
Evaluation of the Sensitivity of Proteomics Methods Using the Absolute Copy Number of Proteins in a Single Cell as a Metric

Abstract
:1. Introduction
2. Materials and Methods
2.1. Obtaining Data
2.2. Compiling the Absolute Copy Numbers
2.3. Visualization of Copy Number Distribution in R/Shiny
3. Results
3.1. Generational Improvements in Proteomics Hardware for Data Dependent Analysis
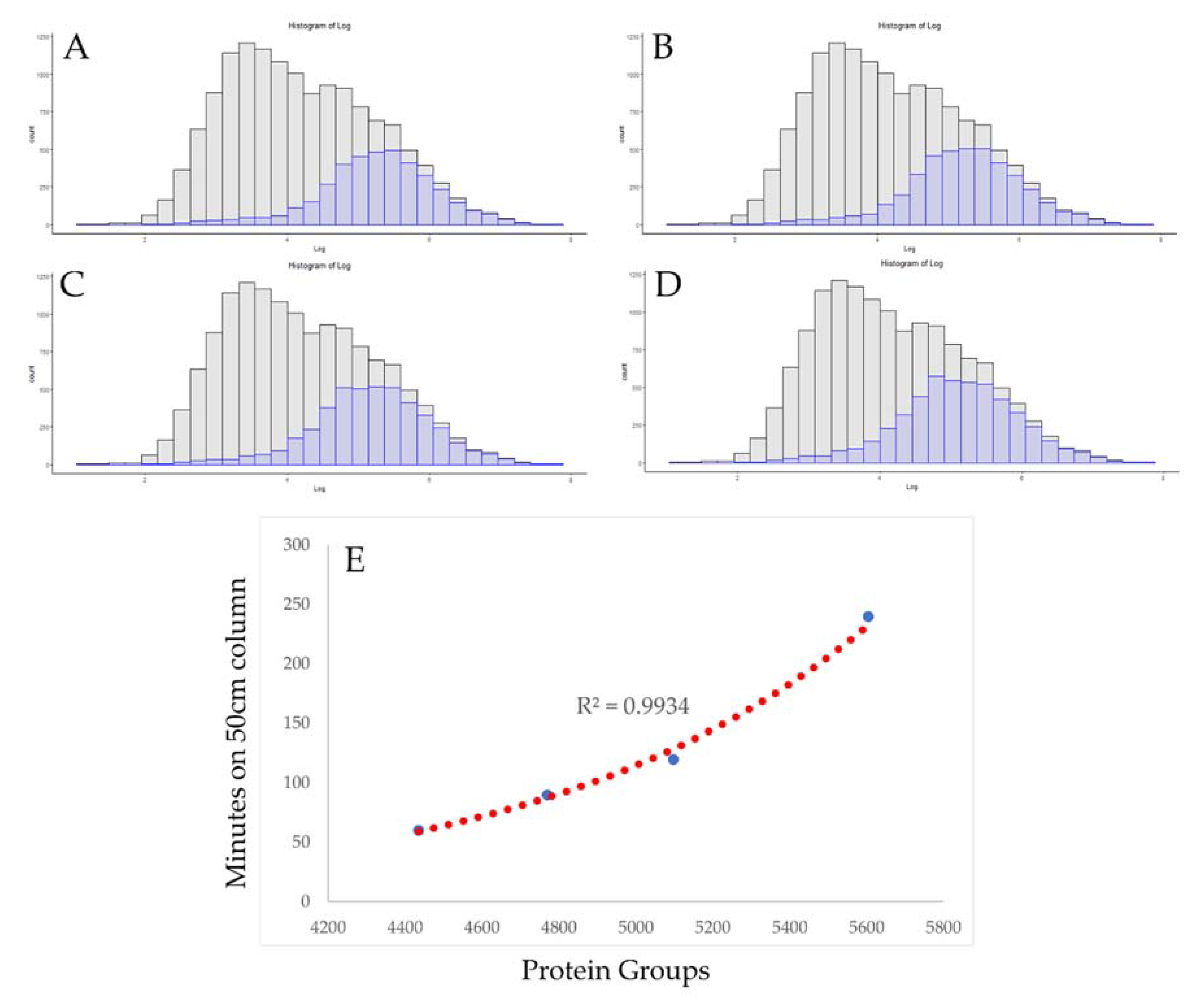
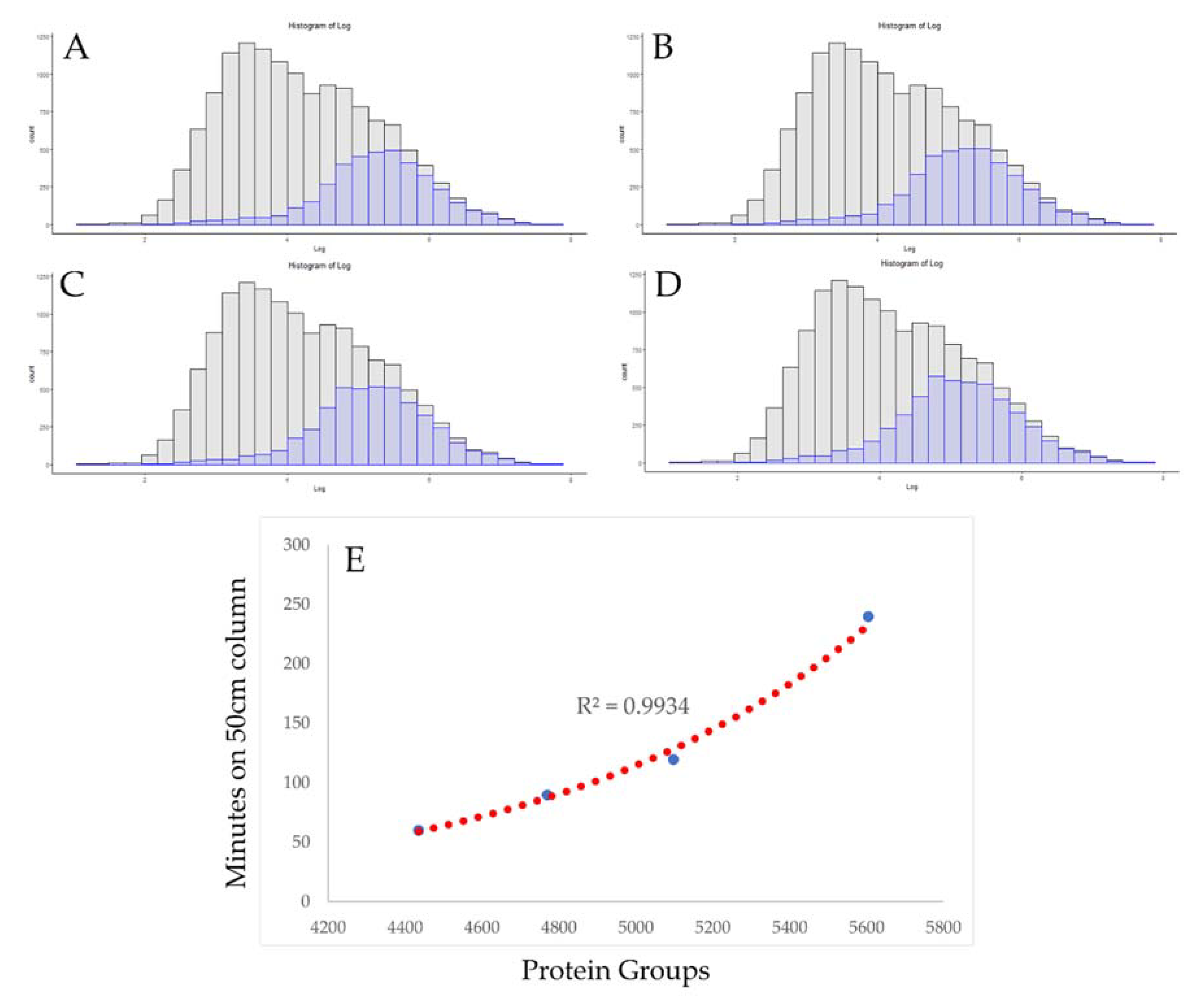
3.2. Use of Absolute Copy Number for Optimization of Chromatographic Conditions
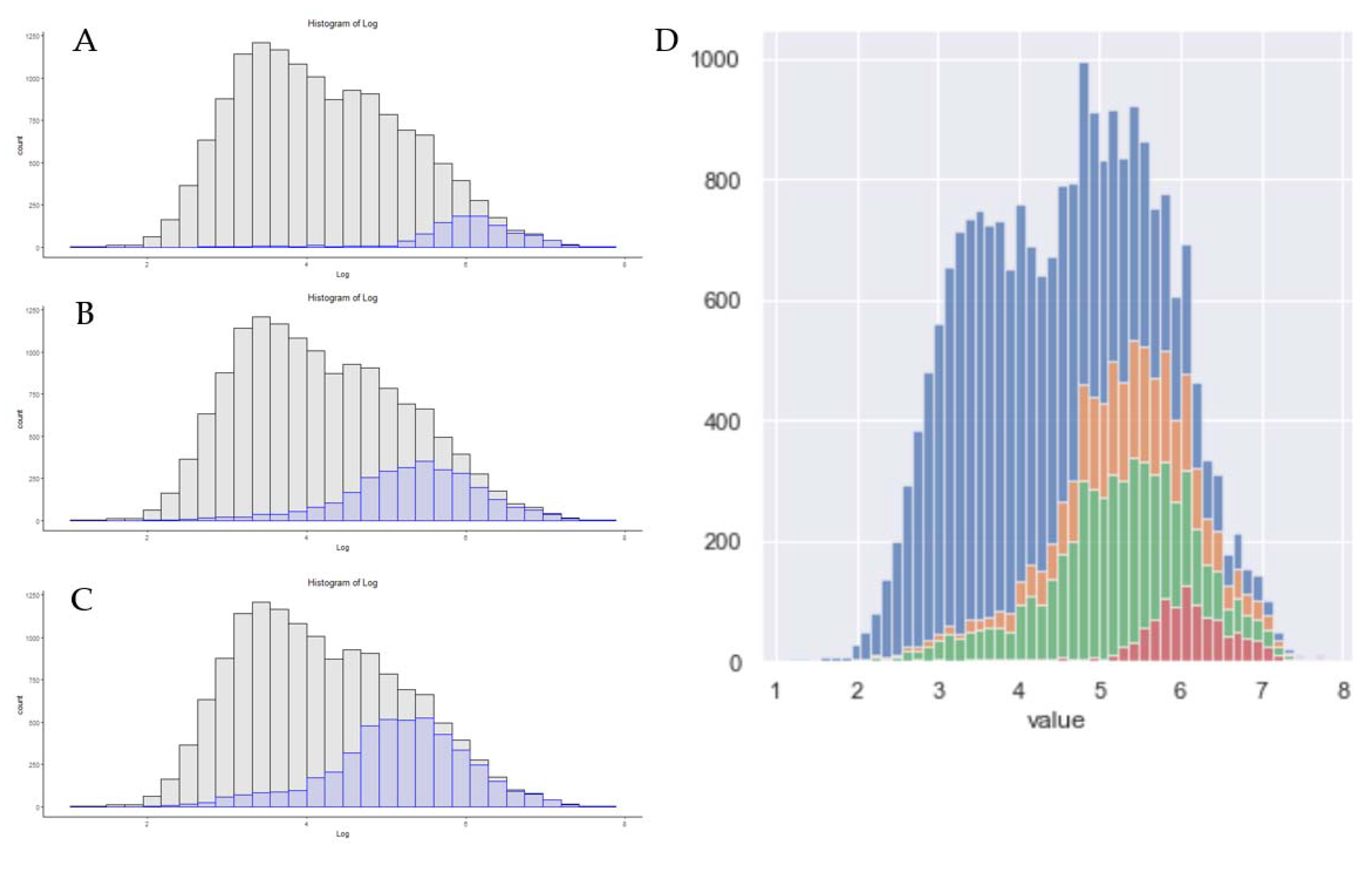
3.3. Rapid Proteomics Methods
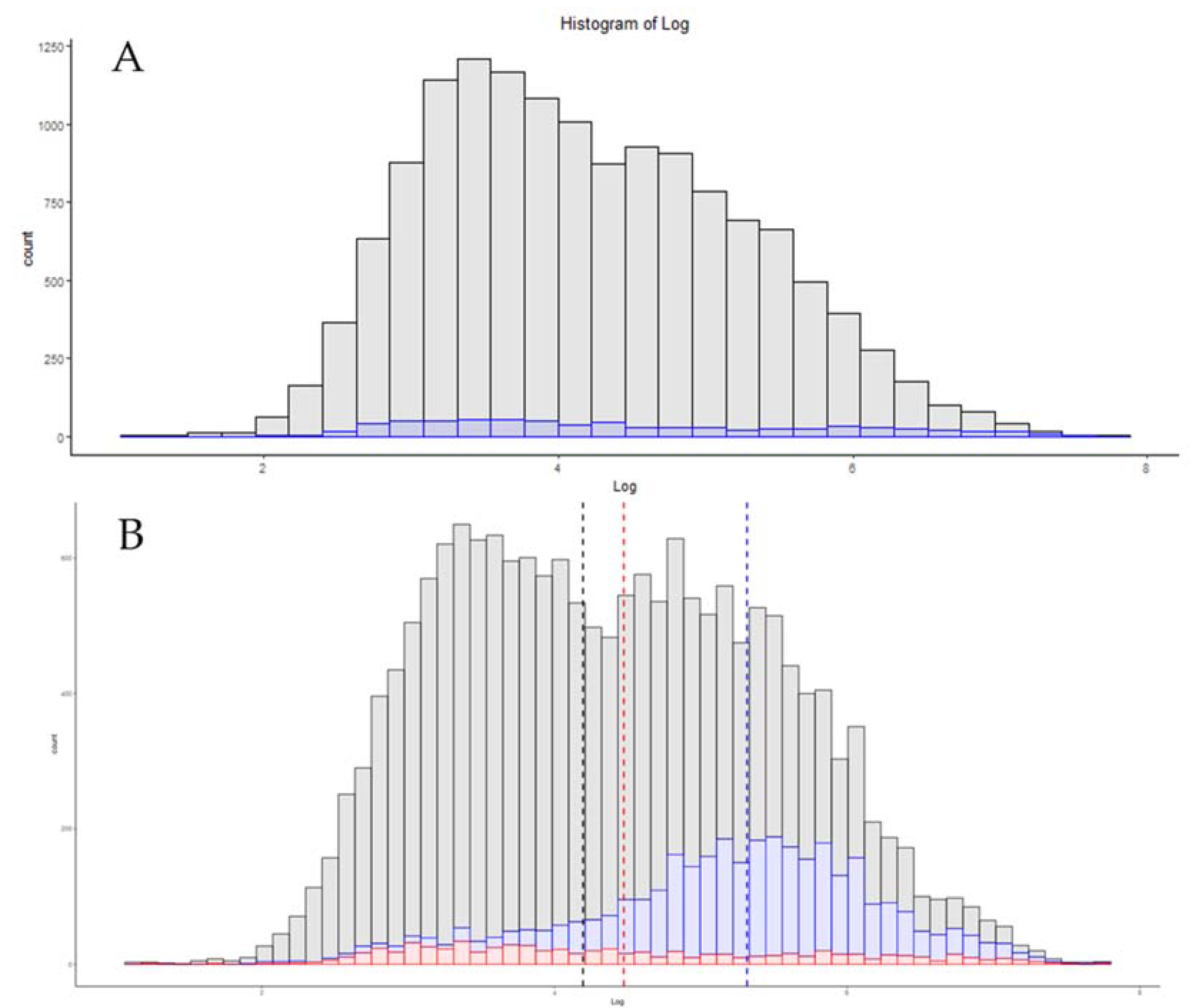
3.4. Absolute Sensitivity in Single-Shot Proteomics Today
3.5. Match between Runs
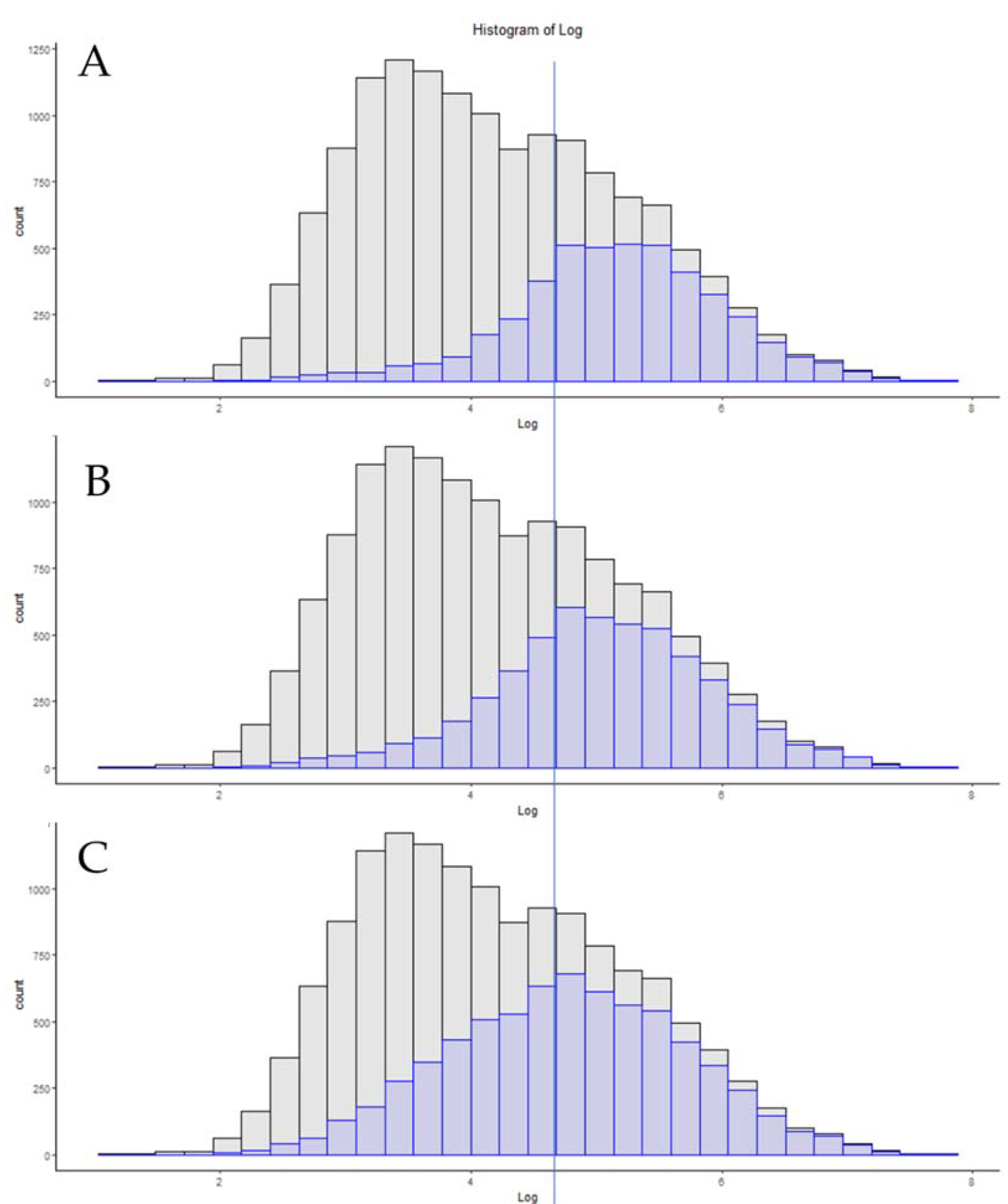
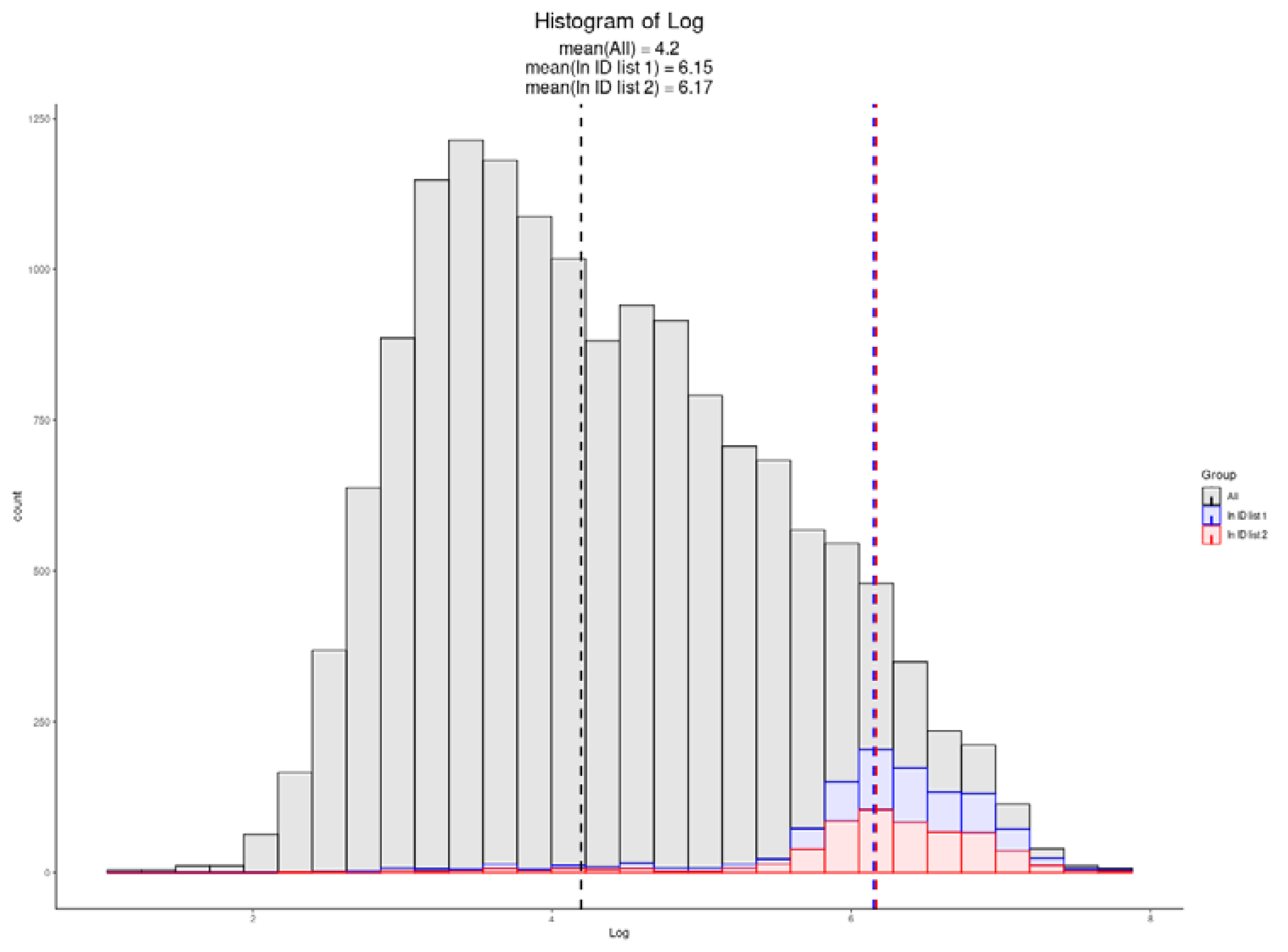
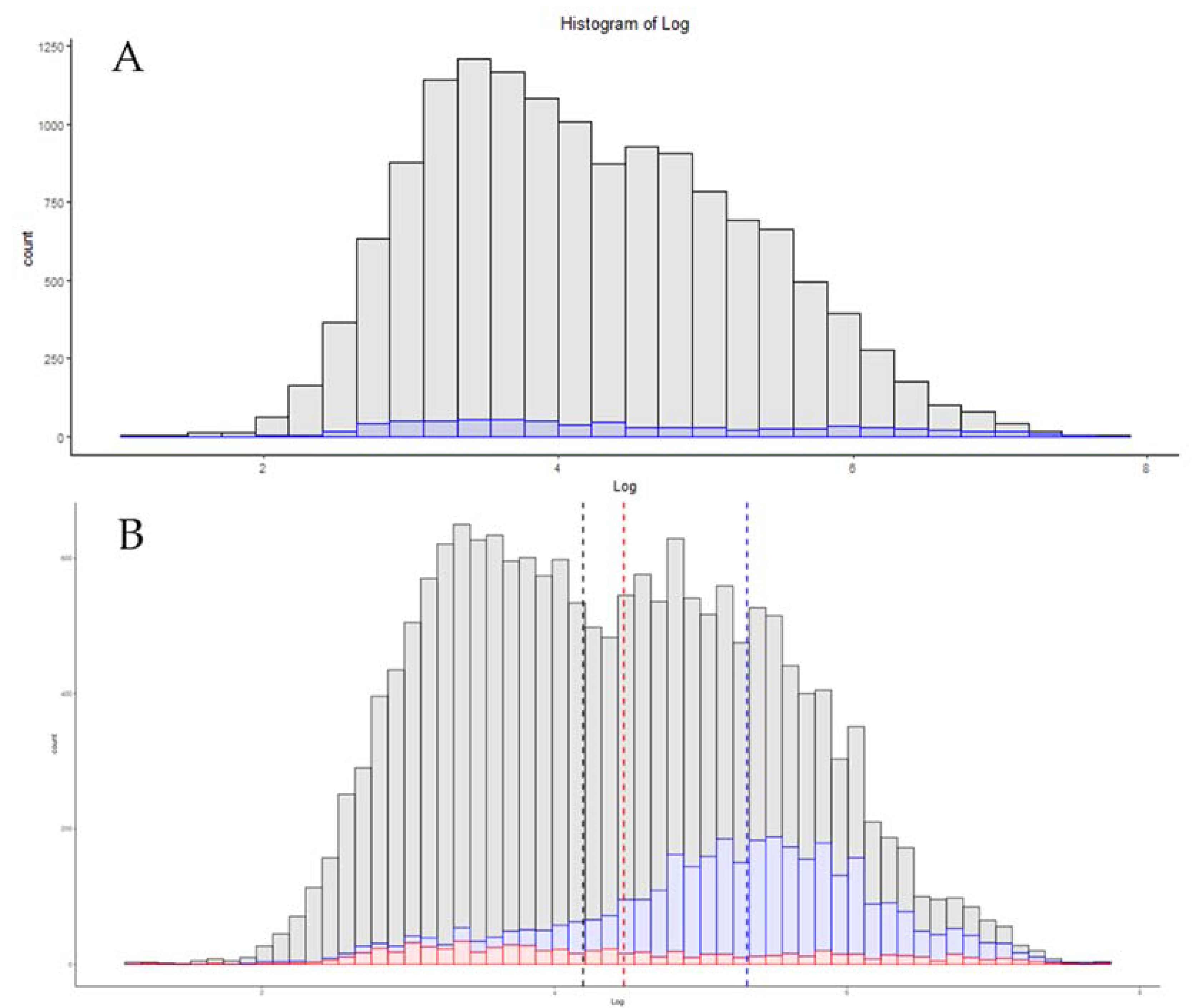
3.6. Single-Cell Proteomics
3.7. Additional Methods
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [Green Version]
- Lössl, P.; van de Waterbeemd, M.; Heck, A.J. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016, 35, 2634–2657. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Majumder, S.; Ahmad, S.; Varkey, M.; Anish, T.A.; Jenkins, C.; Rigby, M.; Orsburn, B. Detection and verification of 2.3 million cancer mutations in NCI60 cancer cell lines with a cloud search engine. J. Proteom. 2019, 209, 103488. [Google Scholar] [CrossRef]
- Prakash, A.; Ahmad, S.; Majumder, S.; Jenkins, C.; Orsburn, B. Bolt: A New Age Peptide Search Engine for Comprehensive MS/MS Sequencing through Vast Protein Databases in Minutes. J. Am. Soc. Mass Spectrom. 2019, 30, 2408–2418. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Solntsev, S.K.; Shortreed, M.R.; Frey, B.L.; Smith, L.M. Enhanced Global Post-translational Modification Discovery with MetaMorpheus. J. Proteome Res. 2018, 17, 1844–1851. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewski, J.R. Label-Free and Standard-Free Absolute Quantitative Proteomics Using the “Total Protein” and “Proteomic Ruler” Approaches. Methods Enzymol. 2017, 585, 49–60. [Google Scholar]
- Köcher, T.; Pichler, P.; De Pra, M.; Rieux, L.; Swart, R.; Mechtler, K. Development and performance evaluation of an ultralow flow nanoliquid chromatography-tandem mass spectrometry set-up. Proteomics 2014, 14, 1999–2007. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Y.; Mueller, T.; Kreibich, S.; Williams, E.G.; Van Drogen, A.; Borel, C.; Germain, P.-L.; Frank, M.; Bludau, I.; et al. Genomic, Proteomic and Phenotypic Heterogeneity in HeLa Cells across Laboratories: Implications for Reproducibility of Research Results. bioRxiv 2018. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sorensen, K.D.; Hoyer, S.; Orntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorfer, V.; Pichler, P.; Stranzl, T.; Stadlmann, J.; Taus, T.; Winkler, S.; Mechtler, K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 2014, 13, 3679–3684. [Google Scholar] [CrossRef]
- Coombs, K.M.; Simon, P.F.; McLeish, N.J.; Zahedi-Amiri, A.; Kobasa, D. Aptamer profiling of A549 cells infected with low-pathogenicity and high-pathogenicity influenza viruses. Viruses 2019, 11, 1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Leeper, T.J.; Tabulizer: Bindings for Tabula PDF Table Extractor Library. R Packag Version 0.2.2. 2018. Available online: https://rdrr.io/cran/tabulizer/ (accessed on 15 July 2021).
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Orsburn, B.C. Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes 2021, 9, 15. [Google Scholar] [CrossRef]
- Matthiesen, R.; Prieto, G.; Beck, H.C. Comparing peptide spectra matches across search engines. Methods Mol. Biol. 2020, 2051, 133–143. [Google Scholar] [PubMed]
- Xiong, H.; Yan, J.; Cai, S.; He, Q.; Peng, D.; Liu, Z.; Liu, Y. Cancer protein biomarker discovery based on nucleic acid aptamers. Int. J. Biol. Macromol. 2019, 132, 190–202. [Google Scholar] [CrossRef]
- Scheltema, R.A.; Hauschild, J.-P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q Exactive HF, a Benchtop Mass Spectrometer with a Pre-filter, High-performance Quadrupole and an Ultra-high-field Orbitrap Analyzer. Mol. Cell. Proteom. 2014, 13, 3698–3708. [Google Scholar] [CrossRef] [Green Version]
- Espadas, G.; Borràs, E.; Chiva, C.; Sabidó, E. Evaluation of different peptide fragmentation types and mass analyzers in data-dependent methods using an Orbitrap Fusion Lumos Tribrid mass spectrometer. Proteomics 2017, 17, 1600416. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online parallel accumulation–serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekker-Jensen, D.B.; Martínez-Val, A.; Steigerwald, S.; Rüther, P.; Fort, K.L.; Arrey, T.N.; Harder, A.; Makarov, A.; Olsen, J.V. A compact quadrupole-orbitrap mass spectrometer with FAIMS interface improves proteome coverage in short LC gradients. Mol. Cell. Proteom. 2020, 19, 716–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, F.; Brunner, A.-D.; Frank, M.; Ha, A.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Aebersold, R.; Collins, B.C.; et al. Parallel accumulation—Serial fragmentation combined with data-independent acquisition (diaPASEF): Bottom-up proteomics with near optimal ion usage. bioRxiv 2019. [Google Scholar] [CrossRef]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef]
- Hartlmayr, D.; Ctortecka, C.; Seth, A.; Mendjan, S.; Tourniaire, G.; Mechtler, K. An automated workflow for label-free and multiplexed single cell proteomics sample preparation at unprecedented sensitivity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Moraga, P.; Moraga, P. Interactive dashboards with flexdashboard and Shiny. In Geospatial Health Data; Chapman and Hall/CRC: Boca Raton, FL, USA, 2019. [Google Scholar]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The One Hour Yeast Proteome. Mol. Cell. Proteom. 2014, 13, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Kelstrup, C.D.; Jersie-Christensen, R.R.; Batth, T.S.; Arrey, T.N.; Kuehn, A.; Kellmann, M.; Olsen, J.V. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field orbitrap mass spectrometer. J. Proteome Res. 2014, 13, 6187–6195. [Google Scholar] [CrossRef]
- Kanawati, B.; Schmitt-Kopplin, P. Fundamentals and Applications of Fourier Transform Mass Spectrometry; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128140147. [Google Scholar]
- Zubarev, R.A. The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics 2013, 13, 723–726. [Google Scholar] [CrossRef]
- Baker, E.S.; Liu, T.; Petyuk, V.A.; Burnum-Johnson, K.E.; Ibrahim, Y.M.; Anderson, G.A.; Smith, R.D. Mass spectrometry for translational proteomics: Progress and clinical implications. Genome Med. 2012, 4, 63. [Google Scholar] [CrossRef] [Green Version]
- Rinas, A.; Jenkins, C.; Orsburn, B. Assessing a commercial capillary electrophoresis interface (ZipChip) for shotgun proteomi applications. bioRxiv 2019. [Google Scholar] [CrossRef]
- Jenkins, C.; Orsburn, B. Standard Flow Multiplexed Proteomics (SFloMPro)—An Accessible and Cost-Effective Alternative to NanoLC Workflows. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grebe, S.K.G.; Singh, R.J. Clinical peptide and protein quantification by mass spectrometry (MS). TrAC Trends Anal. Chem. 2016, 84, 131–143. [Google Scholar] [CrossRef]
- Fernández-Niño, S.M.G.; Smith-Moritz, A.M.; Chan, L.J.G.; Adams, P.D.; Heazlewood, J.L.; Petzold, C.J. Standard flow liquid chromatography for shotgun proteomics in bioenergy research. Front. Bioeng. Biotechnol. 2015, 3, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bache, N.; Geyer, P.E.; Bekker-Jensen, D.B.; Hoerning, O.; Falkenby, L.; Treit, P.V.; Doll, S.; Paron, I.; Müller, J.B.; Meier, F.; et al. A novel LC system embeds analytes in pre-formed gradients for rapid, ultra-robust proteomics. Mol. Cell. Proteom. 2018, 17, 2284–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliuk, S.; Makarov, A. Evolution of Orbitrap Mass Spectrometry Instrumentation. Annu. Rev. Anal. Chem. 2015, 8, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Jenkins, C.; Orsburn, B. BoxCar assisted MS fragmentation (BAMF). bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Scandola, S.; Uhrig, R.G. BoxCar and library-free data-independent acquisition substantially improve the depth, range, and completeness of label-free quantitative proteomics in Arabidopsis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lim, M.Y.; Paulo, J.A.; Gygi, S.P. Evaluating False Transfer Rates from the Match-between-Runs Algorithm with a Two-Proteome Model. J. Proteome Res. 2019, 18, 4020–4026. [Google Scholar] [CrossRef]
- Yu, F.; Haynes, S.E.; Nesvizhskii, A.I. Label-free quantification with FDR-controlled match-between-runs. bioRxiv 2020. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; Kelleher, N.L. Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. [Google Scholar] [CrossRef] [PubMed]
- Specht, H.; Emmott, E.; Koller, T.; Slavov, N. High-throughput single-cell proteomics quantifies the emergence of macrophage heterogeneity. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Abbatiello, S.E.; Schilling, B.; Mani, D.R.; Zimmerman, L.J.; Hall, S.C.; MacLean, B.; Albertolle, M.; Allen, S.; Burgess, M.; Cusack, M.P.; et al. Large-scale interlaboratory study to develop, analytically validate and apply highly multiplexed, quantitative peptide assays to measure cancer-relevant proteins in plasma. Mol. Cell. Proteom. 2015, 14, 2357–2374. [Google Scholar] [CrossRef] [Green Version]
- Levin, Y. The role of statistical power analysis in quantitative proteomics. Proteomics 2011, 11, 2565–2567. [Google Scholar] [CrossRef]
- Ivanov, M.V.; Bubis, J.A.; Gorshkov, V.; Tarasova, I.A.; Levitsky, L.I.; Lobas, A.A.; Solovyeva, E.M.; Pridatchenko, M.L.; Kjeldsen, F.; Gorshkov, M.V. DirectMS1: MS/MS-Free Identification of 1000 Proteins of Cellular Proteomes in 5 Minutes. Anal. Chem. 2020, 92, 4326–4333. [Google Scholar] [CrossRef] [Green Version]
- Slavov, N. Unpicking the proteome in single cells. Science 2020, 367, 512–513. [Google Scholar] [CrossRef]
- Ctortecka, C.; Mechtler, K. The rise of single-cell proteomics. Anal. Sci. Adv. 2021, 2, 84–89. [Google Scholar] [CrossRef]
- Kelly, R.; Zhu, Y.; Liang, Y.; Cong, Y.; Piehowski, P.; Dou, M.; Zhao, R.; Qian, W.-J.; Burnum-Johnson, K.; Ansong, C. Single Cell Proteome Mapping of Tissue Heterogeneity Using Microfluidic Nanodroplet Sample Processing and Ultrasensitive LC-MS. J. Biomol. Tech. 2019, 30, S61. [Google Scholar]
- Specht, H.; Harmange, G.; Perlman, D.H.; Emmott, E.; Niziolek, Z.; Budnik, B.; Slavov, N. Automated sample preparation for high-throughput single-cell proteomics. bioRxiv 2018. [Google Scholar] [CrossRef]
- Huffman, R.G.; Chen, A.; Specht, H.; Slavov, N. DO-MS: Data-Driven Optimization of Mass Spectrometry Methods. J. Proteome Res. 2019, 18, 2493–2500. [Google Scholar] [CrossRef]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. Mass-spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Stejskal, K.; Op de Beeck, J.; Dürnberger, G.; Jacobs, P.; Mechtler, K. Ultra-sensitive nanoLC-MS using second generation micro pillar array LC technology with Orbitrap Exploris 480 and FAIMS PRO to enable single cell proteomics. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cheung, T.K.; Lee, C.Y.; Bayer, F.P.; McCoy, A.; Kuster, B.; Rose, C.M. Defining the carrier proteome limit for single-cell proteomics. Nat. Methods 2021, 18, 76–83. [Google Scholar] [CrossRef]
- Leduc, A.; Huffman, R.G.; Slavov, N. Droplet sample preparation for single-cell proteomics applied to the cell cycle. bioRxiv 2021. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| File Description | Number of Proteins | Mean Log Copy Number |
|---|---|---|
| HeLa HF 2018 (23 h) [11] | 14,179 | 4.2 |
| SomaScan 1300 [19] | 1308 | 4.47 |
| QE Classic 200 ng 120 min [20] | 2016 | 5.72 |
| QE HF 200 ng 120 min [20] | 3487 | 5.4 |
| Lumos HCD-IT 60 min [21] | 4435 | 5.25 |
| Lumos HCD-IT 90 min | 4770 | 5.21 |
| Lumos HCD-IT 120 min | 5098 | 5.17 |
| Lumos HCD-IT 240 min | 5604 | 5.09 |
| Velos OT-IT 30 min (PRIDE PXD011070) | 1171 | 5.98 |
| TIMSTOF Pro pasefDDA 120 min [22] | 5970 | 5.04 |
| Exploris 480 FAIMS 21 min [23] | 3182 | 5.32 |
| pasefDIA 120 min [24] | 7699 | 4.77 |
| QE HF BoxCar 1 ug 60 min (MBR) [25] | 6479 | 5.16 |
| QE HF BoxCar 1 ug 60 min (MS/MS) [25] | 2505 | 5.77 |
| Exploris 480 Single-Cell TMT 20× Carrier [26] | 769 | 6.15 |
| Exploris 480 Single-Cell LFQ [26] | 608 | 6.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orsburn, B.C. Evaluation of the Sensitivity of Proteomics Methods Using the Absolute Copy Number of Proteins in a Single Cell as a Metric. Proteomes 2021, 9, 34. https://doi.org/10.3390/proteomes9030034
Orsburn BC. Evaluation of the Sensitivity of Proteomics Methods Using the Absolute Copy Number of Proteins in a Single Cell as a Metric. Proteomes. 2021; 9(3):34. https://doi.org/10.3390/proteomes9030034
Chicago/Turabian StyleOrsburn, Benjamin C. 2021. "Evaluation of the Sensitivity of Proteomics Methods Using the Absolute Copy Number of Proteins in a Single Cell as a Metric" Proteomes 9, no. 3: 34. https://doi.org/10.3390/proteomes9030034
APA StyleOrsburn, B. C. (2021). Evaluation of the Sensitivity of Proteomics Methods Using the Absolute Copy Number of Proteins in a Single Cell as a Metric. Proteomes, 9(3), 34. https://doi.org/10.3390/proteomes9030034