaptic proteome. Front. Neuroanat. 2021, 15, 627896. [Google Scholar] [CrossRef]
- Madani, R.; Kozlov, S.; Akhmedov, A.; Cinelli, P.; Kinter, J.; Lipp, H.-P.; Sonderegger, P.; Wolfer, D.P. Impaired explorative behavior and neophobia in genetically modified mice lacking or overexpressing the extracellular serine protease inhibitor neuroserpin. Mol. Cell. Neurosci. 2003, 23, 473–494. [Google Scholar] [CrossRef]
- Wu, J.; Cai, Y.; Wu, X.; Ying, Y.; Tai, Y.; He, M. Transcardiac Perfusion of the Mouse for Brain Tissue Dissection and Fixation. Bio Protoc. 2021, 11, e3988. [Google Scholar] [CrossRef]
- Winkler, B.S. Dependence of fast components of the electroretinogram of the isolated rat retina on the ionic environment. Vis. Res. 1973, 13, 457–463. [Google Scholar] [CrossRef]
- Spijker, S. Dissection of Rodent Brain Regions. In Neuroproteomics; Li, K.W., Ed.; Humana Press: New York, NY USA; Heidelberg, Germany, 2011; pp. 23–26. [Google Scholar]
- Ludwig, K.R.; Schroll, M.M.; Hummon, A.B. Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. J. Proteome Res. 2018, 17, 2480–2490. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Mirzaei, M.; Pascovici, D.; Wu, J.X.; Chick, J.; Wu, Y.; Cooke, B.; Haynes, P.; Molloy, M.P. TMT one-stop shop: From reliable sample preparation to computational analysis platform. Proteome Bioinform. 2017, 1549, 45–66. [Google Scholar]
- Akhmedov, M.; Martinelli, A.; Geiger, R.; Kwee, I. Omics Playground: A comprehensive self-service platform for visualization, analytics and exploration of Big Omics Data. NAR Genom. Bioinform. 2020, 2, lqz019. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2019, 36, 2628–2629. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network analysis and visualization of proteomics data. J. Proteome Res. 2018, 18, 623–632. [Google Scholar] [CrossRef]
- Legeay, M.; Doncheva, N.T.; Morris, J.H.; Jensen, L.J. Visualize omics data on networks with Omics Visualizer, a Cytoscape App. F1000Research 2020, 9, 157. [Google Scholar] [CrossRef]
- Mirshahvaladi, S.; Topraggaleh, T.R.; Bucak, M.N.; Rahimizadeh, P.; Shahverdi, A. Quantitative proteomics of sperm tail in asthenozoospermic patients: Exploring the molecular pathways affecting sperm motility. Cell Tissue Res. 2023, 392, 793–810. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Taylor, S.C.; Posch, A. The Design of a Quantitative Western Blot Experiment. BioMed Res. Int. 2014, 2014, 361590. [Google Scholar] [CrossRef]
- Liu, P.; Beer, L.A.; Ky, B.; Barnhart, K.T.; Speicher, D.W. Quantitative comparisons of large numbers of human plasma samples using TMT10plex labeling. Serum/Plasma Proteom. Methods Protoc. 2017, 1619, 319–337. [Google Scholar]
- Brenes, A.; Hukelmann, J.; Bensaddek, D.; Lamond, A.I. Multibatch TMT reveals false positives, batch effects and missing values. Mol. Cell. Proteom. 2019, 18, 1967–1980. [Google Scholar] [CrossRef]
- Myers, S.A.; Klaeger, S.; Satpathy, S.; Viner, R.; Choi, J.; Rogers, J.; Clauser, K.; Udeshi, N.D.; Carr, S.A. Evaluation of advanced precursor determination for tandem mass tag (TMT)-based quantitative proteomics across instrument platforms. J. Proteome Res. 2018, 18, 542–547. [Google Scholar] [CrossRef]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of this Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Rogers, J.C.; Bomgarden, R.D. Sample Preparation for Mass Spectrometry-Based Proteomics; from Proteomes to Peptides. Adv. Exp. Med. Biol. 2016, 919, 43–62. [Google Scholar] [CrossRef]
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Paša-Tolić, L.; Tsybin, Y.O.; Kelleher, N.L.; The Consortium for Top-Down Proteomics. The Human Proteoform Project: Defining the human proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef]
- Majzoub, J.A.; Muglia, L.J. Knockout Mice. N. Engl. J. Med. 1996, 334, 904–906. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Echeverry, R.; Guzman, J.; Yepes, M. Neuroserpin protects neurons from ischemia-induced plasmin-mediated cell death independently of tissue-type plasminogen activator inhibition. Am. J. Pathol. 2010, 177, 2576–2584. [Google Scholar] [CrossRef]
- Ma, J.; Tong, Y.; Yu, D.; Mao, M. Tissue plasminogen activator-independent roles of neuroserpin in the central nervous system. Neural Regen. Res. 2012, 7, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.W.; Tsang, V.W.; Loef, E.J.; Birch, N.P. Physiological and pathological functions of neuroserpin: Regulation of cellular responses through multiple mechanisms. Semin. Cell Dev. Biol. 2017, 62, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Schrimpf, S.P.; Bleiker, A.J.; Brecevic, L.; Kozlov, S.V.; Berger, P.; Osterwalder, T.; Krueger, S.R.; Schinzel, A.; Sonderegger, P. Human neuroserpin (PI12): cDNA cloning and chromosomal localization to 3q26. Genomics 1997, 40, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Sandkvist, M.; Tsui, F.; Moore, E.; Coleman, T.A.; Lawrence, D.A. Identification of a novel targeting sequence for regulated secretion in the serine protease inhibitor neuroserpin. Biochem. J. 2007, 402, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Demb, J.B.; Singer, J.H. Functional circuitry of the retina. Annu. Rev. Vis. Sci. 2015, 1, 263–289. [Google Scholar] [CrossRef]
- Selhorst, J.B.; Chen, Y. The optic nerve. In Seminars in Neurology; Thieme Medical Publishers: Leipzig, Germany, 2009; pp. 29–35. [Google Scholar]
- Ebbesen, C.L.; Insanally, M.N.; Kopec, C.D.; Murakami, M.; Saiki, A.; Erlich, J.C. More than Just a “Motor”: Recent Surprises from the Frontal Cortex. J. Neurosci. 2018, 38, 9402–9413. [Google Scholar] [CrossRef]
- Hübener, M. Mouse visual cortex. Curr. Opin. Neurobiol. 2003, 13, 413–420. [Google Scholar] [CrossRef]
- Reeber, S.L.; Otis, T.S.; Sillitoe, R.V. New roles for the cerebellum in health and disease. Front. Syst. Neurosci. 2013, 7, 83. [Google Scholar] [CrossRef]
- Shi, H.; He, Y.; Zhou, Y.; Huang, J.; Maher, K.; Wang, B.; Tang, Z.; Luo, S.; Tan, P.; Wu, M.; et al. Spatial atlas of the mouse central nervous system at molecular resolution. Nature 2023, 622, 552–561. [Google Scholar] [CrossRef]
- Langlieb, J.; Sachdev, N.S.; Balderrama, K.S.; Nadaf, N.M.; Raj, M.; Murray, E.; Webber, J.T.; Vanderburg, C.; Gazestani, V.; Tward, D.; et al. The molecular cytoarchitecture of the adult mouse brain. Nature 2023, 624, 333–342. [Google Scholar] [CrossRef] [PubMed]
- van Oostrum, M.; Blok, T.M.; Giandomenico, S.L.; tom Dieck, S.; Tushev, G.; Fürst, N.; Langer, J.D.; Schuman, E.M. The proteomic landscape of synaptic diversity across brain regions and cell types. Cell 2023, 186, 5411–5427.e5423. [Google Scholar] [CrossRef]
- Cheng, Y.; Loh, Y.P.; Birch, N.P. Neuroserpin attenuates H 2 O 2-induced oxidative stress in hippocampal neurons via AKT and BCL-2 signaling pathways. J. Mol. Neurosci. 2017, 61, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Subhadra, B.; Schaller, K.; Seeds, N.W. Neuroserpin up-regulation in the Alzheimer’s disease brain is associated with elevated thyroid hormone receptor-β1 and HuD expression. Neurochem. Int. 2013, 63, 476–481. [Google Scholar] [CrossRef]
- Ali, N.H.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alnaaim, S.A.; Alexiou, A.; Papadakis, M.; Saad, H.M.; Batiha, G.E.-S. The probable role of tissue plasminogen activator/neuroserpin axis in Alzheimer’s disease: A new perspective. Acta Neurol. Belg. 2023, 1–12. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Komarov, A.G.; Deng, D.; Craigen, W.J.; Colombini, M. New insights into the mechanism of permeation through large channels. Biophys. J. 2005, 89, 3950–3959. [Google Scholar] [CrossRef]
- Li, J.; Lu, J.; Mi, Y.; Shi, Z.; Chen, C.; Riley, J.; Zhou, C. Voltage-dependent anion channels (VDACs) promote mitophagy to protect neuron from death in an early brain injury following a subarachnoid hemorrhage in rats. Brain Res. 2014, 1573, 74–83. [Google Scholar] [CrossRef]
- Cheng, E.H.-Y.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003, 301, 513–517. [Google Scholar] [CrossRef]
- O’Connell, K.; Ohlendieck, K. Proteomic DIGE analysis of the mitochondria-enriched fraction from aged rat skeletal muscle. Proteomics 2009, 9, 5509–5524. [Google Scholar] [CrossRef]
- Reina, S.; Palermo, V.; Guarnera, A.; Guarino, F.; Messina, A.; Mazzoni, C.; De Pinto, V. Swapping of the N-terminus of VDAC1 with VDAC3 restores full activity of the channel and confers anti-aging features to the cell. FEBS Lett. 2010, 584, 2837–2844. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Oddo, S. Alzheimer’s disease: Aβ, tau and synaptic dysfunction. Trends Mol. Med. 2005, 11, 170–176. [Google Scholar] [CrossRef]
- Abyadeh, M.; Gupta, V.; Paulo, J.A.; Mahmoudabad, A.G.; Shadfar, S.; Mirshahvaladi, S.; Gupta, V.; Nguyen, C.T.; Finkelstein, D.I.; You, Y. Amyloid-beta and tau protein beyond Alzheimer’s disease. Neural Regen. Res. 2024, 19, 1262–1276. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Minthon, L.; Londos, E.; Blennow, K.; Miranda, E.; Perez, J.; Crowther, D.; Lomas, D.; Janciauskiene, S. Plasma and CSF serpins in Alzheimer disease and dementia with Lewy bodies. Neurology 2007, 69, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Ujike, H.; Takaki, M.; Kodama, M.; Kuroda, S. Gene expression related to synaptogenesis, neuritogenesis, and MAP kinase in behavioral sensitization to psychostimulants. Ann. N. Y. Acad. Sci. 2002, 965, 55–67. [Google Scholar] [CrossRef]
- Edfors, F.; Hober, A.; Linderbäck, K.; Maddalo, G.; Azimi, A.; Sivertsson, Å.; Tegel, H.; Hober, S.; Szigyarto, C.A.-K.; Fagerberg, L.; et al. Enhanced validation of antibodies for research applications. Nat. Commun. 2018, 9, 4130. [Google Scholar] [CrossRef] [PubMed]
- Handler, D.C.; Pascovici, D.; Mirzaei, M.; Gupta, V.; Salekdeh, G.H.; Haynes, P.A. The art of validating quantitative proteomics data. Proteomics 2018, 18, 1800222. [Google Scholar] [CrossRef] [PubMed]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the field in multi-omics research: From computational needs to data mining and sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Retina | Optic Nerve | Frontal Cortex | Visual Cortex | Cerebellum | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene ID | FC | Gene ID | FC | Gene ID | FC | Gene ID | FC | Gene ID | FC | |
| Up-Reg (NS−/− vs. WT) | Crybb2 | 5.01 | Col1a2 | 2.58 | Sp8 | 2.47 | Nrgn | 4.42 | Serpina1e | 2.94 |
| Esp6 | 1.82 | Tinagl1 | 1.82 | Ppp1r12a | 2.22 | Psd3 | 3.46 | Zdhhc5 | 2.47 | |
| Nt5dc3 | 1.76 | Pdzd8 | 1.33 | Crtc1 | 2.04 | Tmsb15l | 3.06 | Tnrc6b | 2.34 | |
| Zfyve19 | 1.65 | Psd2 | 1.32 | Slc30a1 | 1.90 | Rnf25 | 2.73 | Mt2 | 2.28 | |
| Sf1 | 1.53 | Cep170b | 1.29 | Psd3 | 1.87 | Prrt2 | 2.62 | Exosc9 | 2.22 | |
| Down-Reg (NS−/− vs. WT) | Hp | 0.70 | Mpz | 0.33 | Pde10a | 0.47 | Plscr4 | 0.28 | Cox4i2 | 0.12 |
| Serpina1e | 0.74 | Prx | 0.44 | HBB1 | 0.53 | Calb2 | 0.41 | Fxyd7 | 0.39 | |
| H1-5 | 0.76 | Serpina1e | 0.55 | Ankrd63 | 0.55 | Pde10a | 0.50 | Atp5pf | 0.50 | |
| Ca1 | 0.81 | Hp | 0.59 | Gng7 | 0.56 | Igh | 0.52 | Igh | 0.50 | |
| Tagln | 0.81 | Arhgef10 | 0.72 | Penk | 0.57 | Ca1 | 0.52 | H1f4 | 0.50 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirshahvaladi, S.; Chitranshi, N.; Amirkhani, A.; Rajput, R.; Basavarajappa, D.; Vander Wall, R.; Pascovici, D.; Godinez, A.; Galliciotti, G.; Paulo, J.A.; et al. Quantitative Proteomics Reveal Region-Specific Alterations in Neuroserpin-Deficient Mouse Brain and Retina: Insights into Serpini1 Function. Proteomes 2024, 12, 7. https://doi.org/10.3390/proteomes12010007
Mirshahvaladi S, Chitranshi N, Amirkhani A, Rajput R, Basavarajappa D, Vander Wall R, Pascovici D, Godinez A, Galliciotti G, Paulo JA, et al. Quantitative Proteomics Reveal Region-Specific Alterations in Neuroserpin-Deficient Mouse Brain and Retina: Insights into Serpini1 Function. Proteomes. 2024; 12(1):7. https://doi.org/10.3390/proteomes12010007
Chicago/Turabian StyleMirshahvaladi, Shahab, Nitin Chitranshi, Ardeshir Amirkhani, Rashi Rajput, Devaraj Basavarajappa, Roshana Vander Wall, Dana Pascovici, Angela Godinez, Giovanna Galliciotti, Joao A. Paulo, and et al. 2024. "Quantitative Proteomics Reveal Region-Specific Alterations in Neuroserpin-Deficient Mouse Brain and Retina: Insights into Serpini1 Function" Proteomes 12, no. 1: 7. https://doi.org/10.3390/proteomes12010007
APA StyleMirshahvaladi, S., Chitranshi, N., Amirkhani, A., Rajput, R., Basavarajappa, D., Vander Wall, R., Pascovici, D., Godinez, A., Galliciotti, G., Paulo, J. A., Gupta, V., Graham, S. L., Gupta, V., & Mirzaei, M. (2024). Quantitative Proteomics Reveal Region-Specific Alterations in Neuroserpin-Deficient Mouse Brain and Retina: Insights into Serpini1 Function. Proteomes, 12(1), 7. https://doi.org/10.3390/proteomes12010007