A Preliminary Metagenome Analysis Based on a Combination of Protein Domains

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Creating Cluster Dendrograms for Combinations of Protein Domains
2.2. Generating a Cluster Dendrogram about Protein Domains Translated from Random 100 bps DNA Fragments
2.3. Generating Phylogenetic Trees from 16S Ribosomal RNA
2.4. Generating Phylogenetic Trees for DNA and Amino Acid Sequences of DNA Gyrase Subunit B
2.5. Comparing the Cluster Dendrograms and Phylogenetic Trees
2.6. Analysis Test on the Environmental Data
3. Results
3.1. Comparing the Cluster Dendrogram of Domain Combinations and the Phylogenetic Tree of DNA Sequences for 16S Ribosomal RNA
3.2. Comparing the Cluster Dendrogram for the Existence of Protein Domains, or Converted into Natural Logarithms, with the Phylogenetic Tree Created from DNA Sequences of 16S Ribosomal RNA
3.3. Comparison of the Cluster Dendrogram Based on Protein Domains Translated from Random 100 bp DNA Fragments and that Created from 16S Ribosomal RNA
3.4. Comparison of the Correlation Coefficients of the Domain Combinations and Pairwise Distances of 16S Ribosomal RNA and DNA Gyrase Subunit B
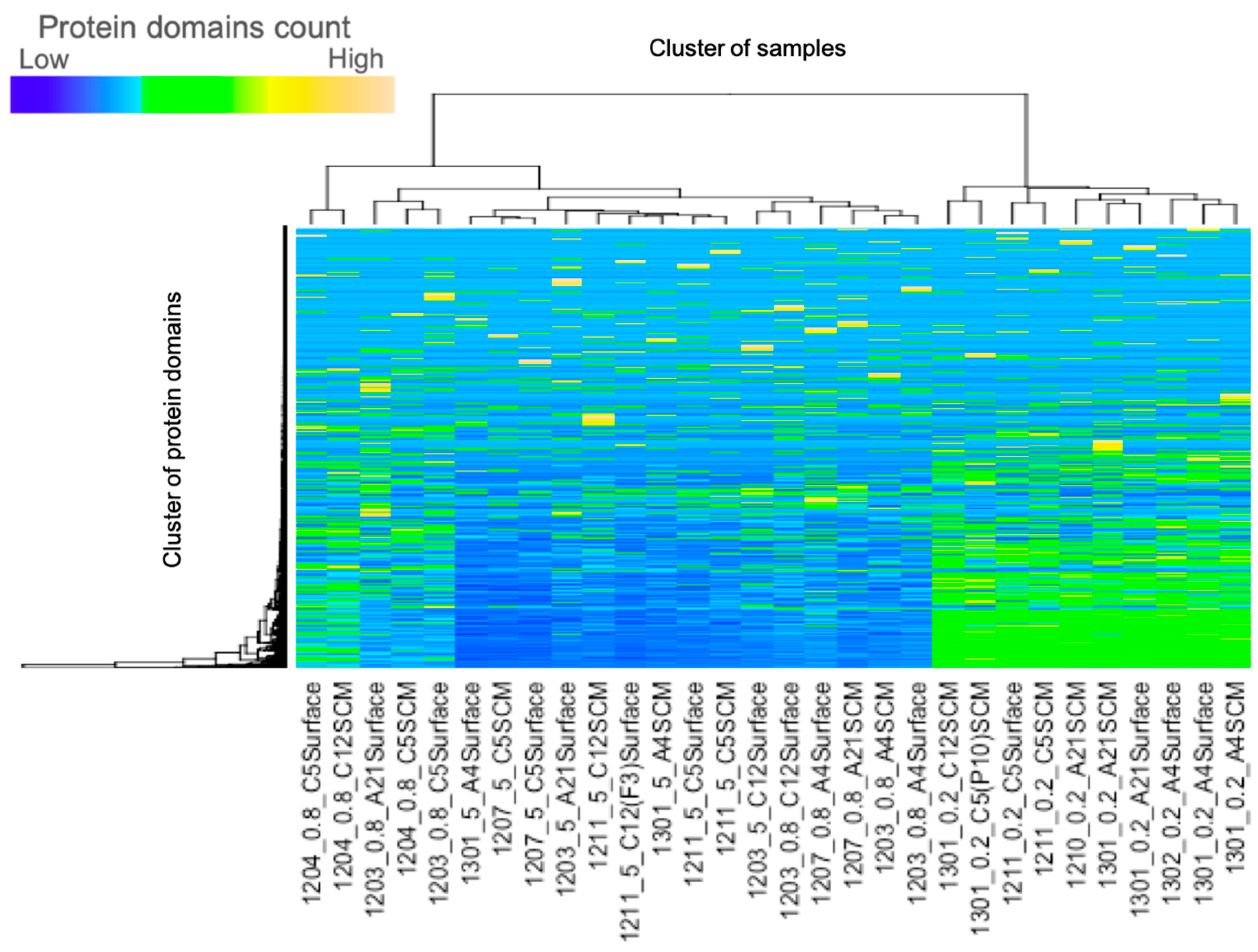
3.5. Cluster Analysis and Principal Component Analysis of the Environmental Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kennedy, J.; Marchesi, J.R.; Dobson, A.D. Marine metagenomics: Strategies for the discovery of novel enzymes with biotechnological applications from marine environments. Microb. Cell Fact. 2008, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugenholtz, P.; Goebel, B.M.; Pace, N.R. Impact of Culture-Independent Studies on the Emerging Phylogenetic View of Bacterial Diversity. J. Bacteriol. 1998, 180, 4765–4774. [Google Scholar] [PubMed]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; de Vargas, C.; Gasol, J.M.; et al. Patterns and ecological drivers of ocean viral communities. Science 2015, 348, 1261498. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef]
- van der Walt, A.J.; van Goethem, M.W.; Ramond, J.B.; Makhalanyane, T.P.; Reva, O.; Cowan, D.A. Assembling metagenomes, one community at a time. BMC Genom. 2017, 18, 521. [Google Scholar] [CrossRef]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5, 209. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; Eddy, S.R.; Durbin, R. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins 1997, 28, 405–420. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Jordan, I.K.; Kondrashov, F.A.; Adzhubei, I.A.; Wolf, Y.I.; Koonin, E.V.; Kondrashov, A.S.; Sunyaev, S. A universal trend of amino acid gain and loss in protein evolution. Nature 2005, 433, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Xie, X.; Chen, C.; Park, J.G.; Stark, C.; James, D.A.; Olhovsky, M.; Linding, R.; Mao, Y.; Pawson, T. Eukaryotic Protein Domains as Functional Units of Cellular Evolution. Sci. Signal. 2009, 2, ra76. [Google Scholar] [CrossRef]
- Fukami-Kobayashi, K.; Minezaki, Y.; Tateno, Y.; Nishikawa, K. A tree of Life Based on Protein Domain Organizations. Mol. Biol. Evol. 2007, 24, 1181–1189. [Google Scholar] [CrossRef]
- Takami, H.; Noguchi, H.; Takaki, Y.; Uchiyama, I.; Toyoda, A.; Nishi, S.; Chee, G.J.; Arai, W.; Nunoura, T.; Itoh, T.; et al. A deeply branching thermophilic bacterium with an ancient acetyl-CoA pathway dominates a subsurface ecosystem. PLoS ONE 2012, 7, e30559. [Google Scholar] [CrossRef]
- Woese, C.R.; Gibson, J.; Fox, G.E. Do genealogical patterns in purple photosynthetic bacteria reflect interspecific gene transfer? Nature 1980, 283, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, M.; Nayer, B.N.; Motavaze, K.; Kharaziha, P.; Chiani, M.; Zali, M.R.; Klena, J.D. Phylogenetic relationship of Salmonella enterica strains in Tehran, Iran, using 16S rRNA and gyrB gene sequences. J. Infect. Dev. Ctries. 2011, 5, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Serdoz, S.; Egri-Nagy, A.; Sumner, J.; Holland, B.R.; Jarvis, P.D.; Tanaka, M.M.; Francis, A.R. Maximum likelihood estimates of pairwise rearrangement distances. J. Theor. Biol. 2017, 423, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. gplots: Various R Programming Tools for Plotting Data. R package version 3.0.1. Available online: https://CRAN.R-project.org/package=gplots (accessed on 9 November 2017).
- Ligges, U.; Mächler, M. Scatterplot3d—An R Package for Visualizing Multivariate Data. J. Stat. Softw. 2003, 8, 1–20. [Google Scholar] [CrossRef]
- Kakehi, S.; Ito, S.-I.; Kuwata, A.; Saito, H.; Tadokoro, K. Phytoplankton distribution during the winter convective season in Sendai Bay, Japan. Cont. Shelf Res. 2015, 97, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, T.; Yamaguchi, H.; Sato, M.; Watanabe, T.; Taniuchi, Y.; Kuwata, A.; Kawachi, M. Seasonal and geographical distribution of near-surface small photosynthetic eukaryotes in the western North Pacific determined by pyrosequencing of 18S rDNA. FEMS Microbiol. Ecol. 2017, 93, fiw229. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Taniuchi, Y.; Kakehi, S.; Sakami, T.; Kuwata, A. Seasonal succession in the diatom community of Sendai Bay, northern Japan, following the 2011 off the Pacific coast of Tohoku earthquake. J. Oceanogr. 2017, 73, 133–144. [Google Scholar] [CrossRef]
- Taniuchi, Y.; Watanabe, T.; Kakehi, S.; Sakami, T.; Kuwata, A. Seasonal dynamics of the phytoplankton community in Sendai Bay, northern Japan. J. Oceanogr. 2017, 73, 1–9. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Igarashi, Y.; Mori, D.; Mitsuyama, S.; Yoshitake, K.; Ono, H.; Watanabe, T.; Taniuchi, Y.; Sakami, T.; Kuwata, A.; Kobayashi, T.; et al. A Preliminary Metagenome Analysis Based on a Combination of Protein Domains. Proteomes 2019, 7, 19. https://doi.org/10.3390/proteomes7020019
Igarashi Y, Mori D, Mitsuyama S, Yoshitake K, Ono H, Watanabe T, Taniuchi Y, Sakami T, Kuwata A, Kobayashi T, et al. A Preliminary Metagenome Analysis Based on a Combination of Protein Domains. Proteomes. 2019; 7(2):19. https://doi.org/10.3390/proteomes7020019
Chicago/Turabian StyleIgarashi, Yoji, Daisuke Mori, Susumu Mitsuyama, Kazutoshi Yoshitake, Hiroaki Ono, Tsuyoshi Watanabe, Yukiko Taniuchi, Tomoko Sakami, Akira Kuwata, Takanori Kobayashi, and et al. 2019. "A Preliminary Metagenome Analysis Based on a Combination of Protein Domains" Proteomes 7, no. 2: 19. https://doi.org/10.3390/proteomes7020019
APA StyleIgarashi, Y., Mori, D., Mitsuyama, S., Yoshitake, K., Ono, H., Watanabe, T., Taniuchi, Y., Sakami, T., Kuwata, A., Kobayashi, T., Ishino, Y., Watabe, S., Gojobori, T., & Asakawa, S. (2019). A Preliminary Metagenome Analysis Based on a Combination of Protein Domains. Proteomes, 7(2), 19. https://doi.org/10.3390/proteomes7020019