Early Responses to Severe Drought Stress in the Arabidopsis thaliana Cell Suspension Culture Proteome

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Growth Conditions
2.2. Stress Treatment and Protein Extraction
2.3. Peptide Labelling with Tandem Mass Tag
2.4. High pH Reversed Phase Chromatography Fractionation
2.5. Protein Identification by Mass Spectrometry and Quantification of Differentially Expressed Proteins
TMT-Labelled Quantification
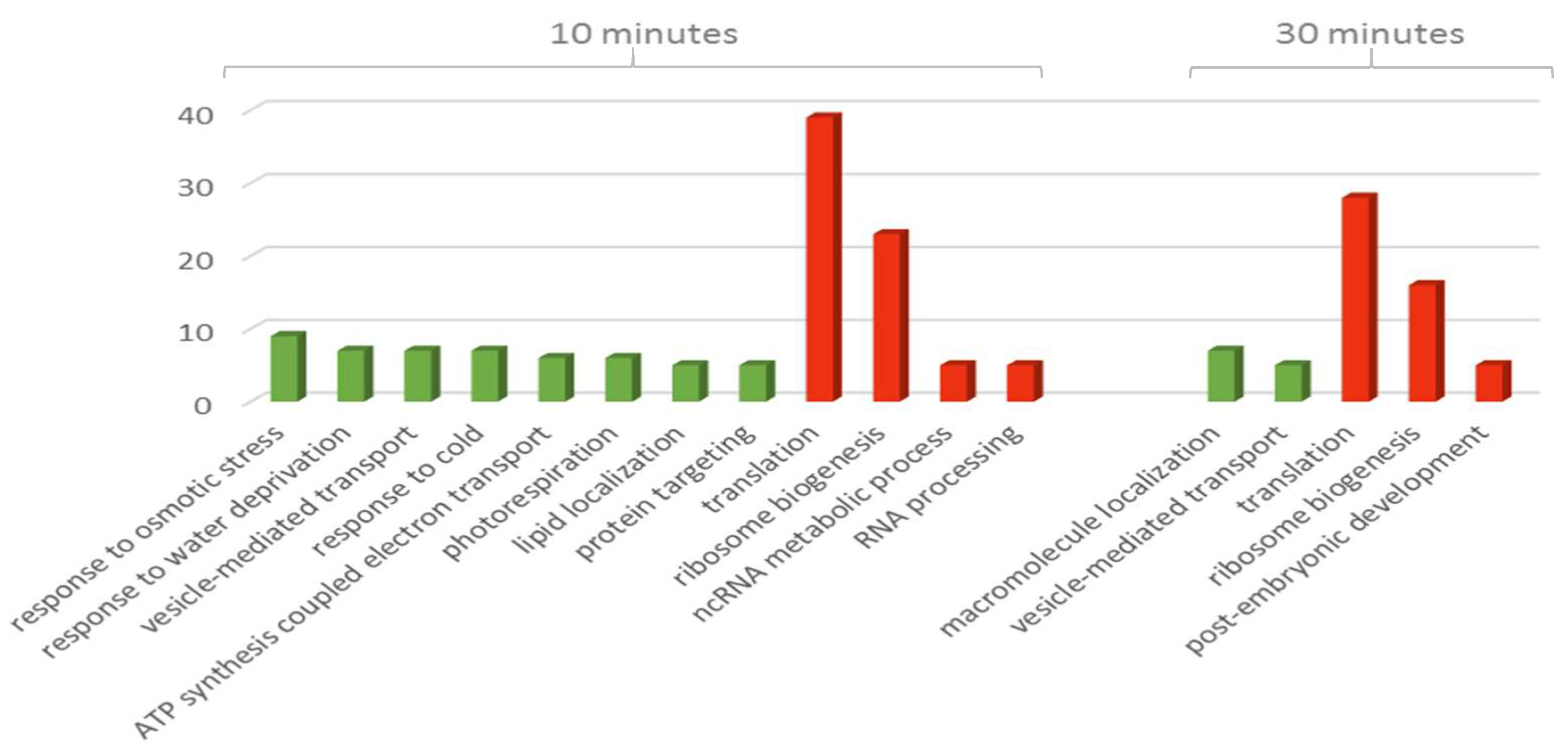
2.6. Computational Analysis of Functional Enrichment
2.7. Cell Viability Test
3. Results
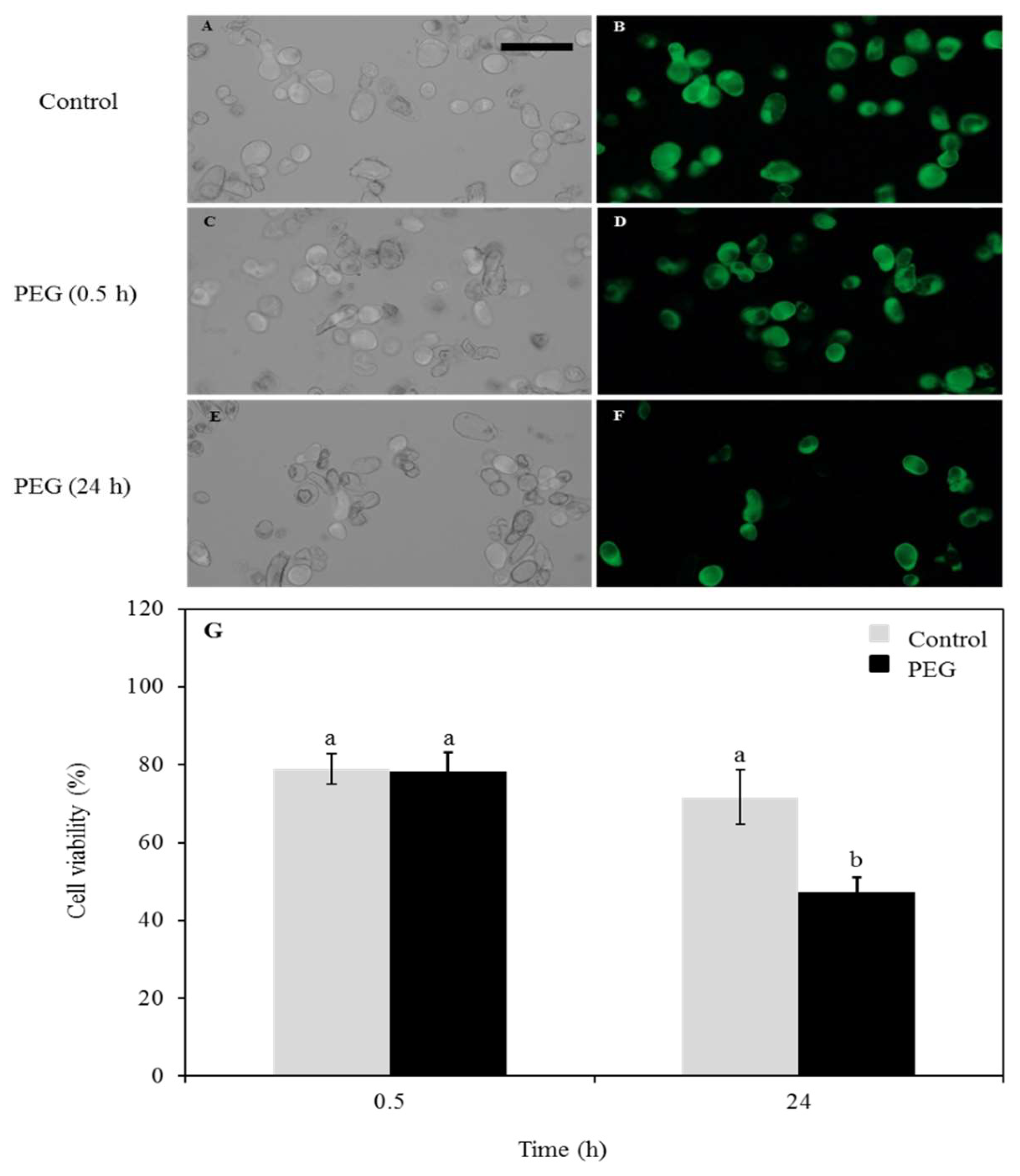
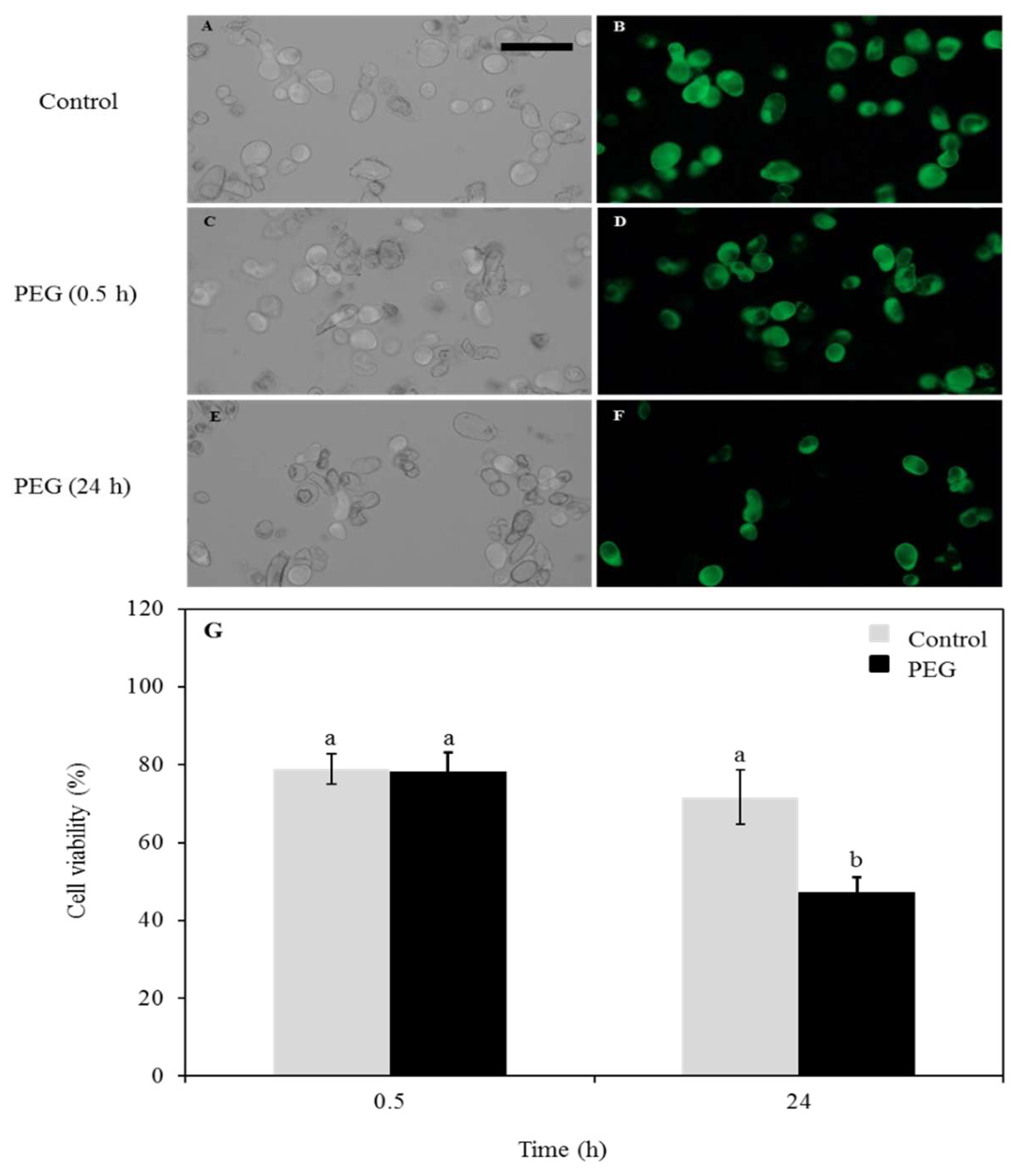
3.1. Cell Viability Test
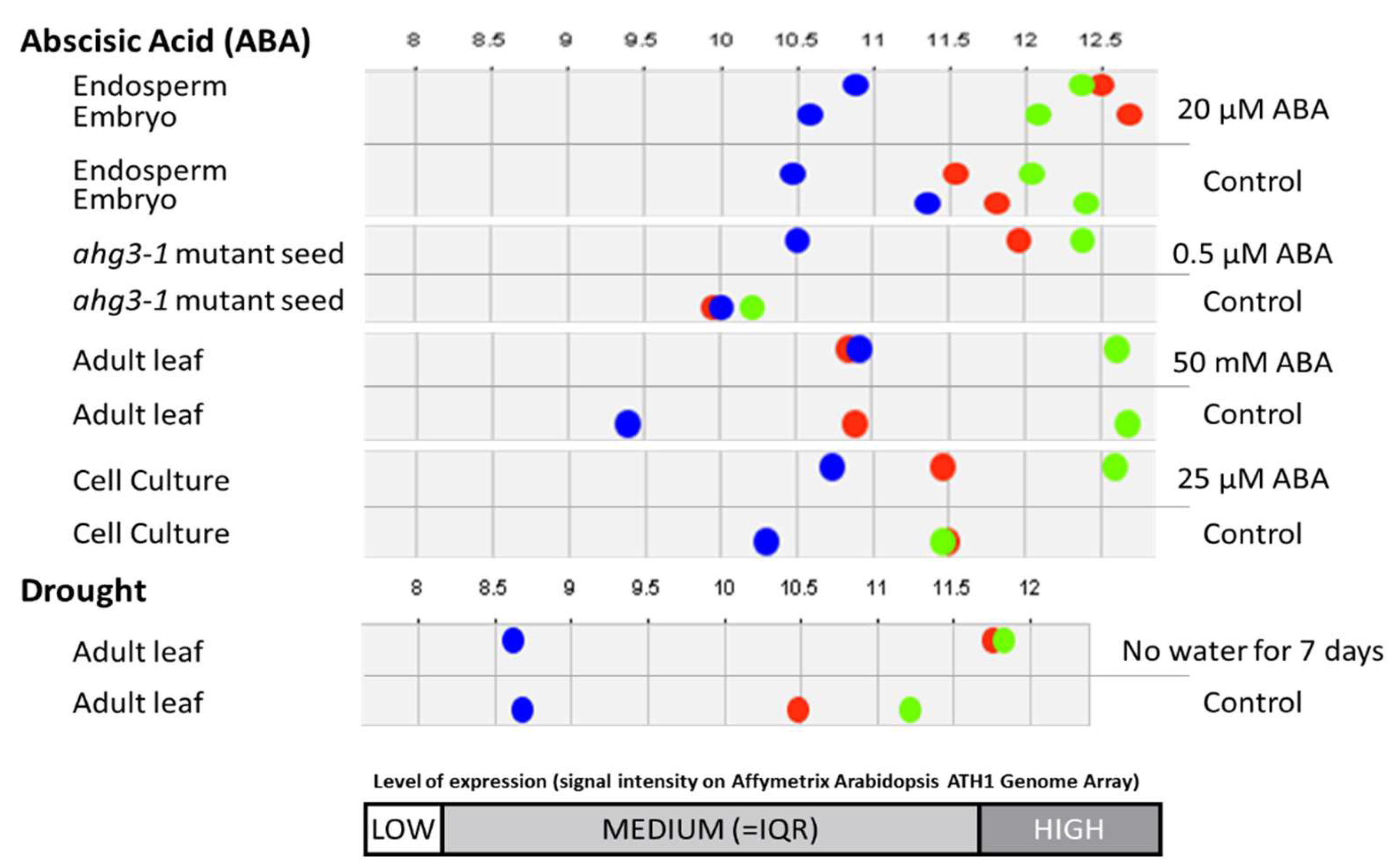
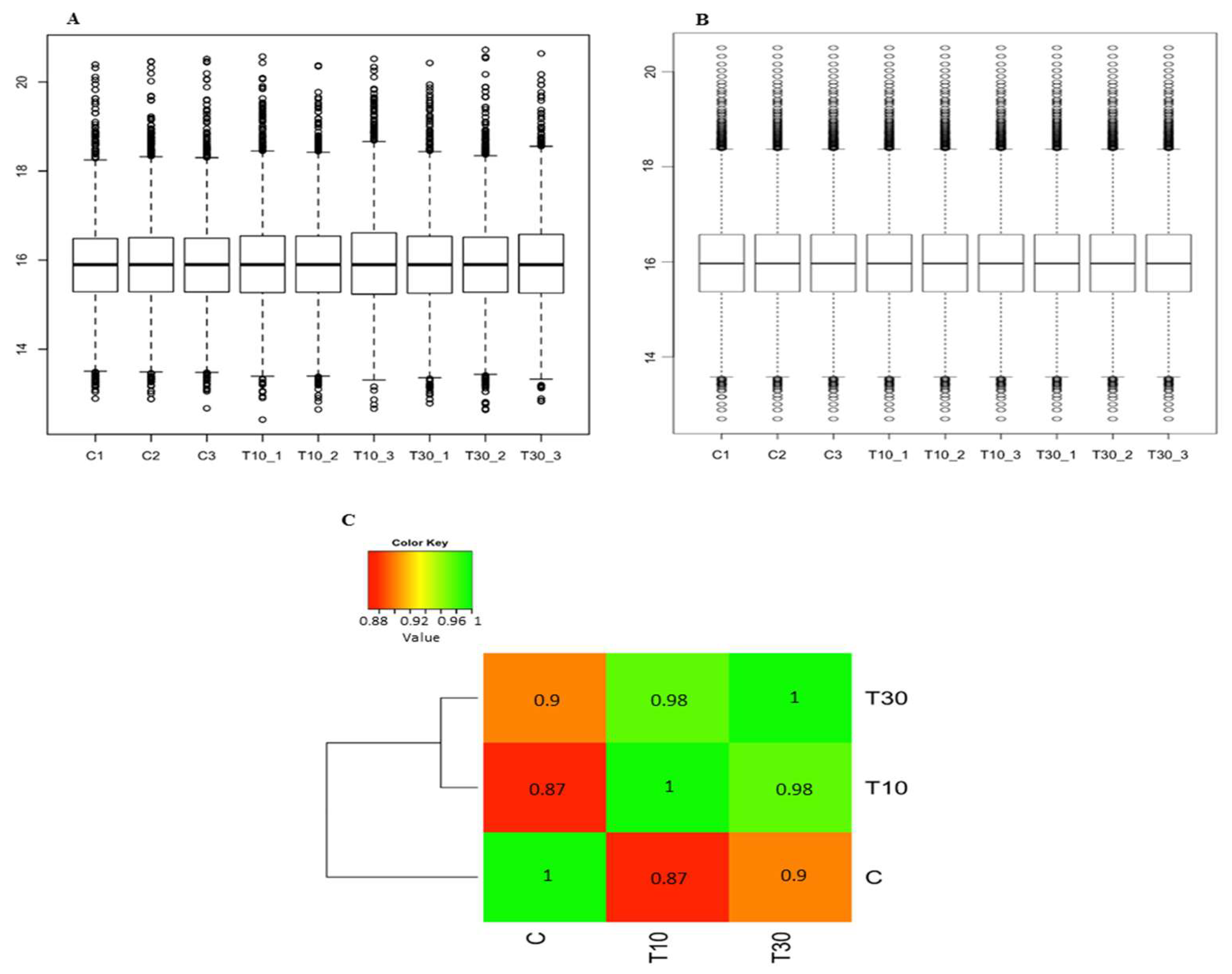
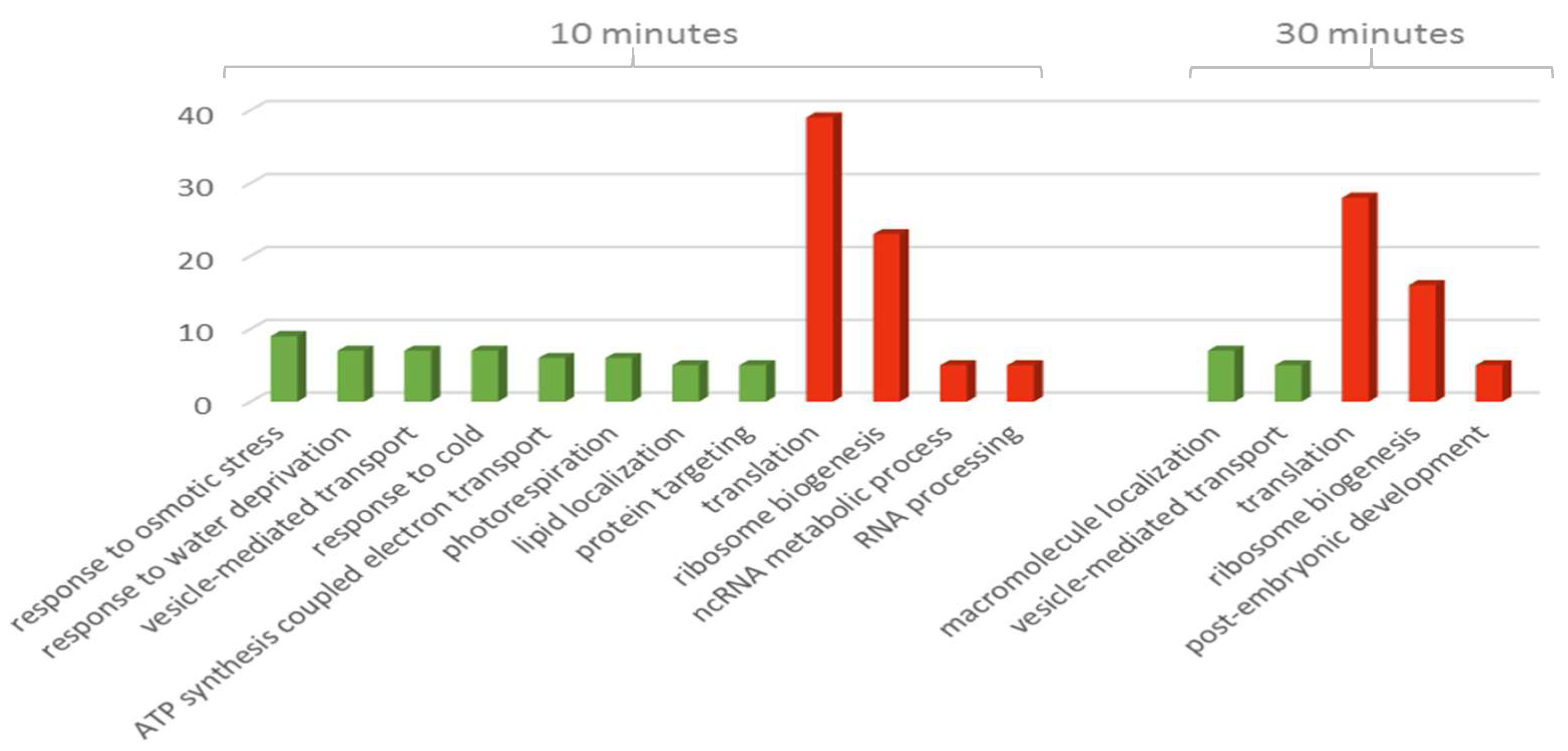
3.2. The Effect of Drought on the Proteomic Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lambers, H.; Chapin, F.S.; Pons, T.L. Plant Physiological Ecology, 2nd ed.; Springer: New York, NY, USA, 2008. [Google Scholar]
- Rosegrant, M.W.; Cline, S.A. Global Food Security: Challenges and Policies. Science 2003, 302, 1917–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, S.; Semiat, R.; Duke, M.; Rahardianto, A.; Cohen, Y. Seawater Use and Desalination Technology. In Treatise on Water Science; Wilderer, P., Ed.; Elsevier: Oxford, UK, 2011; pp. 73–109. [Google Scholar]
- Kiani, S.P.; Talia, P.; Maury, P.; Grieu, P.; Heinz, R.; Perrault, A.; Nishinakamasu, V.; Hopp, E.; Gentzbittel, L.; Paniego, N.; et al. Genetic analysis of plant water status and osmotic adjustment in recombinant inbred lines of sunflower under two water treatments. Plant Sci. 2007, 172, 773–787. [Google Scholar] [CrossRef]
- Farooq, M.; Basra, S.M.A.; Wahid, A.; Ahmad, N.; Saleem, B.A. Improving the Drought Tolerance in Rice (Oryza sativa L.) by Exogenous Application of Salicylic Acid. J. Agron. Crop Sci. 2009, 195, 237–246. [Google Scholar] [CrossRef]
- Taiz, L.; Zeiger, E. Plant Physiology, 5th ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 2010. [Google Scholar]
- Figueiredo, M.V.B.; Burity, H.A.; Martínez, C.R.; Chanway, C.P. Alleviation of drought stress in the common bean (Phaseolus vulgaris L.) by co-inoculation with Paenibacillus polymyxa and Rhizobium tropici. Appl. Soil Ecol. 2008, 40, 182–188. [Google Scholar] [CrossRef]
- Davies, W.J.; Zhang, J. Root Signals and the Regulation of Growth and Development of Plants in Drying Soil. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1991, 42, 55–76. [Google Scholar] [CrossRef]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, R.R.; Rock, C.D. Abscisic acid biosynthesis and response. Arab. Book 2002, 1, e0058. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Lemtiri-Chlieh, F.; Wong, A.; Gehring, C. Direct Modulation of the Guard Cell Outward-Rectifying Potassium Channel (GORK) by Abscisic Acid. Mol. Plant 2017, 10, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Hohl, M.; Schopfer, P. Water Relations of Growing Maize Coleoptiles: Comparison between Mannitol and Polyethylene Glycol 6000 as External Osmotica for Adjusting Turgor Pressure. Plant Physiol. 1991, 95, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Krizek, D.T. Methods of inducing water stress in plants. Hortic. Sci. 1985, 20, 1028–1038. [Google Scholar]
- Cress, W.A.; Johnson, G.V. The effect of three osmotic agents on free proline and amino acid pools in Atriplex canescens and Hilaria jamesii. Can. J. Bot. 1987, 65, 799–801. [Google Scholar] [CrossRef]
- Strauss, J.A.; Agenbag, G.A. A comparison of two methods of inducing water stress in wheat (Triticum aestivum L.). S. Afr. J. Plant Soil 1998, 15, 121–122. [Google Scholar] [CrossRef]
- Ahmad, S.; Garg, M.; Tamboli, E.T.; Abdin, M.Z.; Ansari, S.H. In vitro production of alkaloids: Factors, approaches, challenges and prospects. Pharmacogn. Rev. 2013, 7, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Prime, T.A.; Sherrier, D.J.; Mahon, P.; Packman, L.C.; Dupree, P. A proteomic analysis of organelles from Arabidopsis thaliana. Electrophoresis 2000, 21, 3488–3499. [Google Scholar] [CrossRef]
- Gamborg, O.L.; Miller, R.A.; Ojima, K. Nutrient requirements of suspension cultures of soybean root cells. Exp. Cell Res. 1968, 50, 151–158. [Google Scholar] [CrossRef]
- Gatto, L.; Lilley, K.S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Mockler, T.C.; Michael, T.P.; Priest, H.D.; Shen, R.; Sullivan, C.M.; Givan, S.A.; McEntee, C.; Kay, S.A.; Chory, J. The Diurnal Project: Diurnal and Circadian Expression Profiling, Model-based Pattern Matching, and Promoter Analysis. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator V3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2016, 89, 789–804. [Google Scholar]
- Harb, A.; Krishnan, A.; Ambavaram, M.M.R.; Pereira, A. Molecular and Physiological Analysis of Drought Stress in Arabidopsis Reveals Early Responses Leading to Acclimation in Plant Growth. Plant Physiol. 2010, 154, 1254–1271. [Google Scholar] [CrossRef] [PubMed]
- Kiyosue, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Cloning of cDNAs for genes that are early-responsive to dehydration stress (ERDs) in Arabidopsis thaliana L.: Identification of three ERDs as HSP cognate genes. Plant Mol. Biol. 1994, 25, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Marondedze, C.; Thomas, L.; Serrano, N.L.; Lilley, K.S.; Gehring, C. The RNA-binding protein repertoire of Arabidopsis thaliana. Sci. Rep. 2016, 6, 29766. [Google Scholar] [CrossRef] [PubMed]
- Chavrier, P.; Goud, B. The role of ARF and Rab GTPases in membrane transport. Curr. Opin. Cell Biol. 1999, 11, 466–475. [Google Scholar] [CrossRef]
- O’Mahony, P.J.; Oliver, M.J. Characterization of a desiccation-responsive small GTP-binding protein (Rab2) from the desiccation-tolerant grass Sporobolus stapfianus. Plant Mol. Biol. 1999, 39, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leal, J.B.; Seabra, M.C. Evolution of the rab family of small GTP-binding proteins. J. Mol. Biol. 2001, 313, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Babst, M. A Protein’s Final ESCRT. Traffic 2005, 6, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT Pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Shen, J.; Gao, C.; Zhuang, X.; Wang, J.; Jiang, L. Biogenesis of Plant Prevacuolar Multivesicular Bodies. Mol. Plant 2017, 9, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Dacks, J.B. First and last ancestors: Reconstructing evolution of the endomembrane system with ESCRTs, vesicle coat proteins, and nuclear pore complexes. Curr. Opin. Cell Biol. 2009, 21, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Korbei, B.; Moulinier-Anzola, J.; De-Araujo, L.; Lucyshyn, D.; Retzer, K.; Khan, M.A.; Luschnig, C. Arabidopsis TOL Proteins Act as Gatekeepers for Vacuolar Sorting of PIN2 Plasma Membrane Protein. Curr. Biol. 2017, 23, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, C.; Reyes, F.C.; Buono, R.; Sliwinski, M.K.; Haas, T.J.; Otegui, M.S. The ESCRT-Related CHMP1A and B Proteins Mediate Multivesicular Body Sorting of Auxin Carriers in Arabidopsis and Are Required for Plant Development. Plant Cell 2009, 21, 749–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babst, M.; Katzmann, D.J.; Estepa-Sabal, E.J.; Meerloo, T.; Emr, S.D. Escrt-III: An endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell 2002, 3, 271–282. [Google Scholar] [CrossRef]
- Bowers, K.; Lottridge, J.; Helliwell, S.B.; Goldthwaite, L.M.; Luzio, J.P.; Stevens, T.H. Protein–Protein Interactions of ESCRT Complexes in the Yeast Saccharomyces cerevisiae. Traffic 2004, 5, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Spallek, T.; Beck, M.; Ben Khaled, S.; Salomon, S.; Bourdais, G.; Schellmann, S.; Robatzek, S. ESCRT-I Mediates FLS2 Endosomal Sorting and Plant Immunity. PLoS Genet. 2013, 9, e1004035. [Google Scholar] [CrossRef] [PubMed]
- Geldner, N.; Robatzek, S. Plant Receptors Go Endosomal: A Moving View on Signal Transduction. Plant Physiol. 2008, 147, 1565–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Zhuang, X.; Shen, J.; Jiang, L. Plant ESCRT Complexes: Moving Beyond Endosomal Sorting. Trends Plant Sci. 2017, 22, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Milla, M.A.; Salinas, J. Prefoldins 3 and 5 Play an Essential Role in Arabidopsis Tolerance to Salt Stress. Mol. Plant 2009, 2, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, E.J.; Yang, E.J.; Lee, J.E.; Park, A.R.; Song, W.H.; Park, O.K. Proteomic Identification of Annexins, Calcium-Dependent Membrane Binding Proteins That Mediate Osmotic Stress and Abscisic Acid Signal Transduction in Arabidopsis. Plant Cell 2004, 16, 1378–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.-H.; Jiang, L.-L.; Ma, X.-F.; Xu, Z.-S.; Liu, M.-M.; Shan, S.-G.; Cheng, X.-G. A Soybean C2H2-Type Zinc Finger Gene GmZF1 Enhanced Cold Tolerance in Transgenic Arabidopsis. PLoS ONE 2014, 9, e109399. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, B.; Dong, R.; Hou, B. AtUGT76C2, an Arabidopsis cytokinin glycosyltransferase is involved in drought stress adaptation. Plant Sci. 2015, 236, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Kiyosue, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Characterization of two cDNAs (ERD10 and ERD14) corresponding to genes that respond rapidly to dehydration stress in Arabidopsis thaliana. Plant Cell Physiol. 1994, 35, 225–231. [Google Scholar] [PubMed]
- Heng, K.; Xiaojie, C.; Chao, W.; Aifang, X.; Hui, Z.; Songli, Y.; Zhenzhen, Y.; Danxia, K.; Shaobo, X.; Zonglie, H.; et al. A MYB coiled-coil transcription factor interacts with NSP2 and is involved in nodulation in Lotus japonicus. New Phytol. 2013, 201, 837–849. [Google Scholar]
- Ali, A.M.; Reis, J.M.; Xia, Y.; Rashid, A.J.; Mercaldo, V.; Walters, B.J.; Brechun, K.; Borisenko, V.; Josselyn, S.A.; Karanicolas, J.; et al. Optogenetic inhibitor of the transcription factor CREB. Chem. Biol. 2015, 22, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-J.; Hansen, M.; Troemel, E. Autophagy and innate immunity: Insights from invertebrate model organisms. Autophagy 2018, 14, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, K.C.; Cary, J.W.; Ehrlich, M. A broad bean cDNA clone encoding a DNA-binding protein resembling mammalian CREB in its sequence specificity and DNA methylation sensitivity. Gene 1992, 117, 169–178. [Google Scholar] [CrossRef]
- Shimmen, T.; Ridge, R.W.; Lambiris, I.; Plazinski, J.; Yokota, E.; Williamson, R.E. Plant myosins. Protoplasma 2000, 214, 1–10. [Google Scholar] [CrossRef]
- Kurth, E.G.; Peremyslov, V.V.; Turner, H.L.; Makarova, K.S.; Iranzo, J.; Mekhedov, S.L.; Koonin, E.V.; Dolja, V.V. Myosin-driven transport network in plants. Proc. Natl. Acad. Sci. USA 2017, 114, E1385–E1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiaoliang, H.; Xiaona, H.; Yinzhu, S.; Zhanjing, H. TaSRG, a wheat transcription factor, significantly affects salt tolerance in transgenic rice and Arabidopsis. FEBS Lett. 2011, 585, 1231–1237. [Google Scholar] [Green Version]
- Thomas, L.; Marondedze, C.; Ederli, L.; Pasqualini, S.; Gehring, C. Proteomic signatures implicate cAMP in light and temperature responses in Arabidopsis thaliana. J. Proteom. 2013, 83, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Alqurashi, M.; Gehring, C.; Marondedze, C. Changes in the Arabidopsis thaliana Proteome Implicate cAMP in Biotic and Abiotic Stress Responses and Changes in Energy Metabolism. Int. J. Mol. Sci. 2016, 17, 852. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Annotation | Average 10 min | Average 30 min | Adjusted p-Value (10) | Adjusted p-Value (30) | |
|---|---|---|---|---|---|---|
| Increased Abundance | AT3G52300 | ATP synthase D chain, mitochondrial | 2.25 | 1.60 | 1.51 × 10−4 | 2.93 × 10−3 |
| AT5G16060 | Cytochrome C oxidase biogenesis protein Cmc1-like | 2.24 | 1.48 | 1.04 × 10−4 | 2.93 × 10−3 | |
| AT2G43535 | Defensin-like protein 196 | 1.84 | 1.58 | 2.05 × 10−4 | 2.93 × 10−3 | |
| AT5G59320 | Lipid transfer protein 3 | 1.85 | 1.73 | 3.64 × 10−3 | 1.11 × 10−2 | |
| AT3G51600 | Lipid transfer protein 5 | 2.33 | 2.25 | 2.40 × 10−3 | 7.03 × 10−3 | |
| AT2G02050 | NADH-ubiquinone oxidoreductase B18 subunit, putative | 2.68 | 1.89 | 1.04 × 10−4 | 2.93 × 10−3 | |
| AT1G29990 | Prefoldin 6 | 2.35 | 1.70 | 1.04 × 10−4 | 2.93 × 10−3 | |
| AT5G55125 | Ribosomal protein L31 | 2.18 | 1.40 | 1.04 × 10−4 | 2.93 × 10−3 | |
| AT2G30410 | Tubulin folding cofactor A (KIESEL) | 2.25 | 1.53 | 1.26 × 10−4 | 2.93 × 10−3 | |
| AT5G03660 | Unknown Protein | 2.29 | 1.39 | 1.89 × 10−4 | 6.61 × 10−3 | |
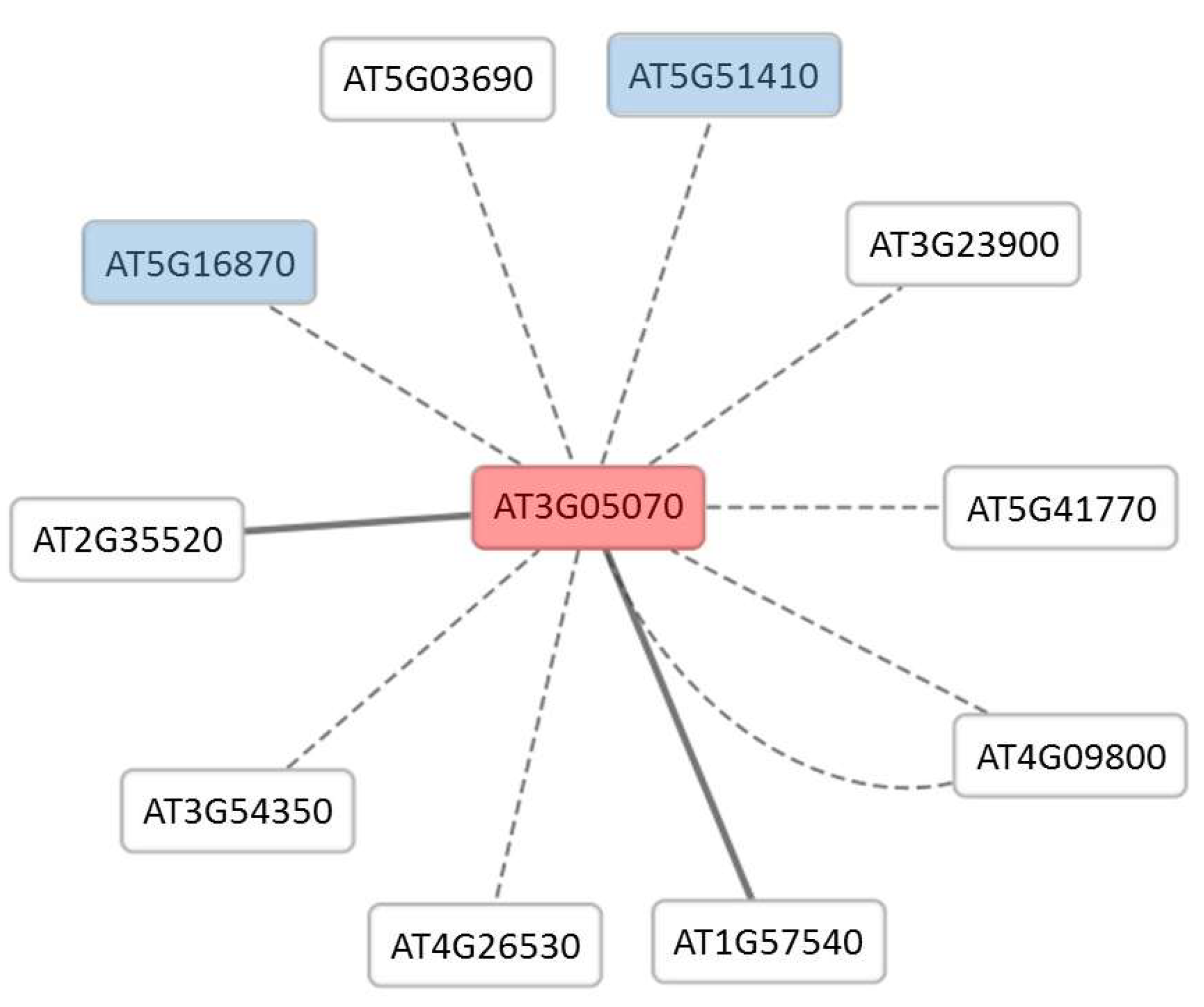
| AT3G05070 | Unknown protein | 2.22 | 1.52 | 8.56 × 10−4 | 1.38 × 10−2 | |
| AT4G15790 | Unknown protein | 2.18 | 1.72 | 1.40 × 10−4 | 2.93 × 10−3 | |
| Decreased Abundance | AT3G49010 | 60S ribosomal protein L13-1 | −3.14 | −2.82 | 6.80 × 10−4 | 3.29 × 10−3 |
| AT5G63550 | DEK domain-containing chromatin associated protein | −1.72 | −1.67 | 4.38 × 10−4 | 2.93 × 10−3 | |
| AT2G18020 | Embryo defective 2296 | −1.99 | −1.63 | 7.50 × 10−4 | 5.76 × 10−3 | |
| AT4G30800 | Nucleic acid-binding, OB-fold-like protein | −2.11 | - | 4.55 × 10−2 | - | |
| AT3G09500 | Ribosomal L29 family protein | −2.28 | −1.97 | 1.76 × 10−3 | 8.87 × 10−3 | |
| AT5G23900 | Ribosomal protein L13e family protein | −2.48 | −2.26 | 1.53 × 10−3 | 6.24 × 10−3 | |
| AT5G64670 | Ribosomal protein L18e/L15 superfamily protein | −1.91 | −1.72 | 7.38 × 10−3 | 2.36 × 10−2 | |
| AT5G46430 | Ribosomal protein L32e | −1.95 | −1.79 | 1.71 × 10−2 | 4.28 × 10−2 | |
| AT5G02450 | Ribosomal protein L36e family protein | −2.85 | −2.41 | 1.69 × 10−3 | 9.10 × 10−3 | |
| AT3G04920 | Ribosomal protein S24e family protein | −2.16 | − | 5.19 × 10−3 | - | |
| AT5G20290 | Ribosomal protein S8e family protein | −2.35 | −1.90 | 1.79 × 10−3 | 1.25 × 10−2 | |
| AT1G52300 | Zinc-binding ribosomal protein family protein | −2.48 | −2.56 | 7.14 × 10−3 | 1.35 × 10−2 |
| Unknown Protein | Score | Accession | Annotation |
|---|---|---|---|
| Protein A | 0.5949 | AT5G51940 | Non-catalytic subunit of nuclear DNA-dependent RNA polymerases |
| 0.5940 | AT1G11240 | Ribosomal RNA-processing protein | |
| 0.5939 | AT3G56510 | RNA-binding (RRM/RBD/RNP motifs) family protein | |
| 0.5907 | AT2G44860 *§ | Ribosomal protein L24e family protein | |
| 0.5886 | AT2G45520 | Coiled-coil protein | |
| 0.5810 | AT1G79200 | Stigma/style cell-cycle inhibitor 1 | |
| 0.5739 | AT4G27380 | Hypothetical protein | |
| 0.5666 | AT5G59460 | Scarecrow-like transcription factor 11 | |
| 0.5617 | AT4G37090 | UDP-N-acetylmuramoyl-l-alanyl-d-glutamate-2,6-diaminopimelate ligase | |
| 0.5563 | AT1G16740 | Ribosomal protein L20 | |
| Protein B | 0.6690 | AT4G30330 | Small nuclear ribonucleoprotein family protein |
| 0.6363 | AT2G19720 | Ribosomal protein S15A B | |
| 0.6255 | AT5G61130 | Plasmodesmata callose-binding protein 1 | |
| 0.6135 | AT4G00810 | 60S acidic ribosomal protein family | |
| 0.6094 | AT3G06680 | Ribosomal L29e protein family | |
| 0.6073 | AT4G15000 *§ | Ribosomal L27e protein family | |
| 0.6070 | AT3G23390 * | Zinc-binding ribosomal protein family protein | |
| 0.6051 | AT4G35950 | A member of ROP GTPases gene family-like | |
| 0.6042 | AT2G27970 | CDK-subunit 2 | |
| 0.6030 | AT3G59650 | Mitochondrial ribosomal protein L51/S25/CI-B8 family protein | |
| Protein C | 0.5496 | AT1G29990*§ | Prefoldin 6 |
| 0.5419 | AT2G18040 * | Peptidylprolyl cis/trans isomerase, NIMA-interacting 1 | |
| 0.4990 | AT1G29850 *§ | Double-stranded DNA-binding family protein | |
| 0.4950 | AT1G66410 | Calmodulin 4 | |
| 0.4877 | AT5G41210 | Glutathione S-transferase theta 1/Glutathione S-transferase tau 12 | |
| 0.4871 | AT4G35570 * | High mobility group B5 | |
| 0.4824 | AT3G50360 *§ | Calmodulin 20/Centrin 2 | |
| 0.4790 | AT2G29960 * | Cyclophilin 5/peptidylprolyl cis/trans-isomerase 19-4 | |
| 0.4570 | AT5G46020 | 28 kDa heat/acid-stable phosphoprotein-like protein | |
| 0.4556 | AT4G30480 | Tetratricopeptide repeat 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqurashi, M.; Chiapello, M.; Bianchet, C.; Paolocci, F.; Lilley, K.S.; Gehring, C. Early Responses to Severe Drought Stress in the Arabidopsis thaliana Cell Suspension Culture Proteome. Proteomes 2018, 6, 38. https://doi.org/10.3390/proteomes6040038
Alqurashi M, Chiapello M, Bianchet C, Paolocci F, Lilley KS, Gehring C. Early Responses to Severe Drought Stress in the Arabidopsis thaliana Cell Suspension Culture Proteome. Proteomes. 2018; 6(4):38. https://doi.org/10.3390/proteomes6040038
Chicago/Turabian StyleAlqurashi, May, Marco Chiapello, Chantal Bianchet, Francesco Paolocci, Kathryn S. Lilley, and Christoph Gehring. 2018. "Early Responses to Severe Drought Stress in the Arabidopsis thaliana Cell Suspension Culture Proteome" Proteomes 6, no. 4: 38. https://doi.org/10.3390/proteomes6040038
APA StyleAlqurashi, M., Chiapello, M., Bianchet, C., Paolocci, F., Lilley, K. S., & Gehring, C. (2018). Early Responses to Severe Drought Stress in the Arabidopsis thaliana Cell Suspension Culture Proteome. Proteomes, 6(4), 38. https://doi.org/10.3390/proteomes6040038